
Abstract
The presence of pulmonary dysfunction after brain injury is well recognized. Acute lung injury (ALI) occurs in 20% of patients with isolated brain injury and is associated with a poor outcome. The “blast injury” theory, which proposes combined “hydrostatic” and “high permeability” mechanisms for the formation of neurogenic pulmonary edema, has been challenged recently by the observation that a systemic inflammatory response may play an integral role in the development of pulmonary dysfunction associated with brain injury. As a result of the primary cerebral injury, a systemic inflammatory reaction occurs, which induces an alteration in blood–brain barrier permeability and infiltration of activated neutrophils into the lung. This preclinical injury makes the lungs more susceptible to the mechanical stress of an injurious ventilatory strategy. Tight CO2 control is a therapeutic priority in patients with acute brain injury, but the use of high tidal volume ventilation may contribute to the development of ALI. Establishment of a therapeutic regimen that allows the combination of protective ventilation with the prevention of hypercapnia is, therefore, required. Moreover, in patients with brain injury, hypoxemia represents a secondary insult associated with a poor outcome. Optimal oxygenation may be achieved by using an adequate FiO2 and by application of positive end-expiratory pressure (PEEP). PEEP may, however, affect the cerebral circulation by hemodynamic and CO2-mediated mechanisms and the effects of PEEP on cerebral hemodynamics should be monitored in these patients and used to titrate its application.
Similar content being viewed by others

Introduction
Acute lung injury (ALI), and its more severe form, acute respiratory distress syndrome (ARDS), are associated with an inflammatory response of the lung following direct or indirect insults, and are characterized by severe hypoxemia, reduced compliance and diffuse radiographic infiltrates [1]. ALI/ARDS occurs in 20–25% of patients with isolated brain injury and is an independent predictor of poor outcome [2]. Understanding of the pathophysiological mechanisms of this syndrome may lead to the development of therapeutic strategies that could improve outcomes for patients with acute brain injury. This review will focus on current knowledge regarding the pathophysiological mechanisms of ALI in patients with severe brain injury and on how ventilatory settings may play a role in the “double hit” model.
Epidemiology
The most recent definition of ARDS is that proposed by the 1994 American-European Consensus Conference Committee [1]: a syndrome of acute onset, with bilateral infiltrates on chest radiography consistent with pulmonary edema, pulmonary-artery wedge pressure <18 mmHg or clinical absence of left atrial hypertension, and hypoxemia with a ratio of partial pressure of arterial oxygen to fraction of inspired oxygen (PaO2/FiO2) <200. Patients meeting the above criteria, but with PaO2/FiO2 ratios <300 are diagnosed as having ALI.
Incidence and Outcome
The incidence of ALI/ARDS in patients with severe brain injury has been reported to be between 5 and 30% [2–12]. This variation may be explained by the specific types of patients included in the study and by different definitions of ALI/ARDS. Most of the studies included patients with traumatic brain injury (TBI) but with different severities of injury as suggested by the Glasgow Coma Scale (GCS) [13] on admission. Restricting the analysis to patients with severe TBI (GCS < 9), gives incidences of 20–30% and 5–10% for ALI and ARDS, respectively [3, 4]. In patients with subarachnoid hemorrhage (SAH) [6, 9, 11, 12], a similar incidence of ALI/ARDS has been observed, suggesting that although the cause of the primary brain injury was different, similar injurious stimuli were present to trigger the development of ALI/ARDS (Table 1).
Regardless of the differences in inclusion criteria, all studies reported that the occurrence of ALI/ARDS was associated with increased mortality and morbidity in patients with severe brain injury [2–12]. Development of ALI/ARDS was also reported to be an independent predictor of increased mortality and poor neurological outcome in patients with TBI and with SAH and was associated with prolonged intensive care unit (ICU) and hospital length of stay and a decreased number of ventilator-free days [5] (Table 1).
Risk Factors
Initially, only the severity of brain injury was evaluated as a potential risk factor for the development of ALI/ARDS: in particular, severely and globally altered initial brain computed tomography (CT) scan [3] and low GCS [2–4] were reported as the most accurate predictors. More recently, extracranial factors, including administration of vasoactive drugs and a history of drug abuse, have been identified as independent predictors of ARDS in patients with TBI [3] (Table 1). The incidence of ALI/ARDS in patients with acute brain injury has been reported to have a bimodal distribution with an early peak on day 2–3 after the initiation of mechanical ventilation and a late peak on day 7–8 [7], the latter often being related to concurrent pneumonia [14]. Ventilator settings (tidal volume and respiratory rate) and a lower PaO2/FiO2 have been identified recently as independent predictors of early ALI/ARDS (within 72 h after mechanical ventilation), among major underlying ALI/ARDS risk factors (aspiration, pneumonia and lung contusion) and treatment variables [5]. These findings support the hypothesis that therapeutic strategies, as well as the severity of injury, contribute to the development of ALI/ARDS in these patients.
Pathophysiology
The presence of pulmonary dysfunction after brain injury is well recognized. Rogers et al. [15], combining a large autopsy database with a database of TBI inpatients, found a significant increase in the weight of the lungs but not of other organs in 50% of patients who died immediately or within 96 h after an isolated head injury. Pathological examination showed the presence of edema, congestion, and hemorrhage, which, associated with the increased lung weight, supported a diagnosis of neurogenic pulmonary edema (NPE), characterized as an increase in extravascular lung water in patients who have sustained an acute neurological injury [16]. In patients who survived more than 96 h, severe deterioration of the PaO2/FiO2 ratio (<300), even in the presence of a normal chest X-ray, was associated with a high initial intracranial pressure (ICP) and low cerebral perfusion pressure (CPP). These data suggest that poor oxygenation, even in the absence of gastric aspiration or direct thoracic injury, may be associated with a greater likelihood of developing late pulmonary complications during the hospital stay.
Blast Injury Theory
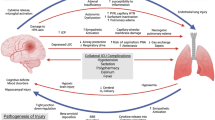
There are various theories regarding the pathophysiology of NPE, the most well known being the “blast injury” theory [17]. This theory proposes that the sympathetic storm caused by an acute increase in ICP induces a transient increase in intravascular pressure, which damages the endothelium enabling protein-rich plasma to escape into the interstitial and alveolar spaces (Fig. 1). The relative contributions of increased pulmonary vascular hydrostatic pressure and increased lung capillary permeability to the development of NPE have been widely studied. Smith and Matthay [18] showed that in 60% of patients with acute brain injury of different etiologies, the edema/plasma protein ratio was not significantly increased suggesting that hydrostatic edema was the principal underlying mechanism for NPE. Conversely, in an experimental model of intracranial hypertension, McClellan et al. [19] showed an increase in the accumulation of pulmonary extravascular protein, which was associated with an increase in systemic and pulmonary pressures. These authors concluded that the extravascular protein accumulation was due to direct adrenergic stimulation triggered by the brain injury while the pressure changes may provide a functional index of the autonomic response to intracranial hypertension. The “blast injury” theory thus explains the coexistence of hydrostatic and high permeability mechanisms of edema: The degree of capillary hypertension determines whether unbalanced Starling forces increase water flux across the endothelium or whether structural damage of the capillary wall allows plasma to escape into the interstitium and alveolar space.

Pathophysiology of acute lung injury in patients with isolated brain injury: double hit model. PEEP positive end expiratory pressure; ALI/ARDS acute lung injury, acute respiratory distress syndrome; VILI ventilator induced lung injury
Inflammatory Reaction
Recently, the blast injury theory has been challenged by the observation that a systemic inflammatory response plays an integral role in the development of pulmonary injury associated with brain damage [20] (Fig. 1). The brain was for many years regarded as an “immune privileged” organ, which was not susceptible to inflammation or immune activation and was thought to be largely unaffected by systemic inflammatory and immune responses; however, this view has recently been re-assessed [21]. In the last decade, it has been suggested that after acute brain injury there is increased intracranial production of pro-inflammatory cytokines resulting in secondary injury to the brain [22] and release of pro-inflammatory mediators into the systemic circulation [23]. McKeating et al. [23] reported an increased transcranial gradient of interleukin (IL)-6 within 48 h after brain injury, suggesting intracranial production, probably involving glial cells. More recently, Hutchinson et al. [24] noted that in TBI patients with a favorable outcome, microdialysis concentrations of IL-1 receptor antagonist (IL-1ra, an anti-inflammatory cytokine) were significantly higher than those of IL-1β (a pro-inflammatory molecule). It is also known that pro-inflammatory cytokines, such as IL-1, IL-6, and tumor necrosis factor (TNF)-α, can modulate the expression of adhesion molecule activity [25]. Interestingly, a strong association between systemic concentrations of soluble intercellular adhesion molecule-1 (ICAM-1) and poor neurological outcome has been demonstrated in patients with TBI [26], suggesting that a systemic inflammatory response after TBI is related to neurological outcome.
Double Hit Model
Based on this evidence, a “double hit” model has been proposed to explain the development of organ failure associated with acute brain injury. First, a traumatic brain insult creates a systemic inflammatory environment [27]. Once primed, the system is then vulnerable to later, normally innocuous secondary inflammatory insults, such as infections, mechanical stress induced by mechanical ventilation, and surgical procedures. These secondary insults, which occur within hours or days after the primary injury, can lead to further damage of the central nervous system and may contribute to the failure of several organs distant from the brain, leading to the development of multiple organ dysfunction syndrome. Recent evidence has suggested that the respiratory system is one of the organs most susceptible to such insults [28].
In an experimental model of cortical impact injury, Kalsotra et al. [29] demonstrated altered lung permeability and marked migration of neutrophils and activated macrophages in the major airways and alveolar spaces at 24 h post-injury, associated with enhanced pulmonary leukotriene B4 production. Although IL-6 and IL-1β concentrations were significantly increased in the lung, there was only a modest increase in IL-1β in the blood, leaving the hypothesis of humoral spillover of these cytokines into the lung unresolved. Yildirim et al. [30], in a different experimental model of brain injury, confirmed ultrastructural damage in type II pneumocytes after 24 h, characterized by the presence of intense intracellular vacuoles and lung tissue lipid peroxidation causing membrane lysis. In patients with fatal brain injury, Fisher et al. [31] reported an increased concentration of pro-inflammatory cytokines in bronchoalveolar lavage (BAL) fluid and an increased expression of IL-8 mRNA in lung tissue from multiple organ donors compared to controls. Moreover, the extent of neutrophil infiltration in donors correlated with the IL-8 concentration in lavage fluid suggesting that preclinical lung injury is already present in brain dead patients and that this may predispose towards an adverse clinical prognosis in lung transplant recipients.
These experimental and clinical data support the hypothesis that after severe brain injury eventually leading to brain death, preclinical lung injury occurs. The catecholamine storm and the systemic production of inflammatory mediators create a systemic inflammatory environment where the lung is more susceptible to further injurious stimuli, such as ventilatory settings, infections, and transfusions.
The Role of Mechanical Ventilation
Mechanical ventilation is the main supportive therapy used to re-establish sufficient oxygen supply and remove carbon dioxide (CO2) produced by peripheral organs during acute respiratory failure in patients with severe brain injury. While tight CO2 control represents a priority in patients with severe brain injury, optimal treatment of ALI/ARDS consists of a protective ventilatory strategy allowing a certain degree of hypercapnia to protect the lung from further injurious stimuli while it recovers from the initial pathological process. The presence of an inflammatory environment involving the lung after brain injury together with the use of a potentially injurious ventilatory strategy to insure tight CO2 control characterize the special challenges of this syndrome in this patient population.
PaCO2 Control
Moderate and forced hyperventilation have been widely used in the past for the treatment of intracranial hypertension. Although this therapeutic intervention is effective in lowering ICP by reducing cerebral blood volume, the risk of reducing cerebral blood flow below a critical threshold has always been a concern. Indeed, several clinical studies showed that hyperventilation may potentially have more deleterious than beneficial effects [32, 33]. Consequently guidelines of the Brain Trauma Foundation (BTF) [34] discourage the use of hyperventilation and suggest the following:
-
(1)
prophylactic hyperventilation to a PaCO2 ≤ 25 mmHg (in the absence of intracranial hypertension) is not recommended (level II evidence).
-
(2)
hyperventilation is only recommended as a temporizing measure for the reduction of elevated ICP (level III evidence).
-
(3)
hyperventilation should be avoided during the first 24 h after injury when cerebral blood flow is often critically reduced.
-
(4)
if hyperventilation is used, cerebral oxygen delivery should be monitored by jugular oxygen saturation (SjO2) or brain oxygen tension (PbrO2).
Nevertheless, in both Europe and the USA, hyperventilation is still used by physicians who do not necessarily follow the BTF guidelines. After publication of the first edition of the BTF guidelines, a survey reported that hyperventilation to a PaCO2 < 30 mmHg was applied by 36% of neurosurgeons in North America, but was much less common than 10 years earlier [35]. More recently, the BrainIT network [36] reported that moderate hyperventilation, resulting in a PaCO2 of 30–35 mmHg, was the most common strategy in Europe and that this level of hyperventilation was applied for half of the total ventilation time. While the first survey reported the attitudes of neurosurgeons to the use of hyperventilation for TBI management as assessed by a questionnaire, the BrainIT network reported the real incidence of hyperventilation in European ICUs including a percentage of “non-intentionally achieved” hypocarbia. We have recently confirmed that sedated TBI patients without ALI require an average minute ventilation of 7.6 l/min to maintain a PaCO2 of 35 mmHg. Those patients who developed ALI were ventilated with a minute ventilation of 10.4 l/min, a tidal volume of 10.6 ml/kg PBW, and a respiratory rate of 15/min to maintain tight CO2 control [5]. Our study highlighted the fact that tight CO2 control was considered the therapeutic priority in patients with TBI who developed ALI/ARDS even though this led to the application of a potentially injurious ventilatory strategy. Indeed the use of a lower tidal volume (6 ml/kg) in patients with ALI/ARDS has been shown in several clinical studies to decrease mortality and increase the number of ventilator-free days [37]. These clinical findings are likely related to the fact that higher tidal volumes exacerbate the pulmonary and systemic inflammatory response in patients with ALI/ARDS [38–40], causing so called ventilator-induced lung injury (VILI), a syndrome that is clinically and morphologically indistinguishable from ARDS. The three basic mechanisms underlying the development of VILI are: increased alveolar-capillary permeability by overdistension of the lung during mechanical ventilation (volutrauma), worsening .llung injury as a result of tidal recruitment-derecruitment of collapsed alveoli (atelectrauma), and more subtle injury manifest by activation of the inflammatory process (biotrauma) [40]. Until recently, only the lungs of patients with ALI/ARDS were regarded as being vulnerable to further inflammatory stimuli. However, Gajic et al. [41] recently showed in a population of general ICU patients with an established inflammatory process (aspiration, sepsis, pneumonia, or trauma), that use of high tidal volumes for the first 48 h of mechanical ventilation was associated with the development of ALI/ARDS. In patients with severe brain injury, the inflammatory process may be triggered by the primary cerebral injury. The already inflamed lungs may have decreased tolerance to subsequent mechanical stress from mechanical ventilation. Indeed, in an experimental model of massive brain injury, isolated lungs subjected to high pressure mechanical ventilation developed more severe injury than those retrieved from non-brain injured animals [28]. Moreover, high tidal volume and respiratory rate have recently been identified as independent predictors of ALI/ARDS in patients with TBI [5]; a dose–response association between the initial tidal volume and development of ALI/ARDS was reported, suggesting that this modifiable risk factor may be a target for future interventional trials.
All these findings support the hypothesis that, after severe brain injury eventually leading to brain death, a preclinical lung injury characterized by an inflammatory response is present. Massive brain injury may act as a preconditioning factor rendering the lung more susceptible to subsequent lung damage induced by mechanical ventilation.
Oxygenation and Positive End-Expiratory Pressure (PEEP)
In patients with TBI, secondary brain injury may occur as a result of hypoxemia as demonstrated in the analysis of a large, prospectively collected data set from the Traumatic Coma Data Bank [42, 43]. Hypoxemia occurred in 22% of patients with severe TBI and was associated with significantly increased morbidity and mortality. These results were also confirmed in the pre-hospital setting [44]. As a result, the BTF guidelines state that: “oxygenation should be monitored and hypoxia (PaO2 < 60 mmHg or O2 saturation < 90%) avoided” [34].
The optimal degree of oxygenation may be reached by using adequate FiO2 and by application of PEEP. Ventilatory support for patients with ALI involves application of PEEP to recruit collapsed alveoli, improve arterial oxygenation, and reduce elastance of the respiratory system [45]. However, in patients with ALI/ARDS, the optimal level of PEEP has not been defined [46]. Ideally, direct assessment of lung recruitability by a dynamic lung imaging technique would allow the best physiological titration of PEEP. Until such an approach is widely available, setting PEEP at the highest level compatible with a plateau pressure of 28 to 30 cm H2O and a tidal volume of 6 ml/kg of predicted body weight seems to be a reasonable alternative [47–49].
The cerebral circulation of patients with acute brain and lung injury is influenced by complex cardiopulmonary interactions [45, 50, 51] and application of PEEP may affect the cerebral circulation by CO2-mediated and hemodynamic mechanisms.
Gas Exchange Mechanism
The increase in PaCO2 directly causes vasodilation of cerebral arteries, thus increasing cerebral blood volume; this may cause a rise in ICP, if intracranial compliance is reduced [52]. We have demonstrated in patients with TBI complicated by ALI/ARDS that when application of PEEP resulted in overdistension of alveolar areas, this contributed to the increase in dead space and in PaCO2, causing cerebral vasodilation [53]. Conversely, if PEEP induced lung recruitment, reduction in shunt with improvement in oxygenation was the predominant effect, while the decrease in PaCO2 due to reduction in dead space was less pronounced; consequently ICP and cerebral perfusion did not change significantly.
Hemodynamic Mechanism
According to the concept of the vasodilatory cascade, the decrease in arterial pressure caused by PEEP may decrease cerebral blood flow in patients whose cerebral autoregulation is impaired, but may cause a compensatory vasodilation if autoregulation is preserved [54]. In the latter case, vasodilation will lead to an increase in cerebral blood volume and ICP, given a reduced intracranial compliance. However, most studies showed that application of PEEP did not induce a significant reduction in arterial and CPP, probably because euvolemia was insured [55].
Application of PEEP may also affect the cerebral circulation through an impairment in local venous return and an increase in right atrial pressure (RAP) due to the passive transmission of pleural pressure to the right atrium [56–59]. The Starling resistor model describes the dynamics of flow in collapsible tubes [60]. In the cerebral circulation, upstream pressure is represented by the arterial pressure and downstream pressure by the ICP, which surrounds collapsible cerebral veins. The application of PEEP increases the intrathoracic pressure leading to an increase in RAP, responsible for an increase in sagittal sinus pressure. The increase in sagittal sinus pressure decreases cerebral venous outflow and increases ICP. However, experimental and clinical studies have demonstrated that the effects of the application of PEEP are more evident if the initial ICP is lower than the applied PEEP [58, 59]. To further minimize the interference with the venous outflow due to the increase in RAP, patients should be managed with a 30° head up tilt [61–63]. Indeed, during head elevation, most of the increase in RAP consequent to the application of PEEP is transmitted through the jugular venous channel, which acts as a Starling resistor where the upstream pressure is the sagittal sinus pressure and the downstream pressure is the RAP. To overcome the collapse in jugular veins occurring at the thoracic inlet and to induce an upward pressure transmission, an increase in RAP equal to 20 mmHg is required [64]. During head elevation, cerebral venous blood also drains through the vertebral venous system [65], which is not subjected to the immediate intrathoracic pressure variations responsible for jugular vein collapse.
Alternative Ventilatory Techniques
The development of ALI/ARDS in patients with acute brain injury is a great challenge for clinicians. As we have seen, in patients with acute brain lesions at risk of cerebral ischemia, mechanical ventilation to obtain tight CO2 control is considered a priority. Conversely, treatment of ALI/ARDS consists of a protective ventilatory strategy in order to insure adequate oxygenation and to protect the lung from further injurious stimuli while recovering from the initial pathological process; this strategy may allow a certain degree of so called “permissive hypercapnia”. Consequently, in patients with acute brain injury who develop ALI/ARDS, the establishment of a therapeutic regimen that allows the combination of protective ventilation with prevention of hypercapnia is required. Alternative ventilatory strategies should, therefore, be considered in order to protect the lung and to insure tight CO2 control.
Tracheal gas insufflation (TGI) is an adjunct to mechanical ventilation that allows ventilation with low tidal volumes while PaCO2 is satisfactorily cleared. Several studies have shown that TGI can be used either to decrease PaCO2 in the setting of hypercapnia or to maintain normocapnia while tidal volume is decreased [66]. In patients with severe head trauma and ALI, Martinez-Perez et al. [67] showed that the application of phasic TGI (at mid-to-end expiration) allowed ventilation with lower tidal volumes and driving pressures while maintaining PaCO2 constant without any deleterious effects on cerebral parameters.
In patients with severe ARDS, extracorporeal membrane oxygenation (ECMO) has been proposed to keep the lung “at rest” while providing adequate gas exchange. More recently, a modified technique (low-frequency positive-pressure ventilation with extracorporeal CO2 removal, LFPPV-ECCO2R) has been proposed in which the lungs are inflated to moderate pressures to maintain functional residual capacity and CO2 removal is ensured by low flow partial VV bypass [68]. Conventional extracorporeal lung assist systems were characterized by the generation of blood flow through a roller or centrifugal pump with systemic heparinization, but a pumpless extracorporeal lung assist system has recently been proposed. This new system is an artero-venous bypass requiring small priming volumes and very low, if any, levels of heparinization because of the use of heparin-coated membranes. Preliminary data have reported good safety and efficacy of this device in patients with ARDS complicating TBI [69, 70].
Prone positioning has been demonstrated to improve oxygenation and to decrease the incidence of ventilator-associated pneumonia (VAP) in patients with acute hypoxic respiratory failure, but it does not improve mortality. Prone positioning is associated with improved ventilation perfusion matching, recruitment of atelectatic areas following a gravitational gradient, and an increase in end-expiratory lung volume [71]. Recently, in a retrospective study, the beneficial effect of prone positioning on cerebral tissue oxygenation as a result of increased arterial oxygenation were reported although this technique induced a significant increase in ICP with a reduction in CPP [72].
An alternative method for protective ventilation with an “open-lung strategy” is the use of high frequency oscillatory ventilation (HFOV) [73]. A retrospective study in brain injured patients reported a significant improvement in oxygenation and a decrease in ICP as a result of a significant reduction in PaCO2 [74].
The use of alternative ventilatory techniques, which combine protective ventilation with tight CO2 control, represents a promising approach to be verified in future clinical trials.
Specific Recommendations for Mechanical Ventilation
Maintenance of a Narrow Range of Minute Ventilation
In patients with severe brain injury, tight CO2 control may be achieved by using controlled mechanical ventilation with a volume-targeted mode. During controlled mechanical ventilation, the driving pressure is provided only by the ventilator with no spontaneous muscle effort by the patient to insure that the targeted minute ventilation is delivered. In volume preset modes, the targeted tidal volume is delivered but the pressure generated by the ventilator and applied to the respiratory system depends on the mechanics of the respiratory system: the higher the elastance (i.e., stiff lungs during ARDS), the higher the ventilation pressure. Consequently, this mode guarantees that the set tidal volume is always delivered and, therefore, that minute ventilation is adequately controlled. However, this setting may expose patients with stiff lungs to a risk of barotrauma because elevated pressures may be applied to the respiratory system to guarantee the delivery of the set tidal volume. Careful monitoring of the pressure applied to the respiratory system is therefore required in order to minimize the risk of barotrauma.
Application of PEEP
PEEP may be safely applied to recruit previously collapsed alveoli if: (a) euvolemia is maintained to minimize the effects of PEEP on arterial pressure and then on CPP; (b) its value is lower than the ICP to minimize interference with venous outflow; (c) effects of PEEP on recruitment/overdistension are carefully monitored by evaluating changes in PaCO2 and PaO2.
Conclusion
Severe brain injury induces a systemic inflammatory reaction, which leads to an alteration in blood–brain barrier permeability and infiltration of activated neutrophils into the alveolar spaces. The lungs of patients with severe brain injury are, therefore, “primed” by the initial cerebral injury. This preclinical injury makes the lungs more susceptible to the mechanical stress of an injurious ventilatory strategy. The exposure of the primed lungs to further insults defines the “double hit” model. The association between the initial tidal volumes and development of ALI/ARDS suggest that this modifiable risk factor may be a target for future interventional trials.
References
Bernard GR, Artigas A, Brigham KL, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial cordination. Am J Respir Crit Care Med. 1994;149:818–24.
Holland MC, Mackersie RC, Morabito D, et al. The development of acute lung injury is associated with worse neurologic outcome in patients with severe traumatic brain injury. J Trauma. 2003;55:106–11.
Contant CF, Valadka AB, Gopinath SP, Hannay HJ, Robertson CS. Adult respiratory distress syndrome: a complication of induced hypertension after severe head injury. J Neurosurg. 2001;95:560–8.
Bratton SL, Davis RL. Acute lung injury in isolated traumatic brain injury. Neurosurgery. 1997;40:707–12.
Mascia L, Zavala E, Bosma K, et al. High tidal volume is associated with the development of acute lung injury after severe brain injury: an international observational study. Crit Care Med. 2007;35:1815–20.
Kahn JM, Caldwell EC, Deem S, Newell DW, Heckbert SR, Rubenfeld GD. Acute lung injury in patients with subarachnoid hemorrhage: incidence, risk factors, and outcome. Crit Care Med. 2006;34:196–202.
Piek J, Chesnut RM, Marshall LF, et al. Extracranial complications of severe head injury. J Neurosurg. 1992;77(6):901–7.
Zygun DA, Kortbeek JB, Fick GH, Laupland KB, Doig CJ. Non-neurologic organ dysfunction in severe traumatic brain injury. Crit Care Med. 2005;33:654–60.
Solenski NJ, Haley EC Jr, Kassell NF, et al. Medical complications of aneurysmal subarachnoid hemorrhage: a report of the multicenter, cooperative aneurysm study. Participants of the Multicenter Cooperative Aneurysm Study. Crit Care Med. 1995;23(6):1007–17.
Salim A, Martin M, Brown C, et al. The presence of the adult respiratory distress syndrome does not worsen mortality or discharge disability in blunt trauma patients with severe traumatic brain injury. Injury. 2008;39(1):30–5.
Gruber A, Reinprecht A, Illievich UM, et al. Extracerebral organ dysfunction and neurologic outcome after aneurysmal subarachnoid hemorrhage. Crit Care Med. 1999;27:505–14.
Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med. 2006;34(3):617–23.
Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2(7872):81–4.
Bronchard R, Albaladejo P, Brezac G, et al. Early onset pneumonia: risk factors and consequences in head trauma patients. Anesthesiology. 2004;100(2):234–9.
Rogers FB, Shackford SR, Trevisani GT, Davis JW, Mackersie RC, Hoyt DB. Neurogenic pulmonary edema in fatal and nonfatal head injuries. J Trauma. 1995;39:860–6.
Touho H, Karasawa J, Shishido H, Yamada K, Yamazaki Y. Neurogenic pulmonary edema in the acute stage of hemorrhagic cerebrovascular disease. Neurosurgery. 1989;25:762–8.
Theodore J, Robin ED. Pathogenesis of neurogenic pulmonary edema. Lancet. 1976;2:749.
Smith WS, Matthay MA. Evidence for a hydrostatic mechanism in human neurogenic pulmonary edema. Chest. 1997;111(5):1326–33.
McClellan MD, Dauber IM, Weil JV. Elevated intracranial pressure increases pulmonary vascular permeability to protein. J Appl Physiol. 1989;67(3):1185–91.
Avlonitis VS, Fisher AJ, Kirby JA, Dark JH. Pulmonary transplantation: the role of brain death in donor lung injury. Transplantation. 2003;75(12):1928–33.
Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147(Suppl 1):S232–40.
Ott L, McClain CJ, Gillespie M, Young B. Cytokines and metabolic dysfunction after severe head injury. J Neurotrauma. 1994;11:447–72.
McKeating EG, Andrews PJ, Signorini DF, Mascia L. Transcranial cytokine gradients in patients requiring intensive care after acute brain injury. Br J Anaesth. 1997;78:520–3.
Hutchinson PJ, O’Connell MT, Rothwell NJ, et al. Inflammation in human brain injury: intracerebral concentrations of IL-1alpha, IL-1beta, and their endogenous inhibitor IL-1ra. J Neurotrauma. 2007;24(10):1545–57.
Kelley BJ, Lifshitz J, Povlishock JT. Neuroinflammatory responses after experimental diffuse traumatic brain injury. J Neuropathol Exp Neurol. 2007;66(11):989–1001.
McKeating EG, Andrews PJ, Mascia L. Leukocyte adhesion molecule profiles and outcome after traumatic brain injury. Acta Neurochir. 1998;Suppl 71:200–2.
Scholz M, Cinatl J, Schädel-Höpfner M, Windolf J. Neutrophils and the blood-brain barrier dysfunction after trauma. Med Res Rev. 2007;27(3):401–16.
Lopez-Aguilar J, Villagra A, Bernabe F, et al. Massive brain injury enhances lung damage in an isolated lung model of ventilator-induced lung injury. Crit Care Med. 2005;33:1077–83.
Kalsotra A, Zhao J, Anakk S, Dash PK, Strobel HW. Brain trauma leads to enhanced lung inflammation and injury: evidence for role of P4504Fs in resolution. J Cereb Blood Flow Metab. 2007;27:963–74.
Yildirim E, Kaptanoglu E, Ozisik K, et al. Ultrastructural changes in pneumocyte type II cells following traumatic brain injury in rats. Eur J Cardiothorac Surg. 2004;25:523–52.
Fisher AJ, Donnelly SC, Hirani N, et al. Enhanced pulmonary inflammation in organ donors following fatal nontraumatic brain injury. Lancet. 1999;353:1412–3.
Muizelaar JP, Marmarou A, Ward JD, et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg. 1991;75(5):731–9.
Coles JP, Fryer TD, Coleman MR, et al. Hyperventilation following head injury: effect on ischemic burden and cerebral oxidative metabolism. Crit Care Med. 2007;35(2):568–78.
Guidelines for the management of severe head injury. Brain Trauma Foundation, American Association of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care. J Neurotrauma. 1996;13:641–734.
Marion DW, Spiegel TP. Changes in the management of severe traumatic brain injury: 1991–1997. Crit Care Med. 2000;28(1):16–8.
Neumann JO, Chambers IR, Citerio G, et al. The use of hyperventilation therapy after traumatic brain injury in Europe: an analysis of the BrainIT database. Intensive Care Med. 2008;34(9):1676–82.
Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome: The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342:1301–8.
Ranieri VM, Suter PM, Tortorella C, et al. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome. A randomised controlled trial. JAMA. 1999;282:54–61.
Ranieri VM, Giunta F, Suter PM, Slutsky AS. Mechanical ventilation as a mediator of multisystem organ failure in acute respiratory distress syndrome. JAMA. 2000;284(1):43–4.
Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest. 1997;99(5):944–52.
Gajic O, Dara SI, Mendez JL, et al. Ventilator-associated lung injury in patients without acute lung injury at the onset of mechanical ventilation. Crit Care Med. 2004;32:1817–24.
Marmarou A, Anderson Rl, Ward JD. Impact of ICP instability and hypotension on outcome in patients with severe head trauma. J Neurosurg. 1991;75:159–66.
Miller JD, Sweet RC, Narayan R, Becker DP. Early insults to the injured brain. JAMA. 1978;240(5):439–42.
Stocchetti N, Furlan A, Volta F. Hypoxemia and arterial hypotension at the accident scene in head injury. J Trauma. 1996;40:764–7.
Ranieri VM, Eissa NT, Corbeil C, Chasse M, Braidy J, Matar N, et al. Effects of positive end-expiratory pressure on alveolar recruitment and gas exchange in patients with the adult respiratory distress syndrome. Am Rev Respir Dis. 1991;144:544–51.
The National Heart, Lung, and Blood Institute ARDS Clinical Trials Network. Higher versus lower positive end-pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004;351:327–36.
Mercat A, Richard JC, Vielle B, et al. Positive end-expiratory pressure setting in adults with acute lung injury and acute respiratory distress syndrome: a randomized controlled trial. JAMA. 2008;299(6):646–55.
Meade MO, Cook DJ, Guyatt GH, et al. Ventilation strategy using low tidal volumes, recruitment maneuvers, and high positive end-expiratory pressure for acute lung injury and acute respiratory distress syndrome: a randomized controlled trial. JAMA. 2008;299(6):637–45.
Gattinoni L, Caironi P. Refining ventilatory treatment for acute lung injury and acute respiratory distress syndrome. JAMA. 2008;299(6):691–3.
Blanch L, Fernandez R, Benito S, Mancebo J, Net A. Effect of PEEP on the arterial minus end-tidal carbon dioxide gradient. Chest. 1987;92:451–4.
Pinsky M, Desmet JM, Vincent JL. Effects of PEEP on right ventricular function in humans. Am Rev Respir Dis. 1991;143:25–31.
Markwalder TM, Grolimund P, Seiler RW, Roth F, Aaslid R. Dependency of blood flow velocity in the middle cerebral artery on end tidal carbon dioxide partial pressure- a transcranial ultrasound Doppler study. J Cerebral Blood Flow Metab. 1984;4:368–72.
Mascia L, Grasso S, Fiore T, Bruno F, Berardino M, Ducati A. Cerebro-pulmonary interactions during the application of low levels of positive end-expiratory pressure. Intensive Care Med. 2005;31(3):373–9.
Rosner MJ, Rosner SD, Johnson AH. Cerebral perfusion pressure: management protocol and clinical results. J Neurosurg. 1995;83(6):949–62.
Doblar DD, Santiago TV, Kahn AU, Edelman NH. The effect of positive end-expiratory pressure ventilation (PEEP) on cerebral blood flow and cerebrospinal fluid pressure in goats. Anesthesiology. 1981;55(3):244–50.
Huseby JS, Luce JM, Cary JM, Pavlin EG, Butler J. Effects of positive end-expiratory pressure on intracranial pressure in dogs with intracranial hypertension. J Neurosurg. 1981;55(5):704–5.
Huseby JS, Pavlin EG, Butler J. Effect of positive end-expiratory pressure on intracranial pressure in dogs. J Appl Physiol. 1978;44(1):25–7.
McGuire G, Crossley D, Richards J, Wong D. Effects of varying levels of positive end-expiratory pressure on intracranial pressure and cerebral perfusion pressure. Crit Care Med. 1997;25(6):1059–62.
Luce JM, Huseby JS, Kirk W, Butler J. A Starling resistor regulates cerebral venous outflow in dogs. J Appl Physiol. 1982;53(6):1496–503.
Permutt S, Riley RL. Hemodynamics of collapsible vessels with tone: the vascular waterfall. J Appl Physiol. 1963;18:924–32.
Lodrini S, Montolivo M, Pluchino F, Borroni V. Positive end-expiratory pressure in supine and sitting positions: its effects on intrathoracic and intracranial pressures. Neurosurgery. 1989;24(6):873–7.
Moraine JJ, Berre J, Melot C. Is cerebral perfusion pressure a major determinant of cerebral blood flow during head elevation in comatose patients with severe intracranial lesions? J Neurosurg. 2000;92:606–14.
Feldman Z, Kanter MJ, Robertson CS, et al. Effect of head elevation on intracranial pressure, cerebral perfusion pressure and cerebral blood flow in head-injured patients. J Neurosurg. 1992;76:207–11.
Toung TJ, Aizawa H, Traystman RJ. Effects of positive end-expiratory pressure ventilation on cerebral venous pressure with head elevation in dogs. J Appl Physiol. 2000;88(2):655–61.
Epstein HM, Linde HW, Crampton AR, Ciric IS, Eckenhoff JE. The vertebral venous plexus as a major cerebral venous outflow tract. Anesthesiology. 1970;32:332–8.
Rossi N, Musch G, Sangalli F, et al. Reverse-thrust ventilation in hypercapnic patients with acute respiratory distress syndrome. Acute physiological effects. Am J Respir Crit Care Med. 2000;162:363–8.
Martinez-Perez M, Bernabe F, Pena R, Fernandez R, Nahum A, Blanch L. Effects of expiratory tracheal gas insufflation in patients with severe head trauma and acute lung injury. Intensive Care Med. 2004;30(11):2021–7.
Gattinoni L, Agostini A, Pesenti A, et al. Treatment of acute respiratory failure with low-frequency positive-pressure ventilation and extracorporeal removal CO2. Lancet. 1980;2:292–4.
Bein T, Weber F, Philipp A, et al. A new pumpless extracorporeal interventional lung assist in critical hypoxemia/hypercapnia. Crit Care Med. 2006;34(5):1372–7.
Bein T, Scherer MN, Philipp A, Weber F, Woertgen C. Pumpless extracorporeal lung assist (pECLA) in patients with acute respiratory distress syndrome and severe brain injury. J Trauma. 2005;58(6):1294–7.
Gattinoni L, Tognoni G, Pesenti A, et al. Effect of prone positioning on the survival of patients with acute respiratory failure. N Engl J Med. 2001;345:568–73.
Reinprecht A, Greher M, Wolfsberger S, Dietrich W, Illievich UM, Gruber A. Prone position in subarachnoid hemorrhage patients with acute respiratory distress syndrome: effects on cerebral tissue oxygenation and intracranial pressure. Crit Care Med. 2003;31(6):1831–8.
Derdak S, Mehta S, Stewart T, et al. High frequency oscillatory ventilation for acute respiratory distress syndrome: a randomized controlled trial. Am J Respir Crit Care Med. 2002;166:801–8.
Salim A, Miller K, Dangleben D, Cipolle M, Pasquale M. High-frequency percussive ventilation: an alternative mode of ventilation for head-injured patients with adult respiratory distress syndrome. J Trauma. 2004;57(3):542–6.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mascia, L. Acute Lung Injury in Patients with Severe Brain Injury: A Double Hit Model. Neurocrit Care 11, 417–426 (2009). https://doi.org/10.1007/s12028-009-9242-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-009-9242-8