
Abstract
Paget’s disease of bone (PDB) is the second most common metabolic bone disorder, after osteoporosis. It is characterised by focal areas of increased and disorganised bone turnover, coupled with increased bone formation. This disease usually appears in the late stages of life, being slightly more frequent in men than in women. It has been reported worldwide, but primarily affects individuals of British descent. Majority of PDB patients are asymptomatic, but clinical manifestations include pain, bone deformity and complications, like pathological fractures and deafness. The causes of the disease are poorly understood and it is considered as a complex trait, combining genetic predisposition with environmental factors. Linkage analysis identified SQSTM1, at chromosome 5q35, as directly related to the disease. A number of mutations in this gene have been reported, pP392L being the most common variant among different populations. Most of these variants affect the ubiquitin-associated (UBA) domain of the protein, which is involved in autophagy processes. Genome-wide association studies enlarged the number of loci associated with PDB, and further fine-mapping studies, combined with functional analysis, identified OPTN and RIN3 as causal genes for Paget’s disease. A combination of risk alleles identified by genome-wide association studies led to the development of a score to predict disease severity, which could improve the management of the disease. Further studies need to be conducted to elucidate other important aspects of the trait, such as its focal nature and the epidemiological changes found in some populations. In this review, we summarize the clinical characteristics of the disease and the latest genetic advances to identify susceptibility genes. We also list current available treatments and prospective options.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Paget’s disease of bone (PDB) is a chronic disorder characterized by focal or multifocal remodelling and disorganized bone structure [1]. It was firstly described as “osteitis deformans” by Sir James Paget in 1876, prior to the discovery of the X-ray [2]. Nowadays, it is considered a common skeletal condition, representing the most frequent metabolic bone disorder after osteoporosis.
Epidemiology
PDB appears usually after the age of 40, being slightly more common in men than in women [1, 3]. It has been described almost worldwide, with an irregular geographical distribution [4]. It primarily affects patients of British descent, being common (around 4%) in England [1], areas of Australia, New Zealand [5] and North America [6] and rare (less than 1‰) in Asia, Scandinavia and Africa [7].
Marked differences in prevalence have been found not only among countries but also between areas within the same country [8]. Some regions show a high prevalence of the disease, like the Lancashire focus in UK, with a prevalence of 7% in the population over 55 years [9] and the Vitigudino-Salamanca region in Spain, with a 5.7% prevalence [10].
Several studies suggest that the prevalence and severity of PDB are declining in most but not all of the studied countries [4, 8, 11, 12]. Although the cause of this reduction is not completely understood, environmental changes, such as different migratory patterns, improved diet, sedentary lifestyle and decrease in the exposure to viral infections and zoonoses, might play a role [4].
Clinical Manifestations
PDB may have a long asymptomatic phase and up to 70% of patients do not present any symptom throughout disease evolution [13].
Clinical manifestations of PDB are pain, bone deformity and features caused by complications, including pathological fractures and deafness [14]. Pain is probably the most common symptom and can be differentiated into primary or secondary. Primary pain is described as dull, deep and predominantly nocturnal. Pain secondary to complications is more frequent than primary pain, especially due to the neurological entrapment or joint deformities [15].
PDB is most commonly located in the pelvis (58–80%), spine (40%), femur (32%) and tibia (16–20%) [16]. In some cases, disease limits to a single bone (monostotic disease), although it often affects several noncontiguous bones (polyostotic disease) [17]. Long bones might bend as a result of the increased bone volume and malleability. Some patients also show skull enlargement and facial deformities, which can transform their physical appearance [14].
The most common complications of PDB comprise arthropathy secondary to an alteration of the subchondral bone, fractures, neurologic compression secondary to bone growth, neurologic dysfunction possibly secondary to vascular steal syndrome, bone hypervascularisation, which may be accompanied by an increased focal heat in the superficial bones as the tibia [18], high output congestive heart failure, hypercalcemia and hypercalciuria in immobilized patients and the tumour transformation of the pagetic bone, commonly into osteosarcoma [19, 20].
Pathology
PDB manifests with a marked increase in bone turnover, which leads to a larger bone volume [21]. Osteoclasts are mainly affected, experimenting an increase in number and size and containing more nuclei than normal osteoclasts. This results in an elevated metabolic osteolytic activity, coupled with increased bone formation by osteoblasts, which are apparently normal [22].
The pagetic bone lesion could be identified in radiographs as a lytic lesion at the first stage (osteolytic pagetic phase). Then, lesions evolve into a mixture of sclerosis, due to new bone formation by osteoblasts, and osteolysis (mixed phase). In the final stages, sclerotic bone is observed, due to a reduction in bone turnover and cells [23]. Different radiographic patterns could be observed in each stage (Table 1) [24].
Bone turnover is greatly accelerated in the pagetic bone. Therefore, new collagen fibres are placed in a chaotic fashion, unlike the laminar distribution of the adult mature bone. This results in the characteristic mosaic pattern of the pagetic bone, combining an abnormal woven bone, with some areas of lamellar bone, and numerous disorganized cement lines from previous osteolytic phases [25]. In the matrix, the osteoid volume is increased in thickness, but usually without mineralization alterations. The increase in bone turnover leads to an increased number of trabeculae, as it has been described in biopsies from the iliac crest. Numerous connective tissue fibres and hypervascularity are observed in the bone marrow. All the above-mentioned changes induce mechanical tissue modifications that facilitate the bowing deformities and cracks; however, a lower mineralization rate and loss of aligned haversian structures may partly compensate these changes, maintaining resistance to crack growth [26].
Diagnosis
A diagnosis of PDB is incidental in most cases, when an elevated level of alkaline phosphatase is detected in the absence of liver disease in analyses that were performed for various reasons or the presence of suggestive radiographic changes ordered by other medical problems [16].
Elevation of bone turnover markers reflects changes in bone metabolism. Due to its wide availability, low variability and price, total alkaline phosphatase (ALP) is the most extended marker for PDB activity. A recent meta-analysis suggests that this activity is better monitored by following procollagen type 1 amino-terminal propeptide (P1NP) levels, considering ALP, bone-specific alkaline phosphatase (bone ALP), and C-terminal telopeptide (CTX) as good alternative markers for disease activity in untreated patients, or when P1NP is not available [27].
Plain radiography is often the basis for diagnosis as its features are easily recognizable. The injury does not usually affect the entire bone and the border between healthy and disease areas appears as a lytic image (blade of grass, or candle flame sign in the shaft of long bones (Table 1)). Computed tomography, magnetic resonance and positron emission tomography images may be useful to detect suspected sarcomatous degeneration [28].
Tc-99 bone scan provides images of increased uptake in areas of increased vascularity and osteoblastic activity. Although it is unspecific, it has a high sensitivity to detect lesions, even at the very early stages of PDB that are still not visible on X-ray [29].
Bone biopsy is rarely required for diagnosis, but it may be useful for tumour differential diagnosis. The most characteristic findings are the presence of abnormal trabeculae, irregular cementation lines with a typical “mosaic” image, increase in vascularity and increased number and size of osteoclasts [21].
Aetiology
The causes of PDB are not well understood and controversies arise with regard to its aetiology [30]. It is considered a complex, multifactorial disease, as a result of a synergistic action between environmental and genetic factors. At an early age, osteoclast precursors could be sensitized by an unknown environmental factor. The genetic conditioning would explain individual susceptibility to finally develop the disease years later [30, 31].
Genetic Predisposition
PDB shows a strong genetic compound. It has been identified in families since 1883 [32] and it is shown that up to 40% of individuals with PDB have affected relatives [33–36]. It is an autosomal dominant condition [36–38] which appears more commonly in first-degree relatives of affected patients [35].
The first genetic approach to identify the causal gene for PDB was performed in a French-Canadian cohort including 11 families with PDB. Linkage analysis identified the 5q35 locus as associated with the disease (LOD score 3.0) [38]. Subsequent studies isolated sequestosome 1 (SQSTM1) as the candidate gene for this locus [39].
In total, seven loci have been associated by linkage analysis with the appearance of classical PDB: 6p21.3 (PDB1 locus) [40, 41], 18q21-22 (PDB2) [42–45], 5q35 (PDB3) [37–39], 5q31 (PDB4) [38], 2q36 (PDB5) [37], 10p13 (PDB6) [37] and 18q23 (PDB7) [46]. The former locus was also associated to rare bone dysplasia familial expansile osteolysis, a rare condition that shares some features with PDB [37].
SQSTM1 Mutations
SQSTM1 gene maps to chromosome 5q35 and contains eight exons. It encodes p62 protein, a 62-kDa scaffolding protein with three functional regions: an N-terminal region which interacts with kinases, a hinge region and a C-terminal area containing the ubiquitin-binding domain (UBA domain). It targets proteins for degradation through the proteasome pathway [47] and mediates the formation of autophagosome by interacting with LC3 protein [48, 49]. SQSTM1 is also important for bone metabolism, since it is involved in the transduction of the NF-κB pathway, which is key in osteoclast differentiation and function [50].
Germline mutations in SQSTM1 have been found in ~40% of the PDB familial cases and in 10% of sporadic cases [39, 51]. p.P392L variant was the first SQTM1 mutation associated with PDB. It has been shown that p.P392 mutation is sufficient to cause PDB in mice, by altering autophagy in osteoclasts [52]. It was initially identified in 46% of familial cases and in 16% of sporadic patients of French-Canadian ascendency [39]. Similarly, it was identified in 19% of familial and 8.9% of sporadic cases in British patients [51], as well as in the Belgian [53], Italian [54] and American populations [55] (Table 2). This mutation was also commonly detected in the Chinese population, where the appearance of Paget’s disease is rare. Reported cases in this population showed similar demographic and clinical features than in Caucasian patients [56, 57]. The above findings suggest that p.P392L is a mutation hotspot. Several other hotspots have been identified in the protein, mainly in the UBA domain [58–61].
To date, 28 different mutations in SQSTM1 have been reported, producing 21 aminoacid substitutions and various truncating mutations affecting the UBA domain of the protein (Table 2). Patients with truncating mutations showed a more severe phenotype than the individuals with missense mutations [61, 62]. Most of the patients present a single mutation in the gene, although several cases have been identified with compound heterozygous mutations [35, 58, 63] and homozygous p.P392L [34].
Only four mutations were identified out of the UBA domain of SQSTM1 (Table 2) [58–61]. These mutations, like p.S349T, also increase NF-κB signalling [61]. This occurs through the reduction in binding SQSTM1 to Keap1, which reduces the activity of Nrf2. Alteration of the Nrf2 function could produce an increase in the oxidative response genes, contributing to the appearance of PDB. Loss of Nrf2 in vivo negatively affects osteoblast differentiation and matrix formation, and it has been proposed that mutations in SQSTM1 could produce alterations in bone remodelling as seen in PDB patients through altering the Nrf2 cellular activity [64].
It is known that PDB is a focal disease showing asymmetric distribution, however, the cause is still unknown. It has been hypothesised that somatic mutations at the early stage of the zygote could be responsible for the mosaicism detected in the patients. Consistently, several studies found p.P392L variant as a somatic mutation in SQSTM1 in the affected bones from two unrelated patients, but not in peripheral blood [65], or restricted to monocytes [66].
Mutations in SQSTM1 have also been reported in other diseases, like amyotrophic lateral sclerosis, in cohorts with familiar, sporadic and frontotemporal dementia—ALS, from Europe, the USA and Japan [67–71]. Among the rare or novel coding mutations found, some of them pathogenic, p.Pro392Leu and p.Glu155Lys, were also identified. The patient carrying p.P392L mutation developed Paget’s disease, as well as the father of the proband carrying p.Glu155Lys mutation [71].
Genome-Wide Association Studies (GWAS)
SQSTM1 mutations have been found in only 20–50% of PDB patients, therefore high-throughput screening techniques, like genome-wide association studies, were used to identify unknown candidate genes [72, 73]. An initial study carried out by Albagha et al. analysed 1250 SQSTM1-ve cases and 1537 controls and identified six SNPs in chromosomes 1, 10 and 18 associated with the disease (p values ranging from 1.86e-11 to 5.38e-24) (Table 2) [74]. Risk allele carriers have ~70% of increase in predisposition to develop the disease [74].
Chromosome 1p13 highlighted a recombination area where only CSF1 gene was located. This gene encodes M-CSF, the macrophage colony-stimulating factor, involved in osteoclast formation and survival [75, 76]. An increase in serum M-CSF has been detected in patients with PDB [77]. The causal variants in this gene that predispose to PDB remain unknown, but it is suggested that they could induce PDB by increasing osteoclast formation, via CSF1 activity [78].
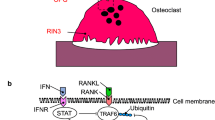
Individuals carrying the risk allele of SNP rs1561570, located in chromosome 10p13, showed an increase of ~60% in developing the disease [74]. This region has been previously detected by linkage analysis, defined as PDB6 locus [37], but the causal gene was not isolated. GWAS allowed to identify a recombination area where Optineurin (OPTN) gene is located. OPTN plays a role in glaucoma [79], but no function has been previously reported in bone metabolism.
Chromosome 18q21.33 corresponds to PDB7 locus, previously identified in some families by linkage analysis [46]. Top GWAS SNPs were located in an intergenic region close to TNFRSF11A gene. It encodes RANK, a receptor protein for RANKL which activates NF-κB signalling. RANK is a key protein for osteoclast differentiation and function, and its disruption leads to an osteopetrotic phenotype in mice [80]. Recent studies have shown that genetic variability of genes such as TNFRSF11A/RANK could increase the severity of the disease in patients carrying a mutation in SQSTM1 [81]. Other syndromes with similar clinical characteristics as PDB were also associated with mutations in RANK gene, like familial expansile osteolysis, early-onset familial PDB and expansile skeletal hyperphosphatasia [44, 45, 82].
Enlarged GWAS analysis in 2223 SQSTM1-ve PDB cases and 4601 controls confirmed the previous GWAS findings and identified four novel signals in chromosomes 7, 8, 14 and 15 (Table 2) [73]. The strongest signal at 7q33 was driven by rs4294134 variant, located in an intronic region of NUP205 gene. It encodes nucleoporin 205 kDa, a component of the nuclear pore involved in transport processes [83]. However, its role in the bone is still unknown.
The signal on chromosome 8q22.3 appointed to an 18-kb LD block covering the whole transmembrane 7 superfamily member 4 (TM7SF4) gene. This gene encodes DC-STAMP protein, involved in the fusion of osteoclasts precursors to form mature osteoclasts [84]. Expression of DC-STAMP is essential for osteoclast formation [85]. Genetic variants predisposing to PDB could enhance the expression of DC-STAMP, to generate the large multinucleated pagetic osteoclasts [73].
SNP on chromosome 14q32.12 also appoints to a novel gene in PDB, RIN3 [73]. It encodes Ras and Rab interactor 3, involved in vesicular trafficking [86, 87]. Its functionality and association with PDB is discussed in “Novel genes associated with PDB”.
Chromosome 15q24.1 also constitutes a new susceptibility locus for PDB. Rs5742915, a missense change (p.Phe645Leu) of promyelocytic leukaemia gene (PML), showed the highest association [73]. This gene is involved in TGF-β signalling and involved in the regulation of bone remodelling [88]. GOLGA6A gene, a member of the golgin family, is located in the same area and could not be completely discarded. Its role in bone metabolism is unknown, but mutations in other members of the same family produce a severe form of osteoporosis [89] and lethal skeletal dysplasia [90].
Novel Genes Associated with PDB
Genome-wide association studies allowed to identify most of the genetic loci involved in the development of the disease. To date, only two GWAS regions have been studied in detail: chromosome 10p13 (OPTN gene) [91] and chromosome 14q32.12 (RIN3 gene) [31].
Chromosome 10p13 highlights Optineurin gene, involved in NF-κB signalling regulation [92], autophagy and immunity [93]. SNP rs1561570 was the strongest signal in GWAS for this locus (p value = 4.37e-38, OR = 1.67 [1.54–1.810]) and was an expression quantitative trait locus (eQTL), reducing the levels of OPTN in T-allele carriers [91]. Mouse knockdown model for optn showed that the gene acts as a negative regulator of osteoclast differentiation in vitro [91]. OptnD477N/D477N knockout mice formed more hypernucleated osteoclasts compared to the wild type. Osteoblasts from these mice showed a reduction in their role to promote osteoclast differentiation. Osteoclast results were supported in vivo. An increase in bone resorption in these mice is thought to be coupled with an increase in bone formation, therefore, no bone loss was found. After RANKL stimulation, an increase in NF-κB activation was detected in these mice. The inhibitory effect of Optn on osteoclasts is mediated by a CYLD-dependent pathway, which is important for the inhibition of NF-κB activation. Optn also inhibits osteoclast differentiation by modulating INF-β signalling pathway. Knockdown and Knockout Optn mice showed enhanced osteoclast differentiation. Rs1561570 SNP in Optn gene increases susceptibility to PDB by reducing OPTN expression [91].
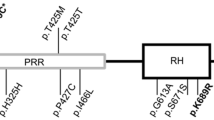
Chromosome 14q32.12 was strongly associated to PDB (p value = 2.55e-11, OR = 1.44 [1.29–1.60]) in the European population, appointing to Ras and Rab interactor 3 (RIN3) as the causal gene, since small GTPases, like Ras and Rab, are important for osteoclast function [94, 95], and molecules involved in vesicular trafficking cause syndromes with PDB-like characteristics, namely inclusion body myopathy with early onset Paget’s disease and frontotemporal dementia [96]. Deep sequencing of the 14q32 locus in 121 PDB patients and 49 controls from the UK identified p.R279C, in strong LD with the GWAS signal rs10498635, as the most probable causal variant for this locus (p value = 1.4e-9, OR = 0.64 [0.55–0.74]). Two other common RIN3 variants (p.H215R and p.T425M) were also detected, but association was inconclusive. In addition, 13 rare missense variants were identified in these patients, affecting either the structured domains (SH2 and VSP9) or the proline-rich domain (Table 2). A combination of these rare variants was associated with an increased risk of presenting the disease [31]. Analysis of mouse tissues showed that RIN3 expresses higher in the lung, followed by bone tissue, with a tenfold expression in osteoclasts compared with that in osteoblasts. These findings suggest that RIN3 could be involved in the pathogenesis of PDB by affecting the osteoclast function in these patients [31].
A missense variant (L408P) in CSF1 gene was detected in a 30-year-old patient with juvenile Paget’s disease, a rare PDB-like syndrome appearing in early stages of life [97]. The patient also carried a missense variant D349G in TMSF4 gene. A rare variant (allele frequency < 0.05) rs62620995 in TM7SF4 gene was identified in a French-Canadian cohort of PDB patients, together with rs62641691 variant in CD276 (Table 2) [98]. Rs62620995 (p.Leu397Phe) could increase the activity of DC-STAMP, altering its expression or its internalization [99].
Other Factors
SQSTM1 protein directs ubiquitinated molecules to degradation in autophagolysosomes. It interacts with autophagy protein LC3, located in the ruffle border of the osteoclasts [48]. Besides, other autophagy proteins regulate osteoclastic bone resorption (ATG5, ATG7 and ATG4B), although the SQSTM1-mediated autophagy role in osteoclasts remains to be confirmed [100]. Alterations in autophagosomes have been found in other diseases with a pagetic component, such as inclusion body myopathy, PDB and FTD, linked to a mutation in the VCP gene [101]. Analysis of a Spanish cohort of 238 PDB patients showed that polymorphisms in genes associated with autophagosome formation, ATG16L1 and ATG5, were linked to an increased risk of developing PDB, whilst a polymorphism in ATG10 decreased the risk of suffering the condition (Table 2) [102].
Splicing site mutation in SQSTM1 have been reported [103, 104] and alternative splicing has been involved in the development of bone diseases, such as TCIRG1-linked autosomal recessive osteopetrosis [105]. Alternative splicing in six genes (LGALS8, RHOT1, CASC4, USP4, TBC1D25 and PIDD), not previously associated with the disease, but associated with TRAF6 ubiquitination [106], apoptosis [107–110] and autophagosome maturation [111], have been associated with PDB.
Genetics of PDB Severity
Results from the genome-wide association analysis helped to build up a risk allele score for severity of disease. In patients without SQSTM1 mutations, a combination of risk GWAS alleles in the highest tertile was associated with a 27% increase in disease extent, defined by the number of affected bones, and 25% increase in disease severity score, which includes complications secondary to the disease. SQSTM1+ve patients showed a highly significant increase in disease extent, severity and number of previous treatments received [112].
In a reduced number of cases, neoplastic transformation appears in the pagetic bones, producing osteosarcoma or, even less frequently, giant cell tumours [113, 114]. This is a serious condition since about 80% of patients diagnosed with GCT die in 10-year time. Analysis of a large family with 14 members affected by PDB, SQSTM1-ve, and four of them presenting giant cell tumours identified a heterozygous missense mutation in the ZNF687 gene (p.P937R) in all unrelated PDB patients and replicated in two families with PDB history. This variation was identified as a founder mutation since it originated from a unique haplotype and segregated in all but one GCT/PDB-affected individual in the study. These results confirmed that p.P937R is necessary and sufficient for the development of GCT in PBD patients. Authors also found a small group of familial PDB patients carrying this mutation, associated with a more severe phenotype than PDB patients without the mutation, with a polyostotic disease and earlier onset of disease. ZNF687 encodes a C2H2 zinc finger protein involved in the transcriptional regulator complex Z3. It is widely expressed, including in the bone, where it is upregulated in osteoclast and osteoblast differentiation in zebrafish model. This gene is located downstream of NF-kB. Mutation p.P937R is a gain-of-function change, producing an accumulation of the protein in the nucleus and subsequent transcription of the pathway downstream. Osteoclast derived from patients carrying this mutation presented an increased size and number of nuclei [115].
Environmental Triggers
Genetic predisposition plays a crucial role in the development of PDB; however, some studies have found that children whose parents carry SQSTM1 mutations do not always develop the disease, or they present a large delay in the appearance of the symptoms [34, 116, 117]. Similarly, mice expressing pP392L SQSTM1 mutation showed an increased number of osteoclasts and progressive bone loss, but osteoblasts were not increased and, therefore, did not present any visible pagetic lesion [118]. These findings, together with the reported changes in the incidence of PDB, support the role of environmental factors in the development of the disease. A persistent viral infection was proposed after observing intracellular inclusions in osteoclasts, similar to measles nucleocapsids (MVNP) [119, 120]. The nature of these bodies is still controversial, since some groups have not found any connexion [121]. It has been suggested lately that they could be protein aggregates resulting from the dysregulation of the autophagy system [122, 123]. However, recent studies have shown that the MVNP protein is associated with the upregulation of IL-6 and IGF1 in osteoclasts from mouse models and PDB patients, which could suggest a role for measles virus in the alteration of bone formation seen in these patients [124, 125].
PDB has also been linked to other factors such as poor calcium and vitamin D intake, consumption of uncontrolled beef meat during childhood [126], consumption of not purified water [10], contact with dogs during early years [127], an excessive mechanical loading on the skeleton and exposure to some environmental toxics [128].
Treatment
The main and the only absolute indication for treatment with clear clinical evidence of PDB is pain in the affected bone [29]. In several clinical trials, bisphosphonates were effective in managing pain in PDB patients. Zoledronic acid is the most potent drug and is currently the first choice of treatment [129]. Calcitonin is effective in reducing pain and expression of bone formation and resorption markers, although its power is clearly lower than that of bisphosphonates; thus, it is rarely used nowadays [130]. Denosumab is a potent inhibitor of bone resorption and has been reported to decrease disease activity in one PDB patient [131], but it has not been tested yet in clinical trials for PDB. Orthopaedic surgery is recommended mainly for bone pagetic fractures, spinal stenosis or pagetic osteoarthritis.
Treatment of PDB patients showing only biochemical activity but no pain is under debate. To date, there is not enough evidence on preventive treatment of complications in asymptomatic patients. The PRISM trial and its extension showed no beneficial effect on the quality of life, fractures, orthopaedic surgery or deafness in patients treated repeatedly with bisphosphonates [33]. An international randomized clinical trial led by Prof Stuart Ralston, at the University of Edinburgh, UK (Zoledronate in the prevention of Paget’s: the ZiPP study, ISRCTN11616770) is currently in progress to detect the effect of bisphosphonate treatment in SQSTM1+ve individuals who have not developed any symptom.
Conclusion
Paget’s disease of bone is a common disorder resulting from a combination of genetic and environmental factors. To date, clinical, laboratory or radiographic features have been used to identify the disease and provide treatment, although the guidelines to prescribe zoledronic acid are still under debate.
SQSTM1 mutations are associated with susceptibility to develop PDB. However, only 40% of familial PDB and 10% of sporadic PDB patients present alterations in this gene. Latest advances in the genetics field identified seven other genes predisposing to the disease. It has been shown that genetic information may constitute a good tool to manage presymptomatic patients. A risk allele score has been developed using the information from all PDB loci, to successfully detect an accumulative risk to develop a more severe disease when carrying a large number of risk alleles. In addition, forthcoming results from the ZiPP study will be crucial to determine prophylactic treatment based on genetic profiling may contribute to prevent skeletal complications associated with PDB.
However, despite the great genetic advances, further research is needed to elucidate other aspects of the disease, including its focal nature, and the changes in severity and prevalence observed in some populations.
References
van Staa TP, Selby P, Leufkens HG, Lyles K, Sprafka JM, Cooper C. Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res. 2002;17(3):465–71.
Paget J. On a form of chronic inflammation of bones (osteitis deformans). Med Chir Trans. 1877;60:37–64.
Cooper C, Dennison E, Schafheutle K, Kellingray S, Guyer P, Barker D. Epidemiology of Paget’s disease of bone. Bone. 1999;24(5 Suppl):3S–5S.
Corral-Gudino L, Borao-Cengotita-Bengoa M, Del Pino-Montes J, Ralston S. Epidemiology of Paget’s disease of bone: a systematic review and meta-analysis of secular changes. Bone. 2013;55(2):347–52.
Reasbeck JC, Goulding A, Campbell DR, Beale LR, Stewart RD. Radiological prevalence of Paget’s disease in Dunedin, New Zealand. Br Med J (Clin Res Ed). 1983;286(6382):1937.
Guyer PB, Chamberlain AT. Paget’s disease of bone in two American cities. Br Med J. 1980;280(6219):985.
Dahniya MH. Paget’s disease of bone in Africans. Br J Radiol. 1987;60(710):113–6.
Corral-Gudino L, Garcia-Aparicio J, Sanchez-Gonzalez MD, Miron-Canelo JA, Blanco JF, Ralston SH, et al. Secular changes in Paget’s disease: contrasting changes in the number of new referrals and in disease severity in two neighboring regions of Spain. Osteoporos Int. 2013;24(2):443–50.
Barker DJ, Chamberlain AT, Guyer PB, Gardner MJ. Paget’s disease of bone: the Lancashire focus. Br Med J. 1980;280(6222):1105–7.
Miron-Canelo JA, Del Pino-Montes J, Vicente-Arroyo M, Saenz-Gonzalez MC. Epidemiological study of Paget’s disease of bone in a zone of the province of Salamanca (Spain). The Paget’s disease of the bone study group of Salamanca. Eur J Epidemiol. 1997;13(7):801–5.
Cundy HR, Gamble G, Wattie D, Rutland M, Cundy T. Paget’s disease of bone in New Zealand: continued decline in disease severity. Calcif Tissue Int. 2004;75(5):358–64.
Poor G, Donath J, Fornet B, Cooper C. Epidemiology of Paget’s disease in Europe: the prevalence is decreasing. J Bone Miner Res. 2006;21(10):1545–9.
Tiegs RD, Lohse CM, Wollan PC, Melton LJ. Long-term trends in the incidence of Paget’s disease of bone. Bone. 2000;27(3):423–7.
Tan A, Ralston SH. Clinical presentation of Paget’s disease: evaluation of a contemporary cohort and systematic review. Calcif Tissue Int. 2014;95(5):385–92.
Vasireddy S, Talwalkar A, Miller H, Mehan R, Swinson DR. Patterns of pain in Paget’s disease of bone and their outcomes on treatment with pamidronate. Clin Rheumatol. 2003;22(6):376–80.
Guma M, Rotes D, Holgado S, Monfort J, Olive A, Carbonell J, et al. Paget’s disease of bone: study of 314 patients. Med Clin (Barc). 2002;119(14):537–40.
Bolland MJ, Cundy T. Paget’s disease of bone: clinical review and update. J Clin Pathol. 2013;66(11):924–7.
Altman RD. Musculoskeletal manifestations of Paget’s disease of bone. Arthritis Rheum. 1980;23(10):1121–7.
Bone HG. Nonmalignant complications of Paget’s disease. J Bone Miner Res. 2006;21(Suppl 2):64–8.
Seton M. Paget disease of bone: diagnosis and drug therapy. Cleve Clin J Med. 2013;80(7):452–62.
Seitz S, Priemel M, Zustin J, Beil FT, Semler J, Minne H, et al. Paget’s disease of bone: histologic analysis of 754 patients. J Bone Miner Res. 2009;24(1):62–9.
Roodman GD, Windle JJ. Paget disease of bone. J Clin Invest. 2005;115(2):200–8.
Ooi CG, Fraser WD. Paget’s disease of bone. Postgrad Med J. 1997;73(856):69–74.
Theodorou DJ, Theodorou SJ, Kakitsubata Y. Imaging of Paget disease of bone and its musculoskeletal complications: review. AJR Am J Roentgenol. 2011;196(6 Suppl):S64–75.
Shankar YU, Misra SR, Vineet DA, Baskaran P. Paget disease of bone: a classic case report. Contemp Clin Dent. 2013;4(2):227–30.
Zimmermann EA, Kohne T, Bale HA, Panganiban B, Gludovatz B, Zustin J, et al. Modifications to nano- and microstructural quality and the effects on mechanical integrity in Paget’s disease of bone. J Bone Miner Res. 2015;30(2):264–73.
Al Nofal AA, Altayar O, BenKhadra K, Qasim Agha OQ, Asi N, Nabhan M, et al. Bone turnover markers in Paget’s disease of the bone: a systematic review and meta-analysis. Osteoporos Int. 2015;26(7):1875–91.
Davis MA, Scalcione LR, Gimber LH, Thompson RB, Avery RJ, Taljanovic MS. Paget sarcoma of the pelvic bone with widespread metastatic disease on radiography, CT, MRI, and 18F-FDG PET/CT with pathologic correlation. Clin Nucl Med. 2014;39(4):371–3.
Selby PL, Davie MW, Ralston SH, Stone MD. Guidelines on the management of Paget’s disease of bone. Bone. 2002;31(3):366–73.
Reddy SV. Etiology of Paget’s disease and osteoclast abnormalities. J Cell Biochem. 2004;93(4):688–96.
Vallet M, Soares DC, Wani S, Sophocleous A, Warner J, Salter DM, et al. Targeted sequencing of the Paget’s disease associated 14q32 locus identifies several missense coding variants in RIN3 that predispose to Paget’s disease of bone. Hum Mol Genet. 2015;24(11):3286–95.
Pick A. Osteitis deformans. Lancet. 1883;2:1125–6.
Langston AL, Campbell MK, Fraser WD, MacLennan GS, Selby PL, Ralston SH. Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget’s disease of bone. J Bone Miner Res. 2010;25(1):20–31.
Morissette J, Laurin N, Brown JP. Sequestosome 1: mutation frequencies, haplotypes, and phenotypes in familial Paget’s disease of bone. J Bone Miner Res. 2006;21(Suppl 2):38–44.
Eekhoff EW, Karperien M, Houtsma D, Zwinderman AH, Dragoiescu C, Kneppers AL, et al. Familial Paget’s disease in the Netherlands: occurrence, identification of new mutations in the sequestosome 1 gene, and their clinical associations. Arthritis Rheum. 2004;50(5):1650–4.
Morales-Piga AA, Rey-Rey JS, Corres-Gonzalez J, Garcia-Sagredo JM, Lopez-Abente G. Frequency and characteristics of familial aggregation of Paget’s disease of bone. J Bone Miner Res. 1995;10(4):663–70.
Hocking LJ, Herbert CA, Nicholls RK, Williams F, Bennett ST, Cundy T, et al. Genomewide search in familial Paget disease of bone shows evidence of genetic heterogeneity with candidate loci on chromosomes 2q36, 10p13, and 5q35. Am J Hum Genet. 2001;69(5):1055–61.
Laurin N, Brown JP, Lemainque A, Duchesne A, Huot D, Lacourciere Y, et al. Paget disease of bone: mapping of two loci at 5q35-qter and 5q31. Am J Hum Genet. 2001;69(3):528–43.
Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet. 2002;70(6):1582–8.
Fotino M, Haymovits A, Falk CT. Evidence for linkage between HLA and Paget’s disease. Transplant Proc. 1977;9(4):1867–8.
Tilyard MW, Gardner RJ, Milligan L, Cleary TA, Stewart RD. A probable linkage between familial Paget’s disease and the HLA loci. Aust NZ J Med. 1982;12(5):498–500.
Cody JD, Singer FR, Roodman GD, Otterund B, Lewis TB, Leppert M, et al. Genetic linkage of Paget disease of the bone to chromosome 18q. Am J Hum Genet. 1997;61(5):1117–22.
Haslam SI, Van HW, Morales-Piga A, Balemans W, San-Millan JL, Nakatsuka K, et al. Paget’s disease of bone: evidence for a susceptibility locus on chromosome 18q and for genetic heterogeneity. J Bone Miner Res. 1998;13(6):911–7.
Hughes AE, Ralston SH, Marken J, Bell C, MacPherson H, Wallace RG, et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat Genet. 2000;24(1):45–8.
Nakatsuka K, Nishizawa Y, Ralston SH. Phenotypic characterization of early onset Paget’s disease of bone caused by a 27-bp duplication in the TNFRSF11A gene. J Bone Miner Res. 2003;18(8):1381–5.
Good DA, Busfield F, Fletcher BH, Duffy DL, Kesting JB, Andersen J, et al. Linkage of Paget disease of bone to a novel region on human chromosome 18q23. Am J Hum Genet. 2002;70(2):517–25.
Geetha T, Wooten MW. Structure and functional properties of the ubiquitin binding protein p62. FEBS Lett. 2002;512(1–3):19–24.
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–45.
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–63.
Vadlamudi RK, Joung I, Strominger JL, Shin J. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem. 1996;271(34):20235–7.
Hocking LJ, Lucas GJ, Daroszewska A, Mangion J, Olavesen M, Cundy T, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet. 2002;11(22):2735–9.
Daroszewska A, van ‘t Hof RJ, Rojas JA, Layfield R, Landao-Basonga E, Rose L, et al. A point mutation in the ubiquitin-associated domain of SQSMT1 is sufficient to cause a Paget’s disease-like disorder in mice. Hum Mol Genet. 2011;20(14):2734–44.
Beyens G, Van HE, Van DK, Fransen E, Devogelaer JP, Vanhoenacker F, et al. Evaluation of the role of the SQSTM1 gene in sporadic Belgian patients with Paget’s disease. Calcif Tissue Int. 2004;75(2):144–52.
Falchetti A, Di SM, Marini F, Del MF, Mavilia C, Strigoli D, et al. Two novel mutations at exon 8 of the sequestosome 1 (SQSTM1) gene in an Italian series of patients affected by Paget’s disease of bone (PDB). J Bone Miner Res. 2004;19(6):1013–7.
Johnson-Pais TL, Wisdom JH, Weldon KS, Cody JD, Hansen MF, Singer FR, et al. Three novel mutations in SQSTM1 identified in familial Paget’s disease of bone. J Bone Miner Res. 2003;18(10):1748–53.
Gu JM, Zhang ZL, Zhang H, Hu WW, Wang C, Yue H, et al. Thirteen Chinese patients with sporadic Paget’s disease of bone: clinical features, SQSTM1 mutation identification, and functional analysis. J Bone Miner Metab. 2012;30(5):525–33.
Tang Z, Liu F, Pang Y. Seven cases of a family with hereditary Paget’s disease of bone. Chin J Endocrinol Metab. 2016;15:372.
Collet C, Michou L, Audran M, Chasseigneaux S, Hilliquin P, Bardin T, et al. Paget’s disease of bone in the French population: novel SQSTM1 mutations, functional analysis, and genotype-phenotype correlations. J Bone Miner Res. 2007;22(2):310–7.
Falchetti A, Di SM, Marini F, Ortolani S, Ulivieri MF, Bergui S, et al. Genetic epidemiology of Paget’s disease of bone in Italy: sequestosome1/p62 gene mutational test and haplotype analysis at 5q35 in a large representative series of sporadic and familial Italian cases of Paget’s disease of bone. Calcif Tissue Int. 2009;84(1):20–37.
Michou L, Morissette J, Gagnon ER, Marquis A, Dellabadia M, Brown JP, et al. Novel SQSTM1 mutations in patients with Paget’s disease of bone in an unrelated multiethnic American population. Bone. 2011;48(3):456–60.
Rea SL, Walsh JP, Ward L, Magno AL, Ward BK, Shaw B, et al. Sequestosome 1 mutations in Paget’s disease of bone in Australia: prevalence, genotype/phenotype correlation, and a novel non-UBA domain mutation (P364S) associated with increased NF-kappaB signaling without loss of ubiquitin binding. J Bone Miner Res. 2009;24(7):1216–23.
Hocking LJ, Lucas GJ, Daroszewska A, Cundy T, Nicholson GC, Donath J, et al. Novel UBA domain mutations of SQSTM1 in Paget’s disease of bone: genotype phenotype correlation, functional analysis, and structural consequences. J Bone Miner Res. 2004;19(7):1122–7.
Visconti MR, Langston AL, Alonso N, Goodman K, Selby PL, Fraser WD, et al. Mutations of SQSTM1 are associated with severity and clinical outcome in paget disease of bone. J Bone Miner Res. 2010;25(11):2368–73.
Wright T, Rea SL, Goode A, Bennett AJ, Ratajczak T, Long JE, et al. The S349T mutation of SQSTM1 links Keap1/Nrf2 signalling to Paget’s disease of bone. Bone. 2013;52(2):699–706.
Merchant A, Smielewska M, Patel N, Akunowicz JD, Saria EA, Delaney JD, et al. Somatic mutations in SQSTM1 detected in affected tissues from patients with sporadic Paget’s disease of bone. J Bone Miner Res. 2009;24(3):484–94.
Guay-Belanger S, Picard S, Gagnon E, Morissette J, Siris ES, Orcel P, et al. Detection of SQSTM1/P392L post-zygotic mutations in Paget’s disease of bone. Hum Genet. 2015;134(1):53–65.
Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68(11):1440–6.
Rubino E, Rainero I, Chio A, Rogaeva E, Galimberti D, Fenoglio P, et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79(15):1556–62.
Teyssou E, Takeda T, Lebon V, Boillee S, Doukoure B, Bataillon G, et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol. 2013;125(4):511–22.
Hirano M, Nakamura Y, Saigoh K, Sakamoto H, Ueno S, Isono C, et al. Mutations in the gene encoding p62 in Japanese patients with amyotrophic lateral sclerosis. Neurology. 2013;80(5):458–63.
Kwok CT, Morris A, de Belleroche JS. Sequestosome-1 (SQSTM1) sequence variants in ALS cases in the UK: prevalence and coexistence of SQSTM1 mutations in ALS kindred with PDB. Eur J Hum Genet. 2014;22(4):492–6.
Estrada K, Styrkarsdottir U, Evangelou E, Hsu YH, Duncan EL, Ntzani EE, et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet. 2012;44(5):491–501.
Albagha OM, Wani SE, Visconti MR, Alonso N, Goodman K, Brandi ML, et al. Genome-wide association identifies three new susceptibility loci for Paget’s disease of bone. Nat Genet. 2011;43(7):685–9.
Albagha OM, Visconti MR, Alonso N, Langston AL, Cundy T, Dargie R, et al. Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget’s disease of bone. Nat Genet. 2010;42(6):520–4.
Tsurukai T, Udagawa N, Matsuzaki K, Takahashi N, Suda T. Roles of macrophage-colony stimulating factor and osteoclast differentiation factor in osteoclastogenesis. J Bone Miner Metab. 2000;18(4):177–84.
Bouyer P, Sakai H, Itokawa T, Kawano T, Fulton CM, Boron WF, et al. Colony-stimulating factor-1 increases osteoclast intracellular pH and promotes survival via the electroneutral Na/HCO3 cotransporter NBCn1. Endocrinology. 2007;148(2):831–40.
Neale SD, Schulze E, Smith R, Athanasou NA. The influence of serum cytokines and growth factors on osteoclast formation in Paget’s disease. QJM. 2002;95(4):233–40.
Albagha OM. Genetics of Paget’s disease of bone. Bonekey Rep. 2015;4:756.
Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002;295(5557):1077–9.
Li J, Sarosi I, Yan XQ, Morony S, Capparelli C, Tan HL, et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci U S A. 2000;97(4):1566–71.
Gianfrancesco F, Rendina D, Di SM, Mingione A, Esposito T, Merlotti D, et al. A nonsynonymous TNFRSF11A variation increases NFkappaB activity and the severity of Paget’s disease. J Bone Miner Res. 2012;27(2):443–52.
Whyte MP, Hughes AE. Expansile skeletal hyperphosphatasia is caused by a 15-base pair tandem duplication in TNFRSF11A encoding RANK and is allelic to familial expansile osteolysis. J Bone Miner Res. 2002;17(1):26–9.
Grandi P, Dang T, Pane N, Shevchenko A, Mann M, Forbes D, et al. Nup93, a vertebrate homologue of yeast Nic96p, forms a complex with a novel 205-kDa protein and is required for correct nuclear pore assembly. Mol Biol Cell. 1997;8(10):2017–38.
Yagi M, Miyamoto T, Sawatani Y, Iwamoto K, Hosogane N, Fujita N, et al. DC-STAMP is essential for cell–cell fusion in osteoclasts and foreign body giant cells. J Exp Med. 2005;202(3):345–51.
Kukita T, Wada N, Kukita A, Kakimoto T, Sandra F, Toh K, et al. RANKL-induced DC-STAMP is essential for osteoclastogenesis. J Exp Med. 2004;200(7):941–6.
Saito K, Murai J, Kajiho H, Kontani K, Kurosu H, Katada T. A novel binding protein composed of homophilic tetramer exhibits unique properties for the small GTPase Rab5. J Biol Chem. 2002;277(5):3412–8.
Kajiho H, Saito K, Tsujita K, Kontani K, Araki Y, Kurosu H, et al. RIN3: a novel Rab5 GEF interacting with amphiphysin II involved in the early endocytic pathway. J Cell Sci. 2003;116(Pt 20):4159–68.
Lin HK, Bergmann S, Pandolfi PP. Cytoplasmic PML function in TGF-beta signalling. Nature. 2004;431(7005):205–11.
Hennies HC, Kornak U, Zhang H, Egerer J, Zhang X, Seifert W, et al. Gerodermia osteodysplastica is caused by mutations in SCYL1BP1, a Rab-6 interacting golgin. Nat Genet. 2008;40(12):1410–2.
Smits P, Bolton AD, Funari V, Hong M, Boyden ED, Lu L, et al. Lethal skeletal dysplasia in mice and humans lacking the golgin GMAP-210. N Engl J Med. 2010;362(3):206–16.
Obaid R, Wani SE, Azfer A, Hurd T, Jones R, Cohen P, et al. Optineurin negatively regulates osteoclast differentiation by modulating NF-kappaB and interferon signaling: implications for Paget’s disease. Cell Rep. 2015;13(6):1096–102.
Zhu G, Wu CJ, Zhao Y, Ashwell JD. Optineurin negatively regulates TNFalpha-induced NF-kappaB activation by competing with NEMO for ubiquitinated RIP. Curr Biol. 2007;17(16):1438–43.
Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333(6039):228–33.
Coxon FP, Rogers MJ. The role of prenylated small GTP-binding proteins in the regulation of osteoclast function. Calcif Tissue Int. 2003;72(1):80–4.
Van WL, Odgren PR, Coxon FP, Frattini A, Moens P, Perdu B, et al. Involvement of PLEKHM1 in osteoclastic vesicular transport and osteopetrosis in incisors absent rats and humans. J Clin Invest. 2007;117(4):919–30.
Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36(4):377–81.
Donath J, Speer G, Kosa JP, Arvai K, Balla B, Juhasz P, et al. Polymorphisms of CSF1 and TM7SF4 genes in a case of mild juvenile Paget’s disease found using next-generation sequencing. Croat Med J. 2015;56(2):145–51.
Beauregard M, Gagnon E, Guay-Belanger S, Morissette J, Brown JP, Michou L. Identification of rare genetic variants in novel loci associated with Paget’s disease of bone. Hum Genet. 2014;133(6):755–68.
Singer FR, Mills BG, Gruber HE, Windle JJ, Roodman GD. Ultrastructure of bone cells in Paget’s disease of bone. J Bone Miner Res. 2006;21(Suppl 2):51–4.
DeSelm CJ, Miller BC, Zou W, Beatty WL, Van ME, Takahata Y, et al. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell. 2011;21(5):966–74.
Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, et al. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. 2010;6(2):217–27.
Usategui-Martin R, Garcia-Aparicio J, Corral-Gudino L, Calero-Paniagua I, Del Pino-Montes J, Gonzalez SR. Polymorphisms in autophagy genes are associated with paget disease of bone. PLoS One. 2015;10(6):e0128984.
Cavey JR, Ralston SH, Hocking LJ, Sheppard PW, Ciani B, Searle MS, et al. Loss of ubiquitin-binding associated with Paget’s disease of bone p62 (SQSTM1) mutations. J Bone Miner Res. 2005;20(4):619–24.
Beyens G, Wuyts W, Cleiren E, De FF, Tiegs R, Van HW. Identification and molecular characterization of a novel splice-site mutation (G1205C) in the SQSTM1 gene causing Paget’s disease of bone in an extended American family. Calcif Tissue Int. 2006;79(5):281–8.
Susani L, Pangrazio A, Sobacchi C, Taranta A, Mortier G, Savarirayan R, et al. TCIRG1-dependent recessive osteopetrosis: mutation analysis, functional identification of the splicing defects, and in vitro rescue by U1 snRNA. Hum Mutat. 2004;24(3):225–35.
Xiao N, Li H, Luo J, Wang R, Chen H, Chen J, et al. Ubiquitin-specific protease 4 (USP4) targets TRAF2 and TRAF6 for deubiquitination and inhibits TNFalpha-induced cancer cell migration. Biochem J. 2012;441(3):979–86.
Fransson A, Ruusala A, Aspenstrom P. Atypical rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem. 2003;278(8):6495–502.
Eshkar SL, Ronen D, Levartovsky D, Elkayam O, Caspi D, Aamar S, et al. The involvement of CD44 and its novel ligand galectin-8 in apoptotic regulation of autoimmune inflammation. J Immunol. 2007;179(2):1225–35.
Norambuena A, Metz C, Vicuna L, Silva A, Pardo E, Oyanadel C, et al. Galectin-8 induces apoptosis in Jurkat T cells by phosphatidic acid-mediated ERK1/2 activation supported by protein kinase A down-regulation. J Biol Chem. 2009;284(19):12670–9.
Huang L, Han D, Yang X, Qin B, Ji G, Yu L. PIDD4, a novel PIDD isoform without the LRR domain, can independently induce cell apoptosis in cytoplasm. Biochem Biophys Res Commun. 2011;407(1):86–91.
Itoh T, Kanno E, Uemura T, Waguri S, Fukuda M. OATL1, a novel autophagosome-resident Rab33B-GAP, regulates autophagosomal maturation. J Cell Biol. 2011;192(5):839–53.
Albagha OM, Visconti MR, Alonso N, Wani S, Goodman K, Fraser WD, et al. Common susceptibility alleles and SQSTM1 mutations predict disease extent and severity in a multinational study of patients with Paget’s disease. J Bone Miner Res. 2013;28(11):2338–46.
Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res. 2006;21(Suppl 2):58–63.
Rendina D, De FG, Ralston SH, Merlotti D, Gianfrancesco F, Esposito T, et al. Clinical characteristics and evolution of giant cell tumor occurring in Paget’s disease of bone. J Bone Miner Res. 2015;30(2):257–63.
Divisato G, Formicola D, Esposito T, Merlotti D, Pazzaglia L, Del FA, et al. ZNF687 mutations in severe Paget disease of bone associated with giant cell tumor. Am J Hum Genet. 2016;98(2):275–86.
Bolland MJ, Tong PC, Naot D, Callon KE, Wattie DJ, Gamble GD, et al. Delayed development of Paget’s disease in offspring inheriting SQSTM1 mutations. J Bone Miner Res. 2007;22(3):411–5.
Cundy T, Rutland MD, Naot D, Bolland M. Evolution of Paget’s disease of bone in adults inheriting SQSTM1 mutations. Clin Endocrinol. 2015;83(3):315–9.
Kurihara N, Hiruma Y, Zhou H, Subler MA, Dempster DW, Singer FR, et al. Mutation of the sequestosome 1 (p62) gene increases osteoclastogenesis but does not induce Paget disease. J Clin Invest. 2007;117(1):133–42.
Rebel A, Malkani K, Basle M. Nuclear anomalies in osteoclasts in Paget’s bone disease. Nouv Press Med. 1974;3(20):1299–301.
Rebel A, Basle M, Pouplard A, Kouyoumdjian S, Filmon R, Lepatezour A. Viral antigens in osteoclasts from Paget’s disease of bone. Lancet. 1980;2(8190):344–6.
Helfrich MH, Hobson RP, Grabowski PS, Zurbriggen A, Cosby SL, Dickson GR, et al. A negative search for a paramyxoviral etiology of Paget’s disease of bone: molecular, immunological, and ultrastructural studies in UK patients. J Bone Miner Res. 2000;15(12):2315–29.
Sieradzan KA, Mechan AO, Jones L, Wanker EE, Nukina N, Mann DM. Huntington’s disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Exp Neurol. 1999;156(1):92–9.
Hocking LJ, Mellis DJ, McCabe PS, Helfrich MH, Rogers MJ. Functional interaction between sequestosome-1/p62 and autophagy-linked FYVE-containing protein WDFY3 in human osteoclasts. Biochem Biophys Res Commun. 2010;402(3):543–8.
Teramachi J, Nagata Y, Mohammad K, Inagaki Y, Ohata Y, Guise T, et al. Measles virus nucleocapsid protein increases osteoblast differentiation in Paget’s disease. J Clin Invest. 2016;126(3):1012–22.
Kurihara N, Hiruma Y, Yamana K, Michou L, Rousseau C, Morissette J, et al. Contributions of the measles virus nucleocapsid gene and the SQSTM1/p62(P392L) mutation to Paget’s disease. Cell Metab. 2011;13(1):23–34.
Piga AM, Lopez-Abente G, Ibanez AE, Vadillo AG, Lanza MG, Jodra VM. Risk factors for Paget’s disease: a new hypothesis. Int J Epidemiol. 1988;17(1):198–201.
Holdaway IM, Ibbertson HK, Wattie D, Scragg R, Graham P. Previous pet ownership and Paget’s disease. Bone Miner. 1990;8(1):53–8.
Lever JH. Paget’s disease of bone in Lancashire and arsenic pesticide in cotton mill wastewater: a speculative hypothesis. Bone. 2002;31(3):434–6.
Reid IR, Lyles K, Su G, Brown JP, Walsh JP, Del Pino-Montes J, et al. A single infusion of zoledronic acid produces sustained remissions in Paget disease: data to 6.5 years. J Bone Miner Res. 2011;26(9):2261–70.
Singer F. 2000 Paget’s Disease of Bone.
Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jorgensen NR. Paget’s disease of the bone after treatment with Denosumab: a case report. Bone. 2012;50(5):1023–5.
Rea SL, Walsh JP, Ward L, Yip K, Ward BK, Kent GN, et al. A novel mutation (K378X) in the sequestosome 1 gene associated with increased NF-kappaB signaling and Paget’s disease of bone with a severe phenotype. J Bone Miner Res. 2006;21(7):1136–45.
Gennari L, Gianfrancesco F, Di SM, Rendina D, Merlotti D, Esposito T, et al. SQSTM1 gene analysis and gene-environment interaction in Paget’s disease of bone. J Bone Miner Res. 2010;25(6):1375–84.
Longato L. Paget’s disease population analysis within Rheumatology Outpatient of the ASL of Biella (Piedmont Region, Italy). Clin Cases Miner Bone Metab. 2014;11(1):49–58.
Cundy T, Naot D, Bava U, Musson D, Tong PC, Bolland M. Familial Paget disease and SQSTM1 mutations in New Zealand. Calcif Tissue Int. 2011;89(3):258–64.
Good DA, Busfield F, Fletcher BH, Lovelock PK, Duffy DL, Kesting JB, et al. Identification of SQSTM1 mutations in familial Paget’s disease in Australian pedigrees. Bone. 2004;35(1):277–82.
Rea SL, Walsh JP, Layfield R, Ratajczak T, Xu J. New insights into the role of sequestosome 1/p62 mutant proteins in the pathogenesis of Paget’s disease of bone. Endocr Rev. 2013;34(4):501–24.
Goode A, Long JE, Shaw B, Ralston SH, Visconti MR, Gianfrancesco F, et al. Paget disease of bone-associated UBA domain mutations of SQSTM1 exert distinct effects on protein structure and function. Biochim Biophys Acta. 2014;1842(7):992–1000.
Acknowledgements
Open access funding provided by University of Edinburgh. We would like to thank Dr Giovanny Rodriguez-Blanco and Prof Stuart H Ralston for their critic revision of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The article does not contain any studies with human or animal subjects performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Alonso, N., Calero-Paniagua, I. & del Pino-Montes, J. Clinical and Genetic Advances in Paget’s Disease of Bone: a Review. Clinic Rev Bone Miner Metab 15, 37–48 (2017). https://doi.org/10.1007/s12018-016-9226-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12018-016-9226-0