
Abstract
Tonic GABAA receptors are a subpopulation of receptors that generate long-lasting inhibition and thereby control network excitability. In recent years, these receptors have been implicated in various neurological and psychiatric disorders, including Parkinson’s disease, schizophrenia, and epilepsy. Their distinct subunit composition and function, compared to phasic GABAA receptors, opens the possibility to specifically modulate network properties. In this review, the role of tonic GABAA receptors in epilepsy and as potential antiepileptic target will be discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Worldwide, more than 50 million people are suffering from epilepsy [1]. Temporal lobe epilepsy (TLE) is the most common type of partial onset epilepsy [2]. Epilepsy has a severe impact on patients’ quality of life, first, because patients experience unpredictable seizures that restrict them in activities of daily living and second, because patients can suffer from several neuropsychiatric comorbidities such as depression or cognitive decline. Current treatment options for epilepsy are insufficient. Approximately 30 % of patients are drug resistant [3], which is defined as failure to achieve seizure freedom despite two tolerated, adequately applied antiepileptic drug (AED) schedules [4]. Furthermore, AEDs can have side effects, including somnolence, behavioral changes, dizziness, and weight gain. Therefore, there is an urgent need for more efficacious AEDs with fewer side effects.
The γ-aminobutyric acid (GABA) type A receptor (GABAAR) is an important target for AEDs, as it is the most important inhibitory receptor in the central nervous system and therefore plays an important role in the development and maintenance of epilepsy. Upon binding of the neurotransmitter GABA to the ionotropic GABAAR, the receptor opens allowing chloride and bicarbonate ions to diffuse into the cell. This results in hyperpolarization and a higher excitation threshold, i.e., an inhibitory postsynaptic potential (IPSP) [5, 6]. GABAARs mediate two different types of inhibition: phasic inhibition characterized by a short-lasting IPSP and tonic inhibition characterized by persistent, long-lasting IPSP. GABAARs mediating tonic inhibition are different from those mediating phasic inhibition. They are located outside the synapse and hence are referred to as perisynaptic or extrasynaptic receptors. Moreover, the subunit composition of tonic GABAARs differs from that of phasic GABAARs.
Due to its long-lasting hyperpolarization, tonic inhibition can be considered as a constant “brake on the system” counterbalancing excitation [7]. Tonic currents are therefore involved in a broad array of vital physiological functions, such as regulating neuronal excitability, network oscillations, synaptic plasticity, neurogenesis, neuronal development, information processing, and cognition [5, 8–14]. The importance of this type of inhibition is further emphasized when realizing that under physiological conditions, the charge carried by tonic GABA currents is bigger than that of synaptic currents. For instance, tonic inhibition is responsible for generating 75 % of the total inhibitory charge received by hippocampal neurons [15].
Considering the important role of tonic GABA signaling in regulating network excitability, alterations in tonic signaling must play a role in epilepsy. The current review discusses physiological tonic GABA signaling, its role in TLE, and describes existing and future strategies using tonic currents as novel targets for antiepileptic treatment.
Physiology of Tonic GABA Signaling
In order to discuss the pathophysiological changes taking place in epilepsy, it is first necessary to understand the physiological principals governing tonic signaling in the central nervous system. The following paragraphs will discuss the molecular composition of GABAARs, determinants influencing tonic signaling including the role of GABAB receptors (GABABRs), the specific role of tonic inhibition in interneurons, and clustering of GABAARs.
Subunit Composition of Tonic GABAARs
GABAARs are heteropentamers assembled from α1-6, β1-3, γ1-3, δ, ε, π, and θ subunits [16, 17]. In theory, these different subunits can combine into thousands of different GABAARs. In reality however, GABAARs have preferential configurations, which means that the number of different GABAARs expressed in vivo is limited. Mostly, two α subunits combine with two β subunits and either a γ or a δ subunit [18]. Some subunits are inhomogeneously distributed over the brain [19, 20]. For instance, α5 is predominantly found in the hippocampal dentate gyrus (DG) and cornu ammonis (CA) 1 and 3 region. In contrast, δ and γ subunits are found in the cortex, hippocampus, thalamus, striatum, and cerebellum. At the subcellular level, some subunits and subunit combinations are preferentially expressed at extrasynaptic sites (Table 1). For instance, α5 subunits and receptors consisting of an α4, α6, and δ subunit are predominantly expressed at extrasynaptic sites [21, 22], whereas, γ2 is mostly expressed at the synaptic site [7, 9, 21, 23–27]. To date, knowledge of the processes guiding these preferential expression sites is incomplete. In general, it is assumed that guidance is driven by the interaction between subunits, anchoring proteins and the cytoskeleton [28].
In addition to a specific distribution, the subunit composition of GABAARs also influences the affinity for GABA. For instance, receptors containing an α3 subunit have a higher affinity for GABA than α5 containing GABAARs [29–32]. Similarly, δ subunit containing GABAARs have a higher affinity than those expressing a γ subunit. This implicates that extrasynaptic GABAARs have a higher affinity for GABA than synaptic ones [5, 33]. Finally, the subunit composition can also affect the kinetic properties of GABAARs, e.g., δ subunit containing GABAARs desensitizes slower [5].
Activation of Tonic GABAARs
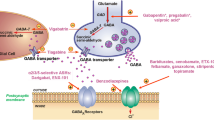
Tonic GABAARs can be activated in several ways (Fig. 1). First of all, repetitive activation of the presynapse results in increased levels of GABA in the synaptic cleft. This is due to insufficient time for clearance by GABA transporters (GATs). As a consequence, GABA can accumulate in the synaptic cleft. Consequently, GABA diffuses to the extracellular space where it activates extrasynaptic receptors [21, 34, 35]. The degree and speed of diffusion are influenced by parameters such as the distance between the synaptic cleft and receptor, diffusional barriers, and geometry of the synapse [36].

Mechanisms modulating the activity of tonic GABAARs. Upon release into the synaptic cleft, GABA is taken up in glia cells by GABA transporter (GAT) type 3 and metabolized by GABA transaminase or taken up presynaptically by GAT-1. Tonic GABAARs can be activated by four mechanisms: (1) spill of synaptically released GABA into the extrasynaptic space due to insufficient clearance by GATs, (2) non-vesicular GABA release by GAT reversal, (3) spontaneous opening in the absence of extracellular GABA, and (4) GABAB receptor activation increases tonic GABAAR signaling via an intracellular mechanism
Second, tonic GABAARs may be activated by GABA that is released from non-vesicular sources [37, 38]. Extracellular GABA may come from glial cells or dendrites (for a review, see Yoon and Lee [39]) [40–44], for instance by GAT reversal [45–51].
Third, opening of tonic GABAARs can occur spontaneously in the absence of synaptic activation and even in the absence of extracellular GABA [52, 53]. This has been shown in vitro for β1 subunit containing GABAARs in the rat [54], the human α1β3ε receptor [55], human α1β1 and α1β1γ2 receptors [56], and in rat hippocampal slices [57].
The three proposed mechanisms of tonic GABAAR activation might be complementary: In case of baseline network activity, tonic activation may be caused by non-vesicular sources of extracellular GABA or by spontaneous opening, while in case of excessive network activity, GABA may spill into the extracellular space.
The magnitude of tonic signaling is influenced by several factors, such as the amount of ambient GABA and the availability of receptors. Moreover, neuromodulators can alter tonic GABA signaling. An example of neuromodulators are neurosteroids, a group of neuromodulators that influences δ subunit containing tonic GABAARs. Binding of these neurosteroids increases tonic inhibition. Next to this acute effect, neurosteroids also cause downregulation of certain subunits such as the γ2 and δ subunits in the DG [58]. Consequently, tonic and phasic currents decreased [59].
The mechanisms governing tonic inhibition are summarized and illustrated in Fig. 1.
The Influence of GABAB Receptors on Tonic GABA Signaling
Metabotropic GABABRs are expressed both, presynaptically and postsynaptically, and are known to modulate seizure activity. At the presynapse, they act as autoreceptors, i.e., activation of this receptor causes a reduced Ca2+ entry, resulting in a decreased GABA release. At the postsynaptic site, GABABRs can modulate tonic signaling. For instance, activation of GABABRs by baclofen increases the tonic current of δ containing GABAARs in thalamocortical cells, DG, and cerebellar granule cells [60]. The mechanisms by which GABAB signaling controls tonic GABAAR currents are not fully understood. It is hypothesized that activation of postsynaptic GABABRs initiates a G-protein coupled signal transduction to δ containing GABAARs (for review, see [61]). The contribution of this pathway versus that of autoreceptors in the proconvulsive or anticonvulsive activity of GABABR-modulating drugs remains to be elucidated.
The spatial expression of GABABRs at the postsynaptic membrane shares striking resemblance with that of extrasynaptic or perisynaptic GABAARs. Most likely GABAARs and GABABRs are activated simultaneously due to spillover of GABA [34, 62, 63]. Furthermore, GABABRs are found on astrocytes. As these cells have an essential buffering capacity, glial GABABRs may be important in regulating the concentration of extracellular GABA [64, 65].
Clustering and Trafficking of GABAAR
The expression of GABAARs is tightly controlled and depends on assembly, maturation, and recycling of different subunits. These processes are regulated by a complex interaction of various proteins such as GABAAR-associated protein (GABARAP) and N-ethylmaleimide-sensitive factor (for a review, see Lorena Arancibia-Cárcamo 2009). Normally, GABAARs are inserted into the cell membrane at the extrasynaptic location and diffuse via lateral trafficking into the postsynaptic density (PSD; Fig. 2). Here, they are clustered by different adhesion and scaffolding proteins, i.e., gephryin, dystrophin, and neurexin [66]. GABAARs can also be clustered at the extrasynaptic site by molecules such as radixin [67, 68]. Receptors are able to migrate back and forth between the synaptic and extrasynaptic site [69, 70] and can rapidly cycle back after endocytosis to the extrasynaptic membrane [28]. It is unclear which mechanisms cause receptors to diffuse away from the synaptic site or facilitate their entry into the PSD.

Receptor trafficking to the membrane and between the extrasynaptic and synaptic site. Additionally, examples of extrasynaptic and synaptic anchoring proteins are shown
An increase in extrasynaptic clustering influences tonic signaling. Surprisingly, not only the amount of channels but also individual channel properties change with increased clustering [71, 72]. Clustered GABAARs have a higher EC50, deactivate faster, and desensitize slower compared to diffuse receptors [72]. The heterogeneity of receptors and preference of certain subunits to cluster extrasynaptically complicate the understanding of this mechanism [71].
Tonic Inhibition in Interneurons
The activity of interneurons paces the hippocampal rhythm. This rhythmogenesis is therefore partly controlled by the tonic GABA conductance in interneurons. This is demonstrated by the finding that δ subunit knockout mice show higher frequency γ oscillations [11]. However, interneurons show a bivalent response to ambient GABA. At low levels of GABA, tonic inhibition is reduced, causing an enhanced excitability of interneurons [24, 73] and consequently an increased inhibition of pyramidal cells. Moreover, this weak tonic conductance imposes regular firing of interneurons that synchronizes the CA 3 network [74]. On the other hand, high concentrations of ambient GABA inhibit interneurons [75]. As a result, they become less excitable and thus release their brake on pyramidal cells. However, under these high extracellular GABA levels, the pyramidal cells themselves also receive more tonic inhibition. This inhibition can counterbalance the loss of interneuronal inhibition. Thus, control of the extracellular GABA concentration provides both a direct and indirect mechanism to regulate pyramidal cell activity [73].
Tonic GABA Signaling in Temporal Lobe Epilepsy
Considering the important role of tonic GABA signaling in regulating network excitability, it is likely that tonic GABA signaling is altered in the occurrence of seizures or in the development of a seizure prone network (epileptogenesis) [76, 77]. Studies on tonic GABA signaling in epilepsy will be discussed in the following paragraph and are summarized in Table 2.
Single nucleotide polymorphisms and mutations in genes coding for tonic subunits are associated with several types of epilepsy [78–80]. For instance, Dibbens et al. [78] and Feng et al. [81] showed that genetic alterations in the GABRD gene, that codes for the tonic δ subunit, cause a decrease in tonic inhibition in complex idiopathic generalized epilepsies. Eugene et al. [80] found that in human epileptic syndromes with febrile seizures, mutations in the γ subunit cause a decrease in tonic currents by reducing the surface expression of α5 containing GABAARs.
Results on the expression levels of tonic subunits in experimental epilepsy are contradictory (for a full overview, see Table 2). Whereas several studies showed an increase of the α5 subunit [82–84] particularly in the DG in the kainate and pilocarpine model, others showed a decrease in CA1 in the pilocarpine model [84], in the DG in the kainate model [85] in CA1, CA2, and CA3 in the pilocarpine model [83], and in the DG, CA1, and CA3 in the hippocampal kindling model and the pilocarpine model [86–88]. The total and surface expression of the δ subunit in the DG of the hippocampus decreases after status epilepticus, both during the latent phase shortly after the induction of a status epilepticus and during the chronic phase, when animals experience spontaneous recurrent seizures [89–91]. As the expression of the δ subunit is concomitantly increased in the microsomal fraction, these results suggest that these subunits do not reach the cell membrane and are retained in the endoplasmatic reticiulum. All together, is seems that there is a quantitative decrease in the amount of tonic subunit expression at the messenger RNA (mRNA) and protein level in different hippocampal regions acutely after an epileptogenic insult but also during the chronic phase.
Nonetheless, electrophysiological studies have shown that alteration in the expression of tonic subunits is not accompanied by a functional loss of tonic inhibition [89, 91–93]. Some studies even report an increase of tonic signaling in experimental epilepsy shortly after status epilepticus and during the epileptogenic phase in the pilocarpine model [88, 94]. If, as animal studies suggest, subunits providing tonic currents are downregulated and tonic inhibition is unchanged or increased, then tonic GABA currents must be maintained by other means. What factors are possibly involved in maintaining tonic currents?
As the amplitude of tonic GABA signaling is determined by the concentration of GABA in the synaptic cleft, an increased GABA concentration could compensate for a decrease in the quantity of tonic receptor subunits. As discussed before, increased extracellular GABA concentrations can result from a reduced activity or number of GATs. Indeed, GAT-1, which is expressed presynaptically, was shown to be upregulated in the molecular layer of the hippocampus acutely after experimental status epilepticus, in the chronic phase in the kainic acid model [95] and in hippocampal specimen from patients with temporal lobe epilepsy [96]. Andre et al. [97] have shown that GAT-1 alterations depend on the time point in the epileptogenic process and the region investigated. Whereas GAT-1 was upregulated in the inner molecular layer of the hippocampus, it was downregulated in CA1 in rats with spontaneous, recurrent seizures compared to controls. Another important regulator of extracellular GABA is GAT-3, which is located on glial processes. The expression of both GAT-1 and GAT-3 are altered in hippocampi from TLE patients. As these changes vary per hippocampal subregion and GATs can reverse, causing non-vesicular GABA release, it is difficult to predict the effect of these alterations to hippocampal physiology [63, 95].
Whether an actual functional reversal of transporters occurs in epilepsy is difficult to establish due to the technical restrictions of measuring intracellular GABA concentrations. GAT reversal is eventually favored by an increased rate of GABA synthesis by GAD [95] that in its turn can be caused by an increase in metabolic rate of neurons as seen in epilepsy. Another possibility of increasing extracellular GABA is by increased activity of glutamic acid decarboxylase (GAD), the enzyme responsible for GABA synthesis. Esclapez and Houser [98] have shown that GAD 65 and 67 are increased in animals with spontaneous, recurrent epileptic activity at the mRNA and protein level. Acutely after status epilepticus, GAD 65 and 67 expression decreased in the hippocampal hilus, whereas GAD 67 expression in the DG is increased [99]. In the chronic phase, GAD accumulated in interneurons of the DG [82, 100]. Interestingly, GAD 65 null mice show spontaneous seizures [101].
Additionally, different channels (e.g., two-pore domain potassium channels) and receptors or GABAARs with a different subunit composition could take over tonic function [74, 102]. Indeed, pharmacological experiments demonstrated that increased tonic GABA currents are not mediated by α5- but δ-containing GABAARs in epilepsy [88]. In several studies, it has been shown that the downregulation of δ subunits in the DG is accompanied with an increase in the expression of receptors containing the α4 and γ2 subunits [86, 89, 90, 103]. As mentioned before, these receptors have a lower affinity for GABA, which implies that tonic GABA currents can only be maintained if there is an increase in extracellular GABA. Possibly, receptors that are typically expressed synaptically under physiological conditions are increasing shifted toward the extrasynaptic site in epilepsy. Evidence for this process comes from the shift of the γ2 subunit that is increasingly expressed at the extrasynaptic site in a murine model of epilepsy [104]. Additional electrophysiological studies are necessary to fully understand the functional consequences of quantitative changes in GABAARs.
Toward Antiepileptic Drugs that Target Tonic GABA Signaling
Considering that tonic currents are functionally preserved in epilepsy, they constitute a potential treatment target [5, 15]. In order to increase tonic currents, there are two strategies: increasing the extracellular concentration of GABA or agonizing GABAARs by ligands binding to tonic subunits. In Fig. 1, the mechanisms modulating tonic signaling are displayed. These mechanisms will be discussed in the light of increasing tonic inhibition in the next paragraph.
Increasing the Extracellular GABA Concentration
Extracellular GABA concentrations are controlled by synthesis, breakdown, and clearance from the synaptic cleft. The synthesis of GABA in the presynaptic terminal is determined by the activity of GAD 65, and the activity of single GAD is influenced by processes such as phosphorylation and the presence of co-factors such as pyridoxal 5′-phosphate. GAD concentrations can be increased by enhancing its transcription by stimulating promoter activity or by influencing epigenetic mechanisms that lead to a higher transcription rate [89, 105]. Alternatively, GAD can be potentiated by viral vector-mediated targeted delivery of therapeutic GAD. This strategy has already proven to be successful in experimental and clinical studies for Parkinson’s disease [106–108]. An advantage of viral vector-mediated targeted delivery therapy is its spatial specificity that potentially eliminates site effects caused by unspecific targeting.
Alternatively, a higher release of GABA could be achieved by increasing the concentration of GABA in individual presynaptic vesicles. GABA is packed into vesicles by vesicular neurotransmitter transporters (VGAT), and their activity depends on electrochemical components. Boosting vesicular GABA transport might be achieved by pharmacologically increasing the activity of VGAT or by increasing its gene expression.
A second mechanism to increase extracellular GABA concentrations is inhibiting breakdown of GABA that is mediated by GABA transaminase (GABA-T). The second-line AED vigabatrin works via irreversibly inhibiting GABA-T [109, 110], increasing the concentration GABA in the synaptic cleft [111]. Furthermore, there is evidence that vigabatrin increases GABA release and constrains glial GABA uptake [112, 113]. Other more potent and novel inhibitors of GABA-T are ethanolamine-O-sulfate (EOS), L-cycloserine, and phenylethylidenehydrazine. Interestingly, the latter is used in various psychiatric disorders and increase extracellular GABA concentrations in vivo and decrease epileptiform activity in the rat ex vivo [114]. However, its anticonvulsant properties in vivo varied depending on the seizure model used. Several other GABA-T inhibitors such as gabaculine, gamma-acetylenic GABA, and gamma-vinyl GABA do not appear suitable for treatment of convulsive disorders in humans due to their severe and sometimes lethal side effects [115].
The last mechanism to increase extracellular GABA concentrations is by decreasing GABA clearance from the synaptic cleft by influencing GAT. Two drugs acting at GATs are SNAP-5114 and tiagabine that inhibit GAT-2 and -3, and GAT-1, respectively [116]. Indeed, they increase the concentration of extracellular GABA in vivo [117]. Tiagabine seems to be efficient as an add-on treatment in partial and secondarily generalized seizures, reducing seizure frequency [118]. However, clinical data show disappointing results with patients suffering from paradoxical proconvulsive effects [119]. So far, no pharmaceutical interventions exits that acts by reversing GATs.
Extrasynaptic GABAAR Agonists
In addition to enhancing extracellular GABA, enhancement of tonic GABAARs can also be achieved by using specific agonists. Ganaxolone is a neurosteroid and a tonic GABAAR agonist, binding to and influencing the δ subunit. At low concentrations, it potentiates GABAARs (positive allosteric modulator), while at higher doses, it acts by directly binding to the δ subunit. Several studies using ganaxolone show promising results, both in rodent seizure models [120–122] as well as in clinical studies [123]. In patients with infantile spasms, seizure frequency was decreased by at least 50 % in one third of the patients [124]. The frequency of occurrence and type of side effects were comparable to classic antiepileptic drugs. Currently, ganoxalone is investigated as an adjunctive treatment in patients with drug-resistant, partial onset seizures (trial identifier NCT01963208).
Also, the anesthetic 4,5,6,7-tetrahydroisoxazolo(5,4-c)pyridin-3-ol (THIP, gaboxadol) agonizes the δ subunit. THIP can reduce spikes in vitro and in vivo [125, 126]. In a clinical study with a small sample size, a trend toward seizure reduction was demonstrated [127]. However, there is conflicting evidence showing no effect on epileptiform activity [128, 129]. Other anesthetics, which selectively target the δ subunit are alphaxalone and propofol. Due to its anesthetic effects, these drugs are not suitable for the treatment of epilepsy.
A relatively new compound binding selectively to the δ subunit is the GABA agonist DS-1 and an enhancer DS-2. DS-2 has been shown to increase tonic currents in the thalamic neurons in vitro [130].
Challenges of Enhancing Tonic Signaling
The development of AEDs aimed at tonic receptors is not straightforward due to several reasons. First of all, the long-term use of tonic GABAAR agonists, in particular neurosteroids, can lead to a downregulation of α4 and δ subunits. The elimination of the binding site by downregulation ceases the potential long-term benefits of this strategy. Furthermore, AEDs can have side effects. Side effects depend on the concentration of the compound and the distribution in the central nervous system. However, one advantage of extrasynaptic GABAARs is their cell type and brain region-specific subunit composition. The advantage of this heterogeneity of receptors is that all subunits differ in their affinity for agonists and antagonists. The aspecificity of drugs can therefore be partially overcome by using different dosages of AEDs. For instance, δ subunit containing receptors have the highest affinity for GABA.
Furthermore, compensatory alterations in other cells or brain regions could be a cause of ineffectiveness of AEDs. For instance, increasing GABA concentrations through inhibition of GAT-3 is not effective in the hippocampus due to the compensatory action of GAT-1. In the thalamus, however, GAT-3 inhibition increases tonic signaling. Related to these region-specific effects is the fact that certain types of epilepsy appear to be more suitable targets for tonic GABA modulation. For instance, in the absence of seizures, tonic inhibition in the thalamus is upregulated rather than downregulated [76]. Enhancing tonic signaling triggers slow wave discharges—a hallmark of this type of epilepsy—and consequently aggravates seizures [131]. In this light, it becomes plausible that an increase in tonic inhibition particularly in the thalamus causes absence-like side effects.
It is important to realize that an increase in extracellular GABA has consequences for synaptic receptors and receptors on which GABA acts as a (partial) ligand (i.e., GABABRs). In this regard, it might be sometimes difficult to disentangle whether the specific increase in tonic currents or a gross enhancement in the inhibitory tone causes antiepileptiform effects.
Future Directions for the Development of Tonic AEDs
To overcome the challenges described above, it is necessary to reveal the complex network effects of tonic signaling and develop more specific and targeted drugs. By targeting specific subunits, one could make use of the particular pharmacokinetic properties such as affinity, channel opening time, refractory period, etc., which would allow fine-tuning of neuronal activity in the presence of varying concentrations of antagonist or agonists [23, 24, 88, 132].
Potential new specific strategies are evolving. An example of a cell-type specific therapy is optogenetics [133]. With optogenetics, certain genetically manipulated neurons are inhibited or excited by light. If one succeeds in selectively inhibiting the principal neuron only in the epileptogenic zone or increasing the activity of interneurons, this would potentially create new treatment opportunities. In the case of epilepsy, it could be beneficial to increase the release of GABA and therefore increase ambient GABA concentrations, which, in turn, can activate extrasynaptic receptors. Considering the important buffering capacity of astroglia cells, those might also form an attractive target with regard to regulating the concentration of extracellular GABA.
Additionally, by using cell-specific approaches, side effects could be limited. Also, designer receptors exclusively activated by designer drugs (DREADDs) might evolve as future treatment strategy [134]. With DREADDs, receptors are administered to the central nervous system via a viral vector into specific cell types. A specific oral drug activates these receptors. In fact, in epilepsy, there are two options with regard to DREADDs [135]. On one hand, inserting the receptor into the presynaptic gabaergic terminal and activating it by the oral drug could increase presynaptic GABA release and therefore augment not only tonic but also phasic inhibition. On the other hand, the precise activation of certain interneuron populations in the epileptogenic zone might contribute to a higher excitation threshold of the principal neuron, which, in turn, might be beneficial at the network level. The advantage of this procedure is the specificity of the treatment and fact that it is less invasive compared to optogenetic techniques.
Another strategy might be to interfere with receptor trafficking. A potential approach would be to enhance tonic signaling by increasing receptor expression and anchoring. This could be achieved by stimulating de novo synthesis or by promoting the migration of receptors from the synaptic to the extrasynaptic site. A potential consequence might be the loss of synaptic receptors, which, in turn, leads to the loss of phasic inhibition, which is detrimental to epilepsy. Next to increasing de novo synthesis, shuttling of ready-made receptors to the cell surface could be enhanced.
Lastly, it might be desirable to improve anchoring at the extrasynaptic side. In this regard, anchoring proteins such as radixin might play a crucial role. Whereas disrupting certain protein-protein interactions becomes more feasible by using virus-derived proteins [136, 137], enhancing these interactions is more difficult.
Conclusion
Tonic inhibition is maintained in epilepsy and therefore serves as an attractive substrate for interventions. Drugs aimed at tonic GABA signaling, such as ganoxalone, show promising anticonvulsive results in rodent studies, but they trigger side effects as well. Future detailed knowledge about receptor trafficking and particular changes in specific brain regions will contribute to more rational drug design leading to more potent drugs with fewer side effects.
Abbreviations
- AED:
-
Antiepileptic drug
- CA:
-
Cornu ammonis region
- DG:
-
Hippocampal dentate gyrus
- GABARAP:
-
γ-Aminobutyric type A receptor-associated protein
- GABAAR:
-
γ-Aminobutyric type A receptor
- IPSP:
-
Inhibitory postsynaptic potential
- PSD:
-
Postsynaptic density
- TLE:
-
Temporal lobe epilepsy
References
World Health Organization (2012) Epilepsy Fact Sheet http://www.who.int/mediacentre/factsheets/fs999/en/. Accessed 30th november 2014
Wiebe S (2000) Epidemiology of temporal lobe epilepsy. Can J Neurol Sci Le J Can Sci Neurol 27(1):S6–10
Kwan P, Brodie MJ (2000) Early identification of refractory epilepsy. N Engl J Med 342(5):314–319. doi:10.1056/NEJM200002033420503
Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, Moshe SL, Perucca E et al (2010) Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 51(6):1069–1077. doi:10.1111/j.1528-1167.2009.02397.x
Farrant M, Nusser Z (2005) Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci 6(3):215–229. doi:10.1038/nrn1625
Glykys J, Mody I (2007) Activation of GABAA receptors: views from outside the synaptic cleft. Neuron 56(5):763–770. doi:10.1016/j.neuron.2007.11.002
Caraiscos VB, Elliott EM, You-Ten KE, Cheng VY, Belelli D, Newell JG, Jackson MF, Lambert JJ et al (2004) Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by alpha5 subunit-containing gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A 101(10):3662–3667. doi:10.1073/pnas.0307231101
Semyanov A, Walker MC, Kullmann DM, Silver RA (2004) Tonically active GABA A receptors: modulating gain and maintaining the tone. Trends Neurosci 27(5):262–269. doi:10.1016/j.tins.2004.03.005
Pavlov I, Savtchenko LP, Kullmann DM, Semyanov A, Walker MC (2009) Outwardly rectifying tonically active GABAA receptors in pyramidal cells modulate neuronal offset, not gain. J Neurosci Off J Soc Neurosci 29(48):15341–15350. doi:10.1523/JNEUROSCI.2747-09.2009
Holter NI, Zylla MM, Zuber N, Bruehl C, Draguhn A (2010) Tonic GABAergic control of mouse dentate granule cells during postnatal development. Eur J Neurosci 32(8):1300–1309. doi:10.1111/j.1460-9568.2010.07331.x
Mann EO, Mody I (2010) Control of hippocampal gamma oscillation frequency by tonic inhibition and excitation of interneurons. Nat Neurosci 13(2):205–212. doi:10.1038/nn.2464
Martin LJ, Zurek AA, MacDonald JF, Roder JC, Jackson MF, Orser BA (2010) Alpha5GABAA receptor activity sets the threshold for long-term potentiation and constrains hippocampus-dependent memory. J Neurosci Off J Soc Neurosci 30(15):5269–5282. doi:10.1523/JNEUROSCI.4209-09.2010
Duveau V, Laustela S, Barth L, Gianolini F, Vogt KE, Keist R, Chandra D, Homanics GE et al (2011) Spatiotemporal specificity of GABAA receptor-mediated regulation of adult hippocampal neurogenesis. Eur J Neurosci 34(3):362–373. doi:10.1111/j.1460-9568.2011.07782.x
Ge S, Goh EL, Sailor KA, Kitabatake Y, Ming GL, Song H (2006) GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 439(7076):589–593. doi:10.1038/nature04404
Mody I, Pearce RA (2004) Diversity of inhibitory neurotransmission through GABA(A) receptors. Trends Neurosci 27(9):569–575. doi:10.1016/j.tins.2004.07.002
Luscher B, Keller CA (2004) Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol Ther 102(3):195–221. doi:10.1016/j.pharmthera.2004.04.003
Olsen RW, Sieghart W (2008) International Union of Pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev 60(3):243–260. doi:10.1124/pr.108.00505
Rudolph U, Mohler H (2004) Analysis of GABAA receptor function and dissection of the pharmacology of benzodiazepines and general anesthetics through mouse genetics. Annu Rev Pharmacol Toxicol 44:475–498. doi:10.1146/annurev.pharmtox.44.101802.121429
Lee V, Maguire J (2014) The impact of tonic GABAA receptor-mediated inhibition on neuronal excitability varies across brain region and cell type. Front Neural Circ 8:3. doi:10.3389/fncir.2014.00003
Sieghart W, Sperk G (2002) Subunit composition, distribution and function of GABA(A) receptor subtypes. Curr Top Med Chem 2(8):795–816
Wei W, Zhang N, Peng Z, Houser CR, Mody I (2003) Perisynaptic localization of delta subunit-containing GABA(A) receptors and their activation by GABA spillover in the mouse dentate gyrus. J Neurosci: Off J Soc Neurosci 23(33):10650–10661
Nusser Z, Sieghart W, Somogyi P (1998) Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci: Off J Soc Neurosci 18(5):1693–1703
Glykys J, Mody I (2006) Hippocampal network hyperactivity after selective reduction of tonic inhibition in GABA A receptor alpha5 subunit-deficient mice. J Neurophysiol 95(5):2796–2807. doi:10.1152/jn.01122.2005
Semyanov A, Walker MC, Kullmann DM (2003) GABA uptake regulates cortical excitability via cell type-specific tonic inhibition. Nat Neurosci 6(5):484–490. doi:10.1038/nn1043
Stell BM, Brickley SG, Tang CY, Farrant M, Mody I (2003) Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by delta subunit-containing GABAA receptors. Proc Natl Acad Sci U S A 100(24):14439–14444. doi:10.1073/pnas.2435457100
Glykys J, Mann EO, Mody I (2008) Which GABA(A) receptor subunits are necessary for tonic inhibition in the hippocampus? J Neurosci: Off J Soc Neurosci 28(6):1421–1426. doi:10.1523/JNEUROSCI.4751-07.2008
Serwanski DR, Miralles CP, Christie SB, Mehta AK, Li X, De Blas AL (2006) Synaptic and nonsynaptic localization of GABAA receptors containing the alpha5 subunit in the rat brain. J Comp Neurol 499(3):458–470. doi:10.1002/cne.21115
Michels G, Moss SJ (2007) GABAA receptors: properties and trafficking. Crit Rev Biochem Mol Biol 42(1):3–14. doi:10.1080/10409230601146219
Knoflach F, Benke D, Wang Y, Scheurer L, Luddens H, Hamilton BJ, Carter DB, Mohler H et al (1996) Pharmacological modulation of the diazepam-insensitive recombinant gamma-aminobutyric acidA receptors alpha 4 beta 2 gamma 2 and alpha 6 beta 2 gamma 2. Mol Pharmacol 50(5):1253–1261
Fisher JL, Macdonald RL (1997) Single channel properties of recombinant GABAA receptors containing gamma 2 or delta subtypes expressed with alpha 1 and beta 3 subtypes in mouse L929 cells. J Physiol 505(Pt 2):283–297
Bohme I, Rabe H, Luddens H (2004) Four amino acids in the alpha subunits determine the gamma-aminobutyric acid sensitivities of GABAA receptor subtypes. J Biol Chem 279(34):35193–35200. doi:10.1074/jbc.M405653200
Minier F, Sigel E (2004) Positioning of the alpha-subunit isoforms confers a functional signature to gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A 101(20):7769–7774. doi:10.1073/pnas.0400220101
Brown N, Kerby J, Bonnert TP, Whiting PJ, Wafford KA (2002) Pharmacological characterization of a novel cell line expressing human alpha(4)beta(3)delta GABA(A) receptors. Br J Pharmacol 136(7):965–974. doi:10.1038/sj.bjp.0704795
Rossi DJ, Hamann M (1998) Spillover-mediated transmission at inhibitory synapses promoted by high affinity alpha6 subunit GABA(A) receptors and glomerular geometry. Neuron 20(4):783–795
Otis TS, Staley KJ, Mody I (1991) Perpetual inhibitory activity in mammalian brain slices generated by spontaneous GABA release. Brain Res 545(1–2):142–150
Barbour B, Hausser M (1997) Intersynaptic diffusion of neurotransmitter. Trends Neurosci 20(9):377–384
Wall MJ, Usowicz MM (1997) Development of action potential-dependent and independent spontaneous GABAA receptor-mediated currents in granule cells of postnatal rat cerebellum. Eur J Neurosci 9(3):533–548
Rossi DJ, Hamann M, Attwell D (2003) Multiple modes of GABAergic inhibition of rat cerebellar granule cells. J Physiol 548(Pt 1):97–110. doi:10.1113/jphysiol.2002.036459
Yoon BE, Lee CJ (2014) GABA as a rising gliotransmitter. Frontiers Neural Circ 8:141. doi:10.3389/fncir.2014.00141
Zilberter Y, Kaiser KM, Sakmann B (1999) Dendritic GABA release depresses excitatory transmission between layer 2/3 pyramidal and bitufted neurons in rat neocortex. Neuron 24(4):979–988
Le Meur K, Mendizabal-Zubiaga J, Grandes P, Audinat E (2012) GABA release by hippocampal astrocytes. Front Comput Neurosci 6:59. doi:10.3389/fncom.2012.00059
Kozlov AS, Angulo MC, Audinat E, Charpak S (2006) Target cell-specific modulation of neuronal activity by astrocytes. Proc Natl Acad Sci U S A 103(26):10058–10063. doi:10.1073/pnas.0603741103
Jimenez-Gonzalez C, Pirttimaki T, Cope DW, Parri HR (2011) Non-neuronal, slow GABA signalling in the ventrobasal thalamus targets delta-subunit-containing GABA(A) receptors. Eur J Neurosci 33(8):1471–1482. doi:10.1111/j.1460-9568.2011.07645.x
Lee S, Yoon BE, Berglund K, Oh SJ, Park H, Shin HS, Augustine GJ, Lee CJ (2010) Channel-mediated tonic GABA release from glia. Science 330(6005):790–796. doi:10.1126/science.1184334
Attwell D, Barbour B, Szatkowski M (1993) Nonvesicular release of neurotransmitter. Neuron 11(3):401–407
Cammack JN, Rakhilin SV, Schwartz EA (1994) A GABA transporter operates asymmetrically and with variable stoichiometry. Neuron 13(4):949–960
Levi G, Raiteri M (1993) Carrier-mediated release of neurotransmitters. Trends Neurosci 16(10):415–419
Lu CC, Hilgemann DW (1999) GAT1 (GABA:Na+:Cl-) cotransport function. Kinetic studies in giant Xenopus oocyte membrane patches. J Gen Physiol 114(3):445–457
O’Malley DM, Sandell JH, Masland RH (1992) Co-release of acetylcholine and GABA by the starburst amacrine cells. J Neurosci: Off J Soc Neurosci 12(4):1394–1408
Pin JP, Bockaert J (1989) Two distinct mechanisms, differentially affected by excitatory amino acids, trigger GABA release from fetal mouse striatal neurons in primary culture. J Neurosci: Off J Soc Neurosci 9(2):648–656
Schwartz EA (1987) Depolarization without calcium can release gamma-aminobutyric acid from a retinal neuron. Science 238(4825):350–355
Wlodarczyk AI, Sylantyev S, Herd MB, Kersante F, Lambert JJ, Rusakov DA, Linthorst AC, Semyanov A et al (2013) GABA-independent GABAA receptor openings maintain tonic currents. J Neurosci: Off J Soc Neurosci 33(9):3905–3914. doi:10.1523/JNEUROSCI.4193-12.2013
McCartney MR, Deeb TZ, Henderson TN, Hales TG (2007) Tonically active GABAA receptors in hippocampal pyramidal neurons exhibit constitutive GABA-independent gating. Mol Pharmacol 71(2):539–548. doi:10.1124/mol.106.028597
Sigel E, Baur R, Malherbe P, Mohler H (1989) The rat beta 1-subunit of the GABAA receptor forms a picrotoxin-sensitive anion channel open in the absence of GABA. FEBS Lett 257(2):377–379
Maksay G, Thompson SA, Wafford KA (2003) The pharmacology of spontaneously open alpha 1 beta 3 epsilon GABA A receptor-ionophores. Neuropharmacology 44(8):994–1002
Lindquist CE, Dalziel JE, Cromer BA, Birnir B (2004) Penicillin blocks human alpha 1 beta 1 and alpha 1 beta 1 gamma 2S GABAA channels that open spontaneously. Eur J Pharmacol 496(1–3):23–32. doi:10.1016/j.ejphar.2004.06.004
Birnir B, Everitt AB, Lim MS, Gage PW (2000) Spontaneously opening GABA(A) channels in CA1 pyramidal neurones of rat hippocampus. J Membrane Biol 174(1):21–29
Maguire J, Mody I (2008) GABA(A)R plasticity during pregnancy: relevance to postpartum depression. Neuron 59(2):207–213. doi:10.1016/j.neuron.2008.06.019
Maguire J, Mody I (2009) Steroid hormone fluctuations and GABA(A)R plasticity. Psychoneuroendocrinology 34(Suppl 1):S84–90. doi:10.1016/j.psyneuen.2009.06.019
Tao W, Higgs MH, Spain WJ, Ransom CB (2013) Postsynaptic GABAB receptors enhance extrasynaptic GABAA receptor function in dentate gyrus granule cells. J Neurosci Off J Soc Neurosci 33(9):3738–3743. doi:10.1523/JNEUROSCI.4829-12.2013
Connelly WM, Fyson SJ, Errington AC, McCafferty CP, Cope DW, Di Giovanni G, Crunelli V (2013) GABAB receptors regulate extrasynaptic GABAA receptors. J Neurosci Off J Soc Neurosci 33(9):3780–3785. doi:10.1523/JNEUROSCI.4989-12.2013
Scanziani M, Gahwiler BH, Thompson SM (1991) Paroxysmal inhibitory potentials mediated by GABAB receptors in partially disinhibited rat hippocampal slice cultures. J Physiol 444:375–396
During MJ, Spencer DD (1993) Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 341(8861):1607–1610
Barbaresi P (2007) Cellular and subcellular localization of the GABA(B) receptor 1a/b subunit in the rat periaqueductal gray matter. J Comp Neurol 505(5):478–492. doi:10.1002/cne.21509
Andersson M, Blomstrand F, Hanse E (2007) Astrocytes play a critical role in transient heterosynaptic depression in the rat hippocampal CA1 region. J Physiol 585(Pt 3):843–852. doi:10.1113/jphysiol.2007.142737
Tretter V, Mukherjee J, Maric HM, Schindelin H, Sieghart W, Moss SJ (2012) Gephyrin, the enigmatic organizer at GABAergic synapses. Front Cell Neurosci 6:23. doi:10.3389/fncel.2012.00023
Jacob TC, Moss SJ, Jurd R (2008) GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci 9(5):331–343. doi:10.1038/nrn2370
Loebrich S, Bahring R, Katsuno T, Tsukita S, Kneussel M (2006) Activated radixin is essential for GABAA receptor alpha5 subunit anchoring at the actin cytoskeleton. EMBO J 25(5):987–999. doi:10.1038/sj.emboj.7600995
Thomas P, Mortensen M, Hosie AM, Smart TG (2005) Dynamic mobility of functional GABAA receptors at inhibitory synapses. Nat Neurosci 8(7):889–897. doi:10.1038/nn1483
Bogdanov Y, Michels G, Armstrong-Gold C, Haydon PG, Lindstrom J, Pangalos M, Moss SJ (2006) Synaptic GABAA receptors are directly recruited from their extrasynaptic counterparts. EMBO J 25(18):4381–4389. doi:10.1038/sj.emboj.7601309
Petrini EM, Marchionni I, Zacchi P, Sieghart W, Cherubini E (2004) Clustering of extrasynaptic GABA(A) receptors modulates tonic inhibition in cultured hippocampal neurons. J Biol Chem 279(44):45833–45843. doi:10.1074/jbc.M407229200
Chen L, Wang H, Vicini S, Olsen RW (2000) The gamma-aminobutyric acid type A (GABAA) receptor-associated protein (GABARAP) promotes GABAA receptor clustering and modulates the channel kinetics. Proc Natl Acad Sci U S A 97(21):11557–11562. doi:10.1073/pnas.190133497
Semyanov A (2003) Cell type specificity of GABA(A) receptor mediated signaling in the hippocampus. Curr Drug Targets CNS Neurol Disord 2(4):240–247
Brickley SG, Cull-Candy SG, Farrant M (1996) Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J Physiol 497(Pt 3):753–759
Song I, Savtchenko L, Semyanov A (2011) Tonic excitation or inhibition is set by GABA(A) conductance in hippocampal interneurons. Nat Commun 2:376. doi:10.1038/ncomms1377
Cope DW, Di Giovanni G, Fyson SJ, Orban G, Errington AC, Lorincz ML, Gould TM, Carter DA et al (2009) Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med 15(12):1392–1398. doi:10.1038/nm.2058
Clarkson AN, Huang BS, Macisaac SE, Mody I, Carmichael ST (2010) Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 468(7321):305–309. doi:10.1038/nature09511
Dibbens LM, Feng HJ, Richards MC, Harkin LA, Hodgson BL, Scott D, Jenkins M, Petrou S et al (2004) GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet 13(13):1315–1319. doi:10.1093/hmg/ddh146
Feng HJ, Kang JQ, Song L, Dibbens L, Mulley J, Macdonald RL (2006) Delta subunit susceptibility variants E177A and R220H associated with complex epilepsy alter channel gating and surface expression of alpha4beta2delta GABAA receptors. J Neurosci: Off J Soc Neurosci 26(5):1499–1506. doi:10.1523/JNEUROSCI.2913-05.2006
Eugene E, Depienne C, Baulac S, Baulac M, Fritschy JM, Le Guern E, Miles R, Poncer JC (2007) GABA(A) receptor gamma 2 subunit mutations linked to human epileptic syndromes differentially affect phasic and tonic inhibition. J Neurosci: Off J Soc Neurosci 27(51):14108–14116. doi:10.1523/JNEUROSCI.2618-07.2007
Feng Y, Kapornai K, Kiss E, Tamas Z, Mayer L, Baji I, Daroczi G, Benak I et al (2010) Association of the GABRD gene and childhood-onset mood disorders. Genes Brain Behav 9(6):668–672. doi:10.1111/j.1601-183X.2010.00598.x
Schwarzer C, Sperk G (1995) Hippocampal granule cells express glutamic acid decarboxylase-67 after limbic seizures in the rat. Neuroscience 69(3):705–709
Fritschy JM, Kiener T, Bouilleret V, Loup F (1999) GABAergic neurons and GABA(A)-receptors in temporal lobe epilepsy. Neurochem Int 34(5):435–445
Bouilleret V, Loup F, Kiener T, Marescaux C, Fritschy JM (2000) Early loss of interneurons and delayed subunit-specific changes in GABA(A)-receptor expression in a mouse model of mesial temporal lobe epilepsy. Hippocampus 10(3):305–324. doi:10.1002/1098-1063(2000)10:3<305::AID-HIPO11>3.0.CO;2-I
Drexel M, Kirchmair E, Sperk G (2013) Changes in the expression of GABAA receptor subunit mRNAs in parahippocampal areas after kainic acid induced seizures. Front Neural Circ 7:142. doi:10.3389/fncir.2013.00142
Nishimura T, Schwarzer C, Gasser E, Kato N, Vezzani A, Sperk G (2005) Altered expression of GABA(A) and GABA(B) receptor subunit mRNAs in the hippocampus after kindling and electrically induced status epilepticus. Neuroscience 134(2):691–704. doi:10.1016/j.neuroscience.2005.04.013
Tsunashima K, Schwarzer C, Kirchmair E, Sieghart W, Sperk G (1997) GABA(A) receptor subunits in the rat hippocampus III: altered messenger RNA expression in kainic acid-induced epilepsy. Neuroscience 80(4):1019–1032
Scimemi A, Semyanov A, Sperk G, Kullmann DM, Walker MC (2005) Multiple and plastic receptors mediate tonic GABAA receptor currents in the hippocampus. J Neurosci: Off J Soc Neurosci 25(43):10016–10024. doi:10.1523/JNEUROSCI.2520-05.2005
Zhang N, Wei W, Mody I, Houser CR (2007) Altered localization of GABA(A) receptor subunits on dentate granule cell dendrites influences tonic and phasic inhibition in a mouse model of epilepsy. J Neurosci: Off J Soc Neurosci 27(28):7520–7531. doi:10.1523/JNEUROSCI.1555-07.2007
Peng Z, Huang CS, Stell BM, Mody I, Houser CR (2004) Altered expression of the delta subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy. J Neurosci: Off J Soc Neurosci 24(39):8629–8639. doi:10.1523/JNEUROSCI.2877-04.2004
Rajasekaran K, Joshi S, Sun C, Mtchedlishvilli Z, Kapur J (2010) Receptors with low affinity for neurosteroids and GABA contribute to tonic inhibition of granule cells in epileptic animals. Neurobiol Dis 40(2):490–501. doi:10.1016/j.nbd.2010.07.016
Scimemi A, Andersson A, Heeroma JH, Strandberg J, Rydenhag B, McEvoy AW, Thom M, Asztely F et al (2006) Tonic GABA(A) receptor-mediated currents in human brain. Eur J Neurosci 24(4):1157–1160. doi:10.1111/j.1460-9568.2006.04989.x
Goodkin HP, Joshi S, Mtchedlishvili Z, Brar J, Kapur J (2008) Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J Neurosci Off J Soc Neurosci 28(10):2527–2538. doi:10.1523/JNEUROSCI.3426-07.2008
Naylor DE, Liu H, Wasterlain CG (2005) Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci Off J Soc Neurosci 25(34):7724–7733. doi:10.1523/JNEUROSCI.4944-04.2005
Sperk G, Schwarzer C, Heilman J, Furtinger S, Reimer RJ, Edwards RH, Nelson N (2003) Expression of plasma membrane GABA transporters but not of the vesicular GABA transporter in dentate granule cells after kainic acid seizures. Hippocampus 13(7):806–815. doi:10.1002/hipo.10133
Mathern GW, Mendoza D, Lozada A, Pretorius JK, Dehnes Y, Danbolt NC, Nelson N, Leite JP et al (1999) Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology 52(3):453–472
Andre V, Marescaux C, Nehlig A, Fritschy JM (2001) Alterations of hippocampal GAbaergic system contribute to development of spontaneous recurrent seizures in the rat lithium-pilocarpine model of temporal lobe epilepsy. Hippocampus 11(4):452–468. doi:10.1002/hipo.1060
Esclapez M, Houser CR (1999) Up-regulation of GAD65 and GAD67 in remaining hippocampal GABA neurons in a model of temporal lobe epilepsy. J Comp Neurol 412(3):488–505
Freichel C, Potschka H, Ebert U, Brandt C, Loscher W (2006) Acute changes in the neuronal expression of GABA and glutamate decarboxylase isoforms in the rat piriform cortex following status epilepticus. Neuroscience 141(4):2177–2194. doi:10.1016/j.neuroscience.2006.05.040
Ding R, Asada H, Obata K (1998) Changes in extracellular glutamate and GABA levels in the hippocampal CA3 and CA1 areas and the induction of glutamic acid decarboxylase-67 in dentate granule cells of rats treated with kainic acid. Brain Res 800(1):105–113
Kash SF, Johnson RS, Tecott LH, Noebels JL, Mayfield RD, Hanahan D, Baekkeskov S (1997) Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A 94(25):14060–14065
Pavlov I, Walker MC (2013) Tonic GABA(A) receptor-mediated signalling in temporal lobe epilepsy. Neuropharmacology 69:55–61. doi:10.1016/j.neuropharm.2012.04.003
Schwarzer C, Tsunashima K, Wanzenbock C, Fuchs K, Sieghart W, Sperk G (1997) GABA(A) receptor subunits in the rat hippocampus II: altered distribution in kainic acid-induced temporal lobe epilepsy. Neuroscience 80(4):1001–1017
Zhan RZ, Nadler JV (2009) Enhanced tonic GABA current in normotopic and hilar ectopic dentate granule cells after pilocarpine-induced status epilepticus. J Neurophysiol 102(2):670–681. doi:10.1152/jn.00147.2009
Lee B, Lee H, Nam YR, Oh JH, Cho YH, Chang JW (2005) Enhanced expression of glutamate decarboxylase 65 improves symptoms of rat parkinsonian models. Gene Ther 12(15):1215–1222. doi:10.1038/sj.gt.3302520
Emborg ME, Carbon M, Holden JE, During MJ, Ma Y, Tang C, Moirano J, Fitzsimons H et al (2007) Subthalamic glutamic acid decarboxylase gene therapy: changes in motor function and cortical metabolism. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab 27(3):501–509. doi:10.1038/sj.jcbfm.9600364
Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, Bland RJ, Young D et al (2007) Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet 369(9579):2097–2105. doi:10.1016/S0140-6736(07)60982-9
LeWitt PA, Guttman M, Tetrud JW, Tuite PJ, Mori A, Chaikin P, Sussman NM, Group USS (2008) Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson’s disease: a double-blind, randomized, multicenter clinical trial (6002-US-005). Ann Neurol 63(3):295–302. doi:10.1002/ana.21315
Grove J, Fozard JR, Mamont PS (1981) Assay of alpha-difluoromethylornithine in body fluids and tissues by automatic amino-acid analysis. J Chromatogr 223(2):409–416
Mumford JP, Dam M (1989) Meta-analysis of European placebo controlled studies of vigabatrin in drug resistant epilepsy. Br J Clin Pharmacol 27(Suppl 1):101S–107S
Jung MJ, Lippert B, Metcalf BW, Bohlen P, Schechter PJ (1977) gamma-Vinyl GABA (4-amino-hex-5-enoic acid), a new selective irreversible inhibitor of GABA-T: effects on brain GABA metabolism in mice. J Neurochem 29(5):797–802
Angehagen M, Ben-Menachem E, Ronnback L, Hansson E (2003) Novel mechanisms of action of three antiepileptic drugs, vigabatrin, tiagabine, and topiramate. Neurochem Res 28(2):333–340
Leach JP, Sills GJ, Majid A, Butler E, Carswell A, Thompson GG, Brodie MJ (1996) Effects of tiagabine and vigabatrin on GABA uptake into primary cultures of rat cortical astrocytes. Seizure 5(3):229–234
Duffy S, Nguyen PV, Baker GB (2004) Phenylethylidenehydrazine, a novel GABA-transaminase inhibitor, reduces epileptiform activity in rat hippocampal slices. Neuroscience 126(2):423–432. doi:10.1016/j.neuroscience.2004.03.007
Loscher W (1980) A comparative study of the pharmacology of inhibitors of GABA-metabolism. Naunyn Schmiedeberg’s Arch Pharmacol 315(2):119–128
Borden LA, Murali Dhar TG, Smith KE, Weinshank RL, Branchek TA, Gluchowski C (1994) Tiagabine, SK&F 89976-A, CI-966, and NNC-711 are selective for the cloned GABA transporter GAT-1. Eur J Pharmacol 269(2):219–224
Fink-Jensen A, Suzdak PD, Swedberg MD, Judge ME, Hansen L, Nielsen PG (1992) The gamma-aminobutyric acid (GABA) uptake inhibitor, tiagabine, increases extracellular brain levels of GABA in awake rats. Eur J Pharmacol 220(2–3):197–201
Wilby J, Kainth A, Hawkins N, Epstein D, McIntosh H, McDaid C, Mason A, Golder S et al (2005) Clinical effectiveness, tolerability and cost-effectiveness of newer drugs for epilepsy in adults: a systematic review and economic evaluation. Health Technol Assess 9(15):1–157, iii-iv
Kellinghaus C, Loddenkemper T, Weitemeyer L, Ludemann P (2001) Experience with tiagabine in the clinical practice; new insights as to the efficacy and safety profile. Nervenarzt 72(10):764–769
Bialer M, White HS (2010) Key factors in the discovery and development of new antiepileptic drugs. Nat Rev Drug Discov 9(1):68–82. doi:10.1038/nrd2997
Reddy DS, Rogawski MA (2012) Neurosteroids—endogenous regulators of seizure susceptibility and role in the treatment of epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV (eds) Jasper’s Basic Mechanisms of the Epilepsies. 4th edn., Bethesda (MD)
Reddy DS, Rogawski MA (2010) Ganaxolone suppression of behavioral and electrographic seizures in the mouse amygdala kindling model. Epilepsy Res 89(2–3):254–260. doi:10.1016/j.eplepsyres.2010.01.009
Bialer M, Johannessen SI, Levy RH, Perucca E, Tomson T, White HS (2013) Progress report on new antiepileptic drugs: a summary of the Eleventh Eilat Conference (EILAT XI). Epilepsy Res 103(1):2–30. doi:10.1016/j.eplepsyres.2012.10.001
Kerrigan JF, Shields WD, Nelson TY, Bluestone DL, Dodson WE, Bourgeois BF, Pellock JM, Morton LD et al (2000) Ganaxolone for treating intractable infantile spasms: a multicenter, open-label, add-on trial. Epilepsy Res 42(2–3):133–139
Wan L, Liu X, Wu Z, Ren W, Kong S, Dargham RA, Cheng L, Wang Y (2014) Activation of extrasynaptic GABAA receptors inhibits cyclothiazide-induced epileptiform activity in hippocampal CA1 neurons. Neurosci Bull 30(5):866–876. doi:10.1007/s12264-014-1466-8
Sun Y, Wu Z, Kong S, Jiang D, Pitre A, Wang Y, Chen G (2013) Regulation of epileptiform activity by two distinct subtypes of extrasynaptic GABAA receptors. Mole Brain 6:21. doi:10.1186/1756-6606-6-21
Petersen HR, Jensen I, Dam M (1983) THIP: a single-blind controlled trial in patients with epilepsy. Acta Neurol Scand 67(2):114–117
Hansen SL, Sperling BB, Sanchez C (2004) Anticonvulsant and antiepileptogenic effects of GABAA receptor ligands in pentylenetetrazole-kindled mice. Prog Neuro-Psychopharmacol Biol Psychiatry 28(1):105–113. doi:10.1016/j.pnpbp.2003.09.026
Loscher W, Schwark WS (1985) Evaluation of different GABA receptor agonists in the kindled amygdala seizure model in rats. Exp Neurol 89(2):454–460
Wafford KA, van Niel MB, Ma QP, Horridge E, Herd MB, Peden DR, Belelli D, Lambert JJ (2009) Novel compounds selectively enhance delta subunit containing GABA A receptors and increase tonic currents in thalamus. Neuropharmacology 56(1):182–189. doi:10.1016/j.neuropharm.2008.08.004
Crunelli V, Leresche N, Cope DW (2012) GABA-A receptor function in typical absence seizures. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV (eds) Jasper’s Basic Mechanisms of the Epilepsies. 4th edn., Bethesda (MD)
Prenosil GA, Schneider Gasser EM, Rudolph U, Keist R, Fritschy JM, Vogt KE (2006) Specific subtypes of GABAA receptors mediate phasic and tonic forms of inhibition in hippocampal pyramidal neurons. J Neurophysiol 96(2):846–857. doi:10.1152/jn.01199.2006
Krook-Magnuson E, Soltesz I (2015) Beyond the hammer and the scalpel: selective circuit control for the epilepsies. Nat Neurosci 18(3):331–338. doi:10.1038/nn.3943
Aston-Jones G, Deisseroth K (2013) Recent advances in optogenetics and pharmacogenetics. Brain Res 1511:1–5. doi:10.1016/j.brainres.2013.01.026
Katzel D, Nicholson E, Schorge S, Walker MC, Kullmann DM (2014) Chemical-genetic attenuation of focal neocortical seizures. Nat Commun 5:3847. doi:10.1038/ncomms4847
Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW et al (2002) Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 298(5594):846–850. doi:10.1126/science.1072873
Cook DJ, Teves L, Tymianski M (2012) Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature 483(7388):213–217. doi:10.1038/nature10841
Conflict of Interest
Sandra Schipper declares that she has no conflict of interest. Marlien Aalbers declares that she has no conflict of interest. Kim Rijkers declares that she has no conflict of interest. Ann Swijsen declares that she has no conflict of interest. Jean-Michel Rigo declares that he has no conflict of interest. Govert Hoogland declares that he has no conflict of interest. Johannes Vles declares that he has no conflict of interest. This article does not contain any studies with human or animal subjects.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s12035-015-9479-5.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Schipper, S., Aalbers, M.W., Rijkers, K. et al. Tonic GABAA Receptors as Potential Target for the Treatment of Temporal Lobe Epilepsy. Mol Neurobiol 53, 5252–5265 (2016). https://doi.org/10.1007/s12035-015-9423-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9423-8