
Abstract
Covalent post-translational modification of proteins by ubiquitin and ubiquitin-like factors has emerged as a general mechanism to regulate myriad intra-cellular processes. The addition and removal of ubiquitin or ubiquitin-like proteins from factors has recently been demonstrated as a key mechanism to modulate DNA damage response (DDR) pathways. It is thus, timely to evaluate the potential for ubiquitin pathway enzymes as DDR drug targets for therapeutic intervention. The synthetic lethal approach provides exciting opportunities for the development of targeted therapies to treat cancer: most tumours have lost critical DDR pathways, and thus rely more heavily on the remaining pathways, while normal tissues are still equipped with all DDR pathways. Here, we review key deubiquitylating enzymes (DUBs) involved in DDR pathways, and describe how targeting DUBs may lead to selective therapies to treat cancer patients.
Similar content being viewed by others
DNA Damage Responses and Cancer
Tumorigenesis is a multistep process, driven by genetic alterations that allow the progressive transformation of normal cells into highly malignant tumours. Genomic instability is fuelled by DNA damage and errors introduced during DNA replication. Many factors—which include endogenously arising agents, such as reactive oxygen species and metabolic by-products together with exogenous agents, such as ultra-violet light, ionising radiation, tobacco smoke chemicals and other genotoxic chemicals—have been identified that generate a range of different damage types or lesions on DNA; see [1] for a recent review. Several complex and interconnected DNA-repair systems have therefore evolved to recognise and correct most of the insults inflicted on the cell’s vital genetic information. Importantly, radiotherapy and most commonly-used anti-cancer chemotherapies operate by generating DNA damage. While, effective tumour eradication by these treatments results from the generation of irreparable DNA damage in tumour cells, resistance mechanisms are provided by tumour cell DNA-repair pathways. DNA damage to normal cells results in the toxicities usually associated with such treatments.
Overlapping DNA repair pathways operate in mammalian cells and, together with DNA-damage signalling processes, they comprise what is often referred to as the cellular “DNA damage response” (DDR), defects in which cause various human diseases [2, 3]. Prime aspects of the DDR are the various DNA repair mechanisms, which encompass: the nucleotide excision repair (NER) and base excision repair (BER) pathways that deal with various DNA helix-distorting lesions and single-strand breaks; mismatch repair pathways that deal with base mismatches and insertions/deletions; while very toxic DNA double-strand breaks (DSBs) are either dealt with by the non-homologous end-joining pathway (NHEJ) and/or less error-prone homologous recombination (HR) pathways. In addition, the Fanconi anaemia (FA) pathway operates in conjunction with certain HR factors to recognise and repair lesions such as inter-strand DNA cross-links.
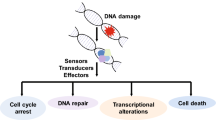
The cellular response to DNA damage is propagated through signal transduction and post-translational modification of proteins involved in the various DNA repair pathways. The amplification of signals from the damage site has an important regulatory function in controlling the cell’s responses to the DNA lesion [4]. Responses to damage can take several forms, depending on where a cell is in its cell cycle, replication status or the level of damage incurred. For instance, signalling can trigger cell-cycle checkpoints that cause the cell to slow or stop cell-cycle progression, thereby preventing replication of damaged DNA templates or mitotic segregation of damaged chromosomes [5–7]. If, however, the level of damage is too high and incompatible with repair, DDR signalling may trigger apoptotic cell death, autophagy or senescence mechanisms, where either the cell is killed or placed into a non-dividing phenotype [8–10]. Central to many of the responses following DNA damage, is the regulation of p53 and the modification of chromatin mediated by various post-translational processes, which are important to trigger various DDR events and maintain genomic integrity [11–13].
Although DDR deficiencies have been linked to a wide range of human pathologies, such as infertility, immune-system defects, neurological defects and developmental disorders, most research has focused on the striking associations that exist between DDR defects and cancer [3, 14]. Indeed, hereditary mutations or epigenetic silencing of a variety of genes that control DNA repair processes are recognised to cause or contribute to early cancer formation in many instances. For example, mutations in the DNA mismatch repair genes MSH2 and MLH1 can lead to non-polyposis colorectal cancers (HNPCC) in a significant number of patients [15, 16]. Similarly, defects in the HR-promoting and DDR-signalling protein kinase ATM, characterised by the syndrome ataxia telangiectasia, are associated with increased incidence of malignancies [17, 18]. Furthermore, mutations in DNA repair genes, such as NBS1, BLM and WRN, all give rise to syndromes associated with high-cancer prevalence [19]. Inherited mutations in the strongly breast cancer predisposing genes BRCA1 and BRCA2, both involved in HR DSB repair processes, are responsible for a considerable proportion of familial breast and ovarian cancers cases [20–22]. BRCA1/2 mutation carriers also show increased risks of developing other cancer types, including prostate, pancreatic and stomach cancers [23, 24]. FA is another disease where mutations in one of fifteen FA genes lead to defects in DNA inter-strand cross-link (ICL) repair, and HR is associated with increased cancer incidence [25, 26].
In addition to DDR factors being linked to cancer through the above hereditary connections, there is strong and growing evidence that DDR defects contribute more widely to sporadic cancers. Indeed, one of the most frequent, early events in tumorigenesis involves abrogation of particular DDR processes. One aspect of such DDR dysfunction is that it can result in increased genomic instability and consequently an increase in mutation rates that, in turn, fosters cancer initiation and progression. In addition, loss of certain DDR components appears to be selected for during early stages of tumorigenesis to dampen genotoxic stress-induced cell death pathways that would otherwise be triggered by heightened levels of DNA damage induction that exists in many cancers and in their precursors. Part of this higher DNA-damage load in cancers arises from factors such as telomere shortening—which triggers DDR activation [27]—and through their growth in non-optimal environments. Moreover, recent study has shown that activation of various oncogenes, such as Ras and Myc, leads to replicative stress, thus leading to DNA damage in S-phase [9, 28]. In light of these factors, cancer cells invariably display differences in their DDR repertoire to normal cells of the patient, and crucially, this often means that cancer cells are more reliant on certain DDR pathways than normal cells. It is this addiction or reliance on particular repair pathway(s) that can be exploited therapeutically in cancer, through the concept of synthetic lethality [29, 30]. In this scenario, a drug inhibiting a particular DDR component will be much more toxic to cancer cells than normal cells (Fig. 1). In other instances, such a DDR targeting drug will enhance the cytotoxicity of standard radiotherapy or chemotherapies much more in cancer cells than in normal cells.

Synthetic lethality relationships. The process of loss of DDR pathways during tumorigenesis is depicted here, and summarises the critical differences between normal and tumour cells. Cancer is in part driven by changes in a cell’s DNA repair capacity and DDR. Inhibiting these pathways can selectively kill cancer cells through a process called synthetic lethality
While there are various potential avenues to drug DDR pathways, most study to date has focused on targeting enzymes that control DNA repair by mediating post-translational modifications. Such modifications operate in many ways in the DDR but often do so by regulating the assembly and disassembly of DDR–protein complexes as well as by controlling the localisation and/or intrinsic activities of DDR components [31, 32]. Indeed, several compounds operating in this way by blocking DDR protein phosphorylation or poly-ADP-ribosylation have been generated and are producing encouraging results in clinical trials. As discussed previously, it has recently become evident that ubiquitylation as well as its reversal by the process of deubiquitylation play key roles in the DDR and associated downstream processes [31–38]. Consequently, enzymes controlling ubiquitylation and related processes offer various new opportunities for therapeutic intervention.
Ubiquitylation and Deubiquitylation
Ubiquitin, a 76 residue polypeptide is used as a post-translational modification to alter intracellular protein functions. Historically, the ubiquitylation system was identified as an ATP-dependent signal for targeting intracellular proteins for proteasomal degradation [39–41]. Ubiquitylation of proteins is a multi-step process requiring the sequential action of three enzymes: the ubiquitin-activating enzymes (E1s) activate ubiquitin that is subsequently loaded onto ubiquitin-conjugating enzymes (E2s) and finally, the ubiquitin is covalently linked to a lysine side-chain from the E2s via specific recruitment of the target protein, and facilitation of the transfer by ubiquitin ligases (E3s). Notably, in addition to ubiquitin being sometimes linked to target proteins singly, to form mono-ubiquitin adducts, in many cases the initial ubiquitin attached is then extended by the covalent attachment (again by E1, E2 and E3 proteins) of additional ubiquitin moieties to form poly-ubiquitin chains. Moreover, because any one of ubiquitin’s seven internal lysine residues or its amino terminus can serve as sites for conjugation, the resulting poly-ubiquitin chains can have various, highly distinct topologies with different biochemical and biological functions. While Lys-48 (K48)-linked poly-ubiquitylation of proteins is widely recognised as a critical pathway for protein degradation, many additional roles have been attributed to either poly-ubiquitylation of proteins via non-K48 chains, linear ubiquitin chains as well as mono-ubiquitylation of proteins [42–45]. In addition to post-translational modification by ubiquitin, a whole family of ubiquitin-like modifications have been described. The degree of conservation between ubiquitin and ubiquitin-like factors is somewhat limited at the protein sequence level; however, all members of the family share similar overall three-dimensional structures and highly related mechanisms of conjugation to their respective targets involving E1, E2 and E3 enzymes [46–48].
DUBs and Their Broad Effects on DDR Processes
As for other protein post-translational modifications, conjugation of ubiquitin or ubiquitin-like factors to target protein is reversible, this being mediated by isopeptidase enzymes that are often collectively referred to as deubiquitylating enzymes or DUBs. DUBs comprise a large class of intra-cellular peptidases that cleave ubiquitin from polypeptide substrates. Their substrates can be ubiquitin precursors, ubiquitin adducts, poly-ubiquitin chains, mono-ubiquitylated proteins or poly-mono-ubiquitylated proteins [45]. If we include ubiquitin-like peptidases in our calculations, over a hundred DUBs are encoded by the human genome.
DUBs can be classified into five families (Fig. 2): ubiquitin carboxyl-terminal hydrolases (UCH), ubiquitin-specific proteases (USPs), ovarian tumour proteases (OTU), MJD (Josephins) and MPN+/JAMM (JAB1/MPN/MOV34 metallo-enzymes). The first four families are cysteine peptidases, while MPN+/JAMMs are metallo-peptidases [40–51]. In addition to processing ubiquitin and ubiquitin adducts, some USPs have been shown to selectively process specific ubiquitin-like proteins (for example, USP18 acts on the ubiquitin-like protein ISG15) [52]. In the case of the SUMO family of ubiquitin-like proteins, however, adducts are reversed by a specialised group of DUBs termed SENPs, all of which are cysteine peptidases [46, 53]. While all DUBs are peptidases, there are considerable differences between their precise mechanisms of action, and there are also major differences in the regulatory mechanisms that modulate DUB selectivity and specificity [54]. In this regard, DUBs can be classified into three main categories according to their type of substrate cleavage activity: some generate free ubiquitin from linear substrates, such as poly-ubiquitin chains or ribosomal protein fusions; others liberate ubiquitin from proteins modified post-transnationally on lysine residues; while, a third class comprises DUBs that edit poly-ubiquitin chains [54]. For in depth discussions of DUB mechanism-of-action, we refer the reader to several excellent reviews on this subject [49, 54–58].

DUB phylogenetic tree. Approximately 100 genes belong to the DUB family of peptidases. Six classes of DUBs have been identified so far in the human genome. Five families belong to the cysteine peptidase class: the ubiquitin carboxyl-terminal hydrolases (UCH); the ubiquitin-specific proteases (USPs); the SENPs or SUMO peptidases; the OTU and the MJD. In addition, the MPN+/JAMM family belongs to the metallo-peptidases class of enzymes. The phylogenetic tree represents only human DUBs and does not cover bacterial or viral DUBs that display additional levels of divergence
Given the prominent role played by ubiquitylation processes in the DDR [31, 38, 59–61], it is not surprising to find multiple DUBs involved in regulating DNA repair and downstream processes (Fig. 3). While there is significant interest in ubiquitin E1, E2 and E3 proteins as DDR regulators and as potential drug targets, in this review we focus on DUBs, a drug target class that we feel has so far been under-appreciated and under-exploited. We survey how many DUBs are intimately connected to the DDR and associated cancer-relevant pathways, and highlight how, and in which contexts, DUB inhibitors may offer exciting new opportunities for treating cancers, eventually through synthetic lethal or related strategies.

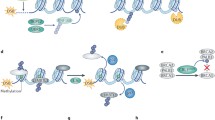
DUBs involved in DNA damage responses. DUBs involved in the DDR can be classified according to their substrates or interaction partners and artificially grouped into DUBs directly interfering with the response to DNA damage at sites of damage, or DUBs that regulate the activities of key DDR proteins involved in the cellular response to the DNA insult
USP1 Functions in Multiple Repair Pathways
The USP1 protein was one of the first ubiquitin hydrolases characterised as a key player in a well-defined DDR pathway. Thus, it was shown that USP1 selectively hydrolyses mono-ubiquitin adducts from the proteins FANCD2 and PCNA [62, 63]. Mono-ubiquitylation of the FA protein FANCD2 is critical for effective ICL DNA repair. The mono-ubiquitylation of FANCD2 does not destabilise the protein: both forms of the protein are equally stable; however, mono-ubiquitylation of FANCD2 is regulated in a cell cycle-dependent manner. Upon mono-ubiquitylation on lysine 561, FANCD2 re-localises to nuclear DNA-damage foci, where it interacts with BRCA1 and the RAD51 recombinase and co-localises with FANCD2 and BRCA2 [64–66]. Interestingly, USP1 was identified in a screen for enzymes that prevent the removal of ubiquitin from FANCD2 [62]. Like mono-ubiquitylated FANCD2, USP1 levels are regulated during the cell cycle, and USP1 has also been shown to interact directly with FANCD2 and to co-localise with chromatin. Notably, however, the isolated USP1 protein does not display strong deubiquitylating activity in vitro. Instead, the co-factor, UAF1 (WDR48), is necessary to form an active USP1 enzyme. UAF1 (WDR48) is a WD40 repeat-containing protein that forms a stoichiometric complex with USP1 in cells and activates the catalytic activity of the USP1 complex [67]. Mechanistically, UAF1 increases the catalytic turnover (k cat), but does not increase the affinity of USP1 for its substrate (K M). Regulation of DUBs by co-factors or upon substrate binding is a common regulatory feature of that class of peptidases. In addition, USP1 has also been shown to remove mono-ubiquitin from PCNA, thus regulating one of the earliest steps of trans-lesion DNA synthesis (TLS)—a process in which specialised DNA polymerases synthesise DNA past a DNA lesion [63, 67]. The USP1/UAF1 complex is also recruited to mono-ubiquitylated PCNA via an interaction between UAF1 and the protein ELG1, a protein involved in replication complexes and in loading PCNA onto DNA for efficient replication, and independently identified as a factor required to suppress genomic instability [68, 69].
Initial observations indicated that reduced levels of USP1 would protect cells from DNA damage [62, 63]; however, follow-up studies evaluating the role of USP1 in gene inactivation studies in mice have clearly demonstrated that USP1 depletion results in genomic instability. Targeted deletion of mouse Usp1 results in elevated perinatal lethality, male infertility, DNA cross-linker hyper-sensitivity, and a FA phenotype. Usp1 −/− mouse embryonic fibroblasts display heightened levels of mono-ubiquitylated FANCD2 in chromatin and exhibit impaired FANCD2 focus assembly and a defect in HR repair. Interestingly, Usp1/Fancd2 double knock-out mice display a higher level of DDR dysfunction than in the Usp1 single knock-out condition, suggesting additional DDR roles for USP1 beyond its effects on FANCD2 [70]. Additional support for a critical role played by USP1 in protecting cells against DNA damage was obtained from a study in chicken DT40 cells: Usp1 disruption resulted in cellular hypersensitivity to DNA damaging agents, strongly supporting a model, whereby, USP1 is a positive regulator of DNA repair [71]. Disrupting the USP1 complex in DT40 cells also leads to increased sensitivity to camptothecin (CPT) (a topoisomerase I inhibitor), poly(ADP-ribose) polymerase inhibitors and the DNA cross-linking agent mitomycin C (MMC), together with defects in HR. These defects were largely rescued by removing Ku70, a key regulator of DSB repair by NHEJ. The USP1 complex is thus, a critical regulator of ICL repair and HR and, together with the FA pathway in general, it has a role in suppressing NHEJ [72].
During replication fork stalling, the G2/M checkpoint response is controlled by CHK1 phosphorylation. Recent evidence suggests that USP1 controls a feedback loop that limits CHK1 activity to rescue DNA-damaged cells. Stimulation of CHK1 degradation by mono-ubiquitylated FANCD2 may thus, represent a feedback mechanism that contributes to the recovery of damaged cells [73]. In addition, USP1 is required to prevent aberrant recruitment of DNA polymerase Κ to replication forks. Lack of recruitment of polymerase Κ to the replication fork results in decreased replication fork speed and enhancement of genomic instability. The process is USP1 driven and generated as a result of elevated PCNA ubiquitylation [74]. PCNA mono-ubiquitylation and trans-lesion synthesis (TLS) polymerase recruitment to UV lesions have also recently been implicated in NER, a DNA repair mechanism that can take place outside of the replication phase [75]. USP1 levels are controlled at the protein level by APC/CCdh1. Low levels of USP1 enable robust UV-induced PCNA mono-ubiquitylation during G1, which is likely to allow recruitment of TLS polymerases to UV lesions [76].
A novel function for USP1 deubiquitylating activity has recently been uncovered: USP1 regulates the stability of ID (inhibitors of DNA binding) proteins [77]. ID proteins antagonise basic-helix–loop–helix (bHLH) transcription factors to inhibit differentiation and maintain stem cell fate [78]. ID ubiquitylation and proteasomal degradation occur in differentiated tissues, but IDs appear to escape degradation in many neoplasms [79]. Whether or not the regulation of ID proteins is linked to the known functions of USP1 in regulating DNA damage responses or not remains to be fully determined. In the same study [77], the authors were able to demonstrate that USP1, through its catalytic DUB activity, promotes in vitro transformation and in vivo tumour formation, thus further supporting models in which USP1 acts as an oncogene. Taken together with its key roles in promoting DNA repair, these findings highlight the potential of USP1 as an attractive target for developing anti-cancer drugs.
USP2 Regulates p53 Activity and Cellular Responses to DNA Damage
USP2 is a DUB with oncogenic properties that regulates cellular levels of fatty acid synthase (FAS) in prostate cells. Furthermore, USP2 over-expression confers resistance to apoptosis induced by chemotherapeutic agents such as cisplatin and paclitaxel in prostate epithelial cells [80]. By virtue of its activity to deubiquitylate the p53 E3 ligase Mdm2, USP2 has been demonstrated to regulate the p53 checkpoint pathway. However, unlike USP7 that is able to target both p53 and Mdm2 (see below), USP2 shows exquisite selectivity towards Mdm2 [81]. Unfortunately, the role played by USP2 in regulating Mdm2 activity under DNA damage conditions has not been described and remains to be characterised. The Mdm2-related protein Mdmx is also a substrate and a partner for USP2a, one of the two USP2 isoforms in human cells, with USP2a catalytic activity being required for Mdmx deubiquitylation. In accord with these findings, USP2a depletion causes destabilisation of Mdmx and results in decreased cellular Mdmx levels [82]. The chemotherapeutic agent cisplatin regulates both USP2a and Mdmx levels, and moreover, USP2a depletion sensitises testicular carcinoma cells to cisplatin, suggesting that USP2 pharmacologic inhibition may lead to anti-tumour activity [82].
The Histone DUB USP3 Affects the S-Phase Checkpoint and Replication
USP3 is a chromatin-associated DUB that regulates ubiquitylation of histones H2A and H2B. Cellular ablation of USP3 leads to checkpoint activation, and delays in S-phase progression associated with the accumulation of DNA breaks and enhanced replication stress [83]. By virtue of its ability to oppose histone H2A ubiquitylation and the fact that USP3 over-expression can block accumulation of the ubiquitin E3 ligase RNF168 at DSB sites, USP3 has been linked to the key RNF8–RNF168 pathway of assembling repair and signalling components at DSB sites [84]. Given, the critical and selective role played by replication stress in tumour survival [3], one might speculate that pharmacological USP3 inhibition might have benefits for cancer treatment. Interestingly, USP3 ablation has also been described as required for hepatocyte-growth factor scattering of epithelial cells [85].
USP4 Regulates p53 Activity and Protects Against Ionising Radiation
USP4 has been recently described as a key regulator of p53 stability: USP4 interacts directly and deubiquitylates the E3 HUWE1 (ARF-BP1; MULE), resulting in reduced p53 levels [86]. While Usp4 deficient mice are viable and developmentally normal, they show enhanced apoptosis in the spleen and thymus upon ionising radiation. Usp4 deficient mouse embryonic fibroblasts recapitulate most of the phenotypes one would expect from a DUB that regulates p53 levels: retarded growth, premature cellular senescence, resistance to oncogenic transformation and hyperactive DNA damage checkpoints [86]. Indeed, it has been suggested that Usp4 is a potential oncogene because it inhibits p53 activity, p53-associated apoptosis and cell-cycle checkpoints; depletion of USP4 promotes cell senescence; loss of USP4 inhibits oncogene-induced primary cell transformation, and finally USP4 is over-expressed in a subset of human cancers [86]. A study has recently described a role for USP4 in modulating the therapeutic efficacy of the topoisomerase II inhibitor doxorubicin through TAK1 ubiquitylation [87] that may link the putative oncogene status of Ups4 with a role in chemo-resistance. Notably, studies have also indicated functions for USP4 in regulating growth factor signalling by the Toll-like receptor/IL1 pathway [88], TGF-β receptor type I [89, 90], TNFα receptor [91], growth factor-activated kinase regulation [92] and Wnt signalling [93], making USP4 a prime target for further evaluation as an oncology drug target with strong potential in DDR contexts.
USP5 Regulates p53 Stability Via Unanchored Ubiquitin Chains
Depletion of USP5 has been reported to cause accumulation of nuclear p53 and increase p53 transcriptional activity. Activation of p53 can be accounted for by the ability of USP5 suppression to inhibit the proteasomal degradation of p53 without affecting the degradation of Mdm2. The differential effect of USP5 depletion on protein stability may be due to the differences in the sensitivities of p53 and Mdm2 to inhibition of proteasomal activity by free/unanchored poly-ubiquitin that accumulates after the USP5 loss [94]. While indirectly linked to the key DNA damage checkpoint factor p53, USP5 has not been strictly identified as a modulator of the DDR and further investigations are needed to assess its roles in DNA damage signalling or repair.
USP7 Regulates Multiple DNA Repair and Checkpoint Pathways
In unstressed cells, p53 levels are kept low via proteasomal targeting, while under stress conditions, p53 is stabilised and contributes to the DDR. USP7 (initially named HAUSP) has a pivotal role in regulating the G2/M checkpoint upon DNA damage [95, 96]. Furthermore, a specific USP7 isoform (USP7S) has recently been described as a downstream effector of the checkpoint pathway controlled by the DSB-responsive kinase ATM, with USP7S activity being down-regulated by the ATM-dependent protein phosphatase PPM1G in response to the DNA damage, thereby impairing Mdm2 and activating the p53 response [97].
FOXO4, a member of the Forkhead box transcription factors that regulates cellular metabolism, cell-cycle progression and cell death, is regulated by mono-ubiquitylation in response to the oxidative stress, resulting in its re-localisation to the nucleus and an increase in its transcriptional activity. Notably, USP7 has been identified as the DUB that deubiquitylates FOXO4 and modulates its transcriptional activity in response to oxidative stress [98], making USP7 a prime controller of oxidative stress responses that are frequent hallmarks of tumours and are often associated with the DDR defects [99]. USP7 has also been reported to modulate BER of oxidative lesions by modulating DNA accessibility and consequently the rate of repair of oxidative lesions through effects on chromatin remodelling [100].
Transcription-coupled nucleotide excision repair (TC-NER) is a sub-pathway of NER that efficiently removes highly toxic RNA polymerase II blocking lesions on DNA. Defective TC-NER gives rise to the human disorders—Cockayne syndrome and UV-sensitive syndrome (UVSS) [101]. Recently, the UVSSA protein was shown to recruit USP7 to ERCC6 TC-NER complexes upon DNA damage, representing a critical regulatory mechanism in restoring gene expression upon damage [102]. As we describe in later sections, the efforts at targeting USP7 with small molecule inhibitors have been undertaken based on the fact that USP7 regulates p53 checkpoint activity [103] and also behaves as a key DUB for multiple DDR pathways.
USP9x Regulates Sensitivity of Tumour Cells to DNA Damaging Agents
Myeloid cell leukaemia sequence 1 (Mcl-1), an anti-apoptotic member of the Bcl-2 family, is often over-expressed in tumour cells and has been demonstrated as a factor limiting therapeutic success. Mcl-1 differs from other Bcl-2 members by its high-turnover rate [104], with its expression being tightly regulated by ubiquitylating and DUBs. Recently, the deubiquitylase ubiquitin-specific protease 9x (USP9x) was described as a factor removing poly-ubiquitin chains from Mcl-1, thereby stabilising Mcl-1 and increasing resistance to apoptosis induced by the Bcl-2/Bcl-xL inhibitor ABT-737 [105]. Notably, increased USP9x and Mcl1 protein expression correlate with prognosis for patients with multiple myeloma [105]. In addition, ionising radiation-induced activation of USP9x inhibits Mcl-1 degradation, resulting in increased radio-resistance and apoptosis [106]. While the exact linkage between Mcl-1 stability and radio-resistance remains to be firmly demonstrated, these findings suggest that the Mcl-1 inhibition might be used in conjunction with radiotherapy in cancer treatment if it can be demonstrated that this would have greater effects on the cancer cells than normal tissues. It is tempting to speculate that cancer with low levels of USP9x expression, such as aggressive pancreatic cancers, might be specifically sensitised to some conventional therapeutic agents [107].
USP10 Affects Homologous Recombination and the p53 Checkpoint
The tumour suppressor functions of p53 are critically regulated via modulation of its stability, with several DUBs implicated in this control. USP10 is a cytoplasmic ubiquitin-specific protease that deubiquitylates p53, reversing Mdm2-induced p53 nuclear export and degradation [108]. After DNA damage, USP10 is stabilised, and a fraction of USP10 translocates to the nucleus to activate p53. The translocation and stabilisation of USP10 is regulated by ATM-mediated phosphorylation of USP10 on Thr-42 and Ser-337. In addition, USP10 suppresses the tumour cell growth in cells with wild-type p53, and USP10 expression is down-regulated in a high percentage of clear cell carcinomas known to have few p53 mutations [108]. A recent study also suggested that USP10 regulates p53 through an additional mode-of-action involving the tumour suppressor Beclin, which regulates the activity of USP13 and USP10 and thus impacts on the p53 stability [109]. The recent identification of USP10 inhibitors may help better understand the DDR roles of USP10 and, in this way, identify the therapeutic opportunities [109].
USP11 Affects Oncogene-Induced Senescence and Homologous Recombination
Ubiquitylation of chromosome-associated proteins is important for many aspects of DNA repair and transcriptional regulation. An important facet of transcriptional repression by polycomb repressive complex 1 (PRC1) is the mono-ubiquitylation of histone H2A by the combined action of the Polycomb group proteins, or PcG [110]. USP11, together with USP7 are recruited at chromatin sites, bind PcG and contribute to the regulation of the tumour suppressor gene locus p16 INK4A by modulating the ubiquitylation status of PcG proteins. Given the importance of the p16 INK4A locus in oncogene-induced mechanisms, the role played by USP11 could be quite critical. Indeed, the central function of ubiquitylation in regulating INK4a and the possibility that USP7 and USP11 can promote bypass of oncogene-induced senescence by stabilising the PRC1 complex suggests exciting opportunities for using specific USP inhibitors in a variety of therapeutic contexts, including in DDR deficient backgrounds [110].
In accord with the above, USP11 was also identified in a screen for factors that trigger the hypersensitivity to PARP inhibitors, with USP11 catalytic activity being needed for effective HR repair at DSB sites [111]. In this regard, mammalian cells lacking the functional BRCA2 are hypersensitive to DNA damaging agents [112], show genomic instability [113, 114], and are deficient in homology-directed DNA repair [115, 116]. Several lines of evidence point towards a critical role for USP11 in regulating BRCA2 stability: USP11 interacts and co-purifies with BRCA2, USP11 deubiquitylates BRCA2, USP11 depletion sensitises cells to DNA damaging agents and finally, mitomycin C(MMC) regulates the stability of BRCA2 in a USP11-dependent manner [117]. Collectively, USP11 has been clearly linked to essential DDR mechanisms and oncogene-induced senescence, highlighting it as a potentially attractive target for therapeutic agents.
USP16 is Implicated in DNA Damage Responses
In a similar manner to USP3, USP16 has been characterised as an enzyme removing ubiquitin from histone H2A and involved in regulating the RNF8–RNF168 pathway [118]. The available evidence suggests that USP16 may regulate the DSB-induced transcription silencing by opposing the activities of RNF8 and RNF168 [119]. The recent identification of histone H2A Lys-13/Lys-15 ubiquitylation as a target of RNF168 adds additional potential substrates for USP3 and USP16 [120]. USP16 characterisation and its potential roles in the DDR are, however, still in their infancy, and further investigations are needed to shed the light on its cellular roles.
USP24 is Connected to Nucleotide Excision Repair
Damage-specific DNA-binding protein 2 (DDB2) was first isolated as a subunit of the UV-DDB heterodimeric complex that is involved in DNA damage recognition in the NER pathway. DDB2 is required for efficient repair of UV lesions in chromatin and is a component of the CRL4 (DDB2) E3 ligase that targets for ubiquitylation histones, DDB2 itself and XPC, a protein that functions in damage detection involved in the first step of global genome NER. USP24 was recently identified as a DUB interacting with DDB2 and involved in controlling the stability of DDB2 [121]. Further investigations into the precise roles played by USP24 and potentially other DUBs in NER, therefore, warrant exploration.
USP28 is Connected to the DDR and Homologous Recombination
In response to DNA damage, effector kinases such as ATM, ATR and DNA-PK, initiate cascades of cellular effects that modulate gene transcription, cell-cycle progression, DNA repair and apoptosis [122]. Factors such as 53BP1, MDC1, and Claspin are then important to link these initial responses to downstream effector DDR pathways [123, 124]. USP28 was identified as an interaction partner of 53BP1 [125], and loss of USP28 has been reported to lead to IR-induced apoptosis in H460 cells, in a similar manner to what has been seen in Chk2, p53 and PUMA null mice. Moreover, the catalytic activity of USP28 has been described as essential for such functions [125]. Independently, USP28 has been reported to modulate the activity of the Myc proto-oncogene [126]. The authors claimed that USP28 controls Myc stability through antagonising the activity of the SCFFBW7 ubiquitin ligase complex, and that the stabilisation of Myc by USP28 is required for proliferation of several tumour cell types and for inhibition of cell differentiation in colon carcinoma. A number of reports have suggested that the steady-state levels of Myc rapidly decline in response to DNA damage [127–129], and Popov et al. have suggested that the USP28 dissociation from Fbw7 in response to DNA damage provides a potential mechanism that couples Myc stability to DNA damage. While USP28 has been firmly connected to the DDR through the above studies, it remains to be determined the extent to which these functions operate in other cell lines, and therefore whether USP28 represents an attractive therapeutic target.
USP29 Regulates p53 Stability Upon Oxidative Stress
Cellular networks involving c-Myc and p53 control proliferation, differentiation and apoptosis, and are responsive to, and cross-regulate a variety of stress, metabolic and biosynthetic processes. At the c-Myc gene, the far upstream element binding protein (FBP) and FBP-interacting repressor (FIR) program transcription by looping to RNA polymerase II complexes engaged at the promoter. Another FBP partner, JTV1/AIMP2, a structural subunit of a multi-aminoacyl-tRNA synthetase (ARS) complex, has also been reported to stabilise p53 via an apparently independent mechanism [130]. In response to the oxidative stress, JTV1 dissociates from the ARS complex, translocates to the nucleus, associates with FBP and co-activates the transcription of the FBP target, ubiquitin-specific peptidase 29 (USP29). A previously uncharacterised deubiquitylating enzyme, USP29 binds to, cleaves poly-ubiquitin chains from, and stabilises p53. This accumulated p53 quickly induces apoptosis. Thus, FBP and JTV1 help coordinate the molecular and cellular responses to oxidative stress mediated by USP29 [130].
USP44, a Tumour Suppressor Regulating Chromosomal Stability
To preserve genomic integrity, the cells must ensure the accurate and timely segregation of chromosomes to daughter cells in mitosis. A complex pathway known as the spindle assembly checkpoint or mitotic checkpoint ensures that the transition to anaphase is delayed until all the chromosome kinetochores are properly attached to the mitotic spindle [131]. While not directly involved in the DDR, defects in the mitotic checkpoint result in mis-segregation of chromosomes and can thereby lead to genomic instability. USP44 has been identified as a DUB critical for spindle assembly checkpoint activity in RNAi screens, where USP44 depletion was found to cause bypass of this checkpoint [132, 133]. Recently, Usp44 null mice were generated and display cellular and biochemical features consistent with a role for USP44 in the mitotic checkpoint but also unravelled a function for USP44 in centrosome regulation [134, 135]. Additionally, Usp44 deficient mice were found to be prone to spontaneous tumours of the lung, supporting a role for Usp44 as a tumour suppressor gene and underscoring its role in cancer.
USP47 Regulates BER and is Involved is DNA Damage Sensitisation
BER, is an essential cellular mechanism for maintaining the genome stability, mediating the repair of DNA lesions arising due to the chemical instability of DNA molecules or base changes induced by endogenous or environmental mutagens [136]. Polymerase β (Pol-β) is a critical BER enzyme possessing abasic site lyase activity that removes the 5′-sugar phosphate and also functions as a DNA polymerase, adding one nucleotide to the 3′-end of the arising single-nucleotide gap [137]. USP47 has been identified as the major enzyme that deubiquitylates Pol-β, mainly through the stabilisation of newly synthesised Pol-β, and consequently regulates BER activity [138]. Reduced levels of cytoplasmic Pol-β following USP47 depletion are unable to effectively respond to DNA damage induced by exogenous agents, such as MMS, and are insufficient to elevate the levels of nuclear Pol-β protein required for efficient DNA repair [138]. USP47 has also been described as an interaction partner for the ubiquitin E3 ligase complex, β-TrCP, with the authors of this study showing that silencing of USP47 expression inhibits cell survival and sensitises cells to chemotherapeutic agent-induced apoptosis [139]. A hypomorphic Usp47 mouse model was generated, and fibroblasts derived from the Usp47 deficient mice exhibited UV hypersensitivity, thus further supporting a role for USP47 in the DDR [139]. Along with USP7, USP47 is thus so far one of the very few DUBs described as regulating BER [100].
UCHL1 a DUB Regulating p53 Stability and Tumorigenesis
Ubiquitin C-terminal hydrolase L1 (UCHL1), a member of the UCH class of DUBs, is probably the most studied DUB: its association with neurodegenerative conditions, including Parkinson’s disease, and a wide range of malignancies have generated an abundant literature. Despite this, many aspects of the molecular mechanisms and functions of UCHL1 remain either contradictory or poorly understood. While UCHL1 is normally almost exclusively expressed in neurones, the neuroendocrine system and the gonads, its aberrant expression has been described in many tumour types (reviewed in [51]). While clear roles for UCHL1 in the DDR have not been formally demonstrated, UCHL1 can be included in the list of DUBs that have been reported to modulate the ubiquitylation status of proteins in the p53-mdm2 checkpoint pathways. Thus, in breast cancer models, UCHL1 can induce the levels of p53 and reduce mdm2 protein levels. UCHL1 also induced G0/G1 cell-cycle arrest and apoptosis of breast tumour cells, an activity that requires its catalytic activity and depends on the accumulation of p53 [140]. Similar observations have also been made in head and neck cancer [141], as well as in prostate cancer [142]. The above mentioned publications all support a role for UCHL1 as a tumour suppressor gene; although it is also important to mention that several reports have demonstrated oncogenic properties for UCHL1 in other cancer types such as lymphoma [143] and colorectal cancer [144]. UCHL1 therefore seems to play important roles in tumorigenesis but a clear understanding of a mechanism for this or for a DDR role(s) remains to be firmly established. It is also quite difficult to reconcile the roles played by UCHL1 in neurons and neuro-endocrine cells, with its functions in cancer cells.
BRCC36 is a Key Player in Cellular Responses to DSBs
BRCC36, which belongs to the JAMM (JAB1/MPN/Mov34 metallo-enzyme) family of DUBs, is a lysine 63-ubiquitin (K63-Ub)-specific DUB and a member of two protein complexes: the DNA damage-responsive BRCA1–RAP80 complex, and the cytoplasmic BRCC36 isopeptidase complex (BRISC). BRCC36 was initially isolated from a multi-protein complex containing the HR-promoting factors BRCA1, BRCA2 and Rad51 [145]. Notably, BRCC36 depletion has been shown to impair activation of BRCA1 and to sensitise breast cancer cells to IR-induced apoptosis [146]. The interaction of the BRCA1 BRCT domain with RAP80, a ubiquitin-binding protein, targets a complex containing the BRCA1–BARD1 (BRCA1-associated ring domain protein 1) E3 ligase and BRCC36 to MDC1–γH2AX-dependent lysine(6)- and lysine(63)-linked ubiquitin polymers at sites of DSBs [147]. Interestingly, it appears that concomitant and opposing RNF8–Ubc13 ubiquitin ligase and RAP80–BRCC36 ubiquitin hydrolysis activities are responsible for determining steady-state ubiquitin levels at DSB sites [148, 149].
Recent discoveries have shed some light on the mode-of-action of the BCC36-containing complex. Thus, in striking contrast to other BRCA1-containing complexes that are known to promote homology-directed repair, the BRCA1–RAP80 complex restricts DNA end resection in S/G2 phase of the cell cycle, thereby limiting HR. Consequently, RAP80 or BRCC36 deficiency was found to result in elevated MRE11–CtIP-dependent 5′ DNA end resection with a concomitant increase in HR mechanisms that rely on 3′ single-stranded overhangs. In this way, the BRCA1–RAP80 complex limits nuclease activities at DSB sites, preventing excessive end resection and potentially deleterious homology-directed DSB repair mechanisms that can impair genome integrity [150]. Notably, the human BRCC36 gene is located at the Xq28 locus, a chromosomal break-point in patients with prolymphocytic T cell leukaemia [151]. Furthermore, BRCC36 is aberrantly expressed in the majority of breast tumours, indicating a potential role in the pathogenesis of this disease [145]. Taking these findings together, BRCC36 is clearly a key player in controlling cellular DSB responses through regulating the ubiquitylation status of repair factors at DNA damage sites.
MYSM1 is Involved in Maintaining Genome Stability and histone Ubiquitylation
MYSM1 is a member of the MPN+/JAMM family of DUBs. It was initially identified as a histone H2A deubiquitylating enzyme involved in regulating transcriptional programmes and epigenetic regulation of B cell differentiation [152, 153]. Furthermore, analyses of MYSM1 deficient mice uncovered a role for MYSM1 in bone marrow stem cell maintenance, control of oxidative stress and genomic stability in hematopoietic progenitors, and in the development of lymphoid and erythroid lineages [154]. Given the intricate links between epigenetic regulation of histone ubiquitylation and DNA damage responses [13], it will be interesting to further explore the roles played by MYSM1 in such processes.
PSMD14, a Proteasome Subunit Involved in the DNA Damage Response
The sequential recruitment of multiple protein complexes at sites of damage involves multiple post-translational modifications, with several key steps requiring modification of DDR proteins by ubiquitin or the ubiquitin-like protein SUMO [32]. ATM-dependent phosphorylation signalling that leads to ubiquitin conjugation at DSB sites is promoted by RNF8 and RNF168, with resulting ubiquitin-conjugated histones H2A, H2AX and other proteins promoting the recruitment of 53BP1 and other proteins to potentiate repair and DSB signalling [35, 61, 155, 156]. Recently, the proteasome-associated DUB POH1 (PSMD14) was shown to function in the DDR, at least in part through opposing RNF8/RNF168-mediated formation of Lys-63 linked ubiquitin chains at DSB sites, thereby restricting 53BP1 accumulation [157]. In addition, these authors found that POH1 also enhances RAD51 loading at DNA damage sites, thereby facilitating HR repair. Intriguingly, POH1 and BRCC36 (see above section) are two members of the JAMM family with K63-ubiquitin chain linkage specificity that are now known to affect formation of 53BP1 foci, with co-depletion experiments suggesting that they act on the same repair pathways [158]. Additional connections between DSB responses and the proteasome have been established recently through studies on the SUMO-targeted ubiquitin E3 ligase, RNF4, which together with RNF8 mediates proteasome recruitment to DNA damage sites to promote DSB repair [159].
OTUB1, an OTU Family Member Regulates DSB Repair
The OTU-family DUB OTUB1 was identified as a negative regulator of RNF168-dependent ubiquitylation, through it inhibiting DSB-induced chromatin ubiquitylation [160]. Surprisingly, this function of OTUB1 was found to be independent of its isopeptidase activity. Instead, OTUB1 is able to bind and inhibit a subset of E2 conjugating enzymes that comprises UBC13 and UbcH5 proteins, E2s that are employed by RNF168 [160–163]. Notably, OTUB1 uses a highly unusual mechanism to bind and inhibit ubiquitin-charged E2s through the assembly of a pseudo-cleavage product in its catalytic site [161–163]. OTUB1 has also been described as a regulator of p53 ubiquitylation in cells and in vitro [164], with OTUB1 inhibition markedly impairing p53 activation induced by DNA damaging agents such as etoposide, UV light or 5-FU. OTUB1 appears to directly suppress MDM2-mediated p53 ubiquitylation, independently of its catalytic activity by suppressing the activity of UbcH5 proteins that function with MDM2. Interestingly, unlike USP7 that regulates p53/MDM2 ubiquitylation in the nucleus, OTUB1 does so in the cytoplasm upon DNA damage [164] in a similar manner to USP10 (see above).
While OTUB1’s roles in regulating the DDR are strongly supported by multiple publications, making OTUB1 an attractive therapeutic target, it seems to be clear that the catalytic activity of OTUB1 is not required for such processes, thus preventing the possibility of applying classical enzyme assay-based drug discovery for OTUB1 inhibition.
SENP1 Regulates DSB Responses and Modulates p53-Dependent Cell Senescence
Apart from its role in metabolism and energy homeostasis, the protein SIRT1 has been shown to regulate the DDR. SIRT1 de-acetylates protein substrates with established roles in the DDR. These proteins include p53, FOXO factors and Ku70, and deacetylation of p53 reduces both its transcriptional and apoptotic activities. SIRT1 has been shown to be sumoylated, and its desumoylation by SENP1 regulates cellular responses to genotoxic stress [165]. Multiple SENPs (SENP1, SENP2 and SENP7) play critical roles in the modulation of p53-dependent premature senescence and suggests that inhibition of some SENPs may be attractive for anti-tumour activities [166]. In addition, SENP1 levels correlate with HIF1 levels in human prostate carcinoma. SENP1 expression correlates with the severity of the disease, as high levels of SENP1 are observed in more aggressive prostate cancer [167]. SENP1 has also been reported as a critical regulator of the activity of the KAP1; an essential downstream effector of the ATM-dependent DDR [168]. However, the detailed mechanism-of-action of SENP1-dependent regulation of the DDR is still not clearly established.
SENP2 Regulates Survival Upon Genotoxic Stress and p53 Checkpoint Activity
Multiple lines of evidence have linked the desumoylating enzyme SENP2 to the regulation of various aspects of the DDR. For example, in response to DNA damage, sumoylation of NEMO, an essential NF-κB modulator, is critical for NF-κB activation. Notably, only SENP2 can efficiently associate with NEMO, desumoylate NEMO and inhibit NF-κB activation induced by DNA damage [169]. Interestingly, SENP2 is also an NF-κB inducible gene, with NF-κB-dependent SENP2 induction preventing the second phase of NF-κB activation to significantly limit cell survival in response to genotoxic stress [169]. Another DDR link for SENP2 comes from it desumoylating the protein hnRNP K: under DNA-damage conditions, hnRNP-K is transiently stabilised and serves as a transcriptional co-activator of p53 for cell-cycle arrest [170]. UV-induced sumoylation of hnRNP-K has been shown to prevent its ubiquitylation for stabilisation. These findings indicate that SUMO modification plays a crucial role in the control of hnRNP-K function as a p53 co-activator in response to DNA damage [68]. By reversing this modification, SENP2 likely also affects cellular responses to UV and perhaps other DNA damaging agents. SENP2-mediated regulation of Mdm2 is also reported to be critical for promoting genome integrity via p53-dependent stress responses, with an isoform of SENP2 associating with Mdm2, regulating its cellular localisation and DDR function [171].
SENP6 Regulates Replication and Responses to Genotoxic Stress
The replication protein A complex (RPA) plays a crucial role in DNA replication and various DDR processes. Notably, the 70 kDa subunit of RPA (RPA70) associates in the nucleus with the SUMO-specific protease SENP6, to maintain RPA70 in a hypo-sumoylated state during S phase. CPT, an inducer of replication stress, dissociates SENP6 from RPA70, allowing RPA70 to be modified by SUMO-2/3, which in turn facilitates recruitment of RAD51 to the DNA damage sites to promote DNA repair through HR [172]. These results thus support a key role for SENP6 in regulating HR. Another connection between SENP6 and the DDR comes through the histone acetyl-transferase TIP60 that regulates the DDR by acetylating histones and remodelling chromatin as well as targeting and activating the DDR kinase ATM. Using a proteomic approach, SENP6 has been identified as a TIP60 binding partner. Interestingly, the association between the two proteins was reported to be induced by UV. The reversible modification of TIP60 by sumoylation has a critical impact on the acetyl-transferase activity of TIP60 and illustrates how an ubiquitin-like modification is able to act as an essential regulator of DDR pathways [173].
Viral DUBs and the DDR
Ubiquitin or ubiquitin-like systems exist or are hijacked by pathogens, viruses or bacteria to interfere with the proper functioning of human cells [174], with several viral DUBs having been linked to the DDR. For instance, in addition to the cellular DUB USP1, the Epstein–Barr virus DUB-like protein BPLF1 has recently been described to deubiquitylate PCNA and attenuates polymerase η recruitment to DNA damage sites [175]. Interestingly, the DUB activity of BPLF1 is conserved amongst other members of the herpes virus family.
Conclusions on DUB Therapeutic Opportunities
Inhibiting the proteasome and selected E1, E2 or E3 enzymes are providing exciting new therapeutic opportunities [176–180]. Many DUBs have been described as being attractive therapeutic targets and the level of biological target validation is becoming very clear for many DUBs [181–183]. Indeed, most DDR pathways have already been allocated at least one DUB, which clearly highlights the large number of ubiquitin modifications involved in responses to DNA damage. It is also interesting to note that DUBs from most families of DUBs have been reported to play critical roles in the DDR (Fig. 4). DUBs regulating the processes of HR, NHEJ, NER, BER, TLS, FA, mismatch repair and checkpoint control have been identified (Table 1). Moreover, examples of synthetic lethality interaction between DDR pathways clearly identified as mutated in tumours but fully functional in normal cells have triggered a plethora of screening approaches for the identification of novel therapeutic opportunities [26, 184–187], which may include DUBs. In addition, many DUBs have been linked to cancer pathways that have not been linked to DDR pathways, thus further broadening the scope for DUB inhibitors in oncology [37, 51, 57]. Having said this, the drug discovery aspects of DUB biology remain largely untapped and challenging, with very few selective inhibitors of DUBs being described so far. Nonetheless, a limited number of studies have been reported and will be summarised below.

DUBs are involved in all DDR pathways. A picture is now emerging of the fine control that certain DUBs play in all DNA repair processes of a cell. By identifying these DUBs and through their selective inhibition certain DDR pathways can be targeted in cancer cells
Selective and cell active inhibitors of the USP1/UAF1 deubiquitylase complex seem to be able to reverse the cisplatin-resistance of non-small cell lung cancer cells in vitro [188]. However, a clear recapitulation of a USP1 phenotype expected from the biochemical and genetic studies with the use of the USP1 inhibitors remains to be demonstrated (see USP1 section above). As described previously, USP7 seems to play critical roles in many DNA repair events as well as being a regulator of p53-mdm2 pathways; and unsurprisingly, USP7 has received much attention as a possible drug target for the development of anti-tumour therapies [189–192] and remains the DUB that has been most used as a paradigm for tackling DUBs for drug discovery. Selective USP7 inhibitors have been identified recently and will very likely help characterise the potential for USP7 inhibitors in preclinical settings [193–195]. However, the number of attractive chemical series selectively inhibiting USP7 still remains limited. Innovative drug discovery breakthroughs would benefit the field and indeed, novel technological advances are currently being employed to tackle the problem [196].
In addition, a number of DUB inhibitors with various degrees of selectivity but whose links with the DDR have not yet been evaluated have been described [197–206]. Probe inhibitors of SENPs have also been described recently [207, 208]. As described above, we now know of many DUBs that modulate or are connected to the DDR, and which deserve closer attention as potential therapeutic targets. Given the growing interest for this enzyme class by academic groups, pharma and biotech such as MISSION Therapeutics, it seems likely that the next few years will witness significant advances in our appreciation of therapeutic opportunities for DUBs as well as development of compounds that can begin to exploit this potential in the clinic as recently illustrated for other DDR inhibitors [209].
Abbreviations
- 5FU:
-
5-Fluoro-uracil
- BER:
-
Base excision repair
- CPD:
-
Cyclobutane pyridine dimer
- DDR:
-
DNA damage response
- DSB:
-
DNA double-strand break
- FA:
-
Fanconi anaemia
- H2O2 :
-
Hydrogen peroxide
- HR:
-
Homologous recombination
- HU:
-
Hydroxyurea
- IR:
-
Ionising radiation
- MMC:
-
Mitomycin C
- NER:
-
Nucleotide excision repair
- NHEJ:
-
Non-homologous end-joining
- TLS:
-
Trans-lesion synthesis
- UV:
-
Ultra-violet light
References
Curtin, N. J. (2012). DNA repair dysregulation from cancer driver to therapeutic target. Nature Reviews Cancer, 12, 801–817.
Ciccia, A., & Elledge, S. J. (2010). The DNA damage response: Making it safe to play with knives. Molecular Cell, 40, 179–204.
Jackson, S. P., & Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature, 461, 1071–1078.
Bradbury, J. M., & Jackson, S. P. (2003). The complex matter of DNA double-strand break detection. Biochemical Society Transactions, 31, 40–44.
Hartwell, L. H., & Kastan, M. B. (1994). Cell cycle control and cancer. Science, 266, 1821–1828.
Branzei, D., & Foiani, M. (2008). Regulation of DNA repair throughout the cell cycle. Nature Reviews Molecular Cell Biology, 9, 297–308.
Reinhardt, H. C., & Yaffe, M. B. (2009). Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Current Opinion in Cell Biology, 21, 245–255.
Bartek, J., & Lukas, J. (2007). DNA damage checkpoints: From initiation to recovery or adaptation. Current Opinion in Cell Biology, 19, 238–245.
Halazonetis, T. D., Gorgoulis, V. G., & Bartek, J. (2008). An oncogene-induced DNA damage model for cancer development. Science, 319, 1352–1355.
Kroemer, G., Mariño, G., & Levine, B. (2010). Autophagy and the integrated stress response. Molecular Cell, 40, 280–293.
Vousden, K. H., & Prives, C. (2009). Blinded by the light: The growing complexity of p53. Cell, 137, 413–431.
van Attikum, H., & Gasser, S. M. (2009). Crosstalk between histone modifications during the DNA damage response. Trends in Cell Biology, 19, 207–217.
Miller, K. M., & Jackson, S. P. (2012). Histone marks: Repairing DNA breaks within the context of chromatin. Biochemical Society Transactions, 40, 370–376.
Hoeijmakers, J. H. (2009). DNA damage, aging, and cancer. New England Journal of Medicine, 361, 1475–1485.
Fishel, R., Lescoe, M. K., Rao, M. R., Copeland, N. G., Jenkins, N. A., Garber, J., et al. (1993). The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell, 75, 1027–1038.
Lynch, H. T., & de la Chapelle, A. (1999). Genetic susceptibility to non-polyposis colorectal cancer. Journal of Medical Genetics, 36, 801–818.
Shiloh, Y. (1997). Ataxia-telangiectasia and the Nijmegen breakage syndrome: Related disorders but genes apart. Annual Review of Genetics, 31, 635–662.
Yuille, M. A., & Coignet, L. J. (1998). The ataxia telangiectasia gene in familial and sporadic cancer. Recent Results in Cancer Research, 154, 156–173.
McKinnon, P. J., & Caldecott, K. W. (2007). DNA strand break repair and human genetic disease. Annual Review of Genomics and Human Genetics, 8, 37–55.
Futreal, P. A., Liu, Q., Shattuck-Eidens, D., Cochran, C., Harshman, K., Tavtigian, S., et al. (1994). BRCA1 mutations in primary breast and ovarian carcinomas. Science, 266(5182), 120–122.
Wooster, R., Bignell, G., Lancaster, J., et al. (1995). Identification of the breast cancer susceptibility gene BRCA2. Nature, 378(6559), 789–792.
Jazaeri, A. A., Yee, C. J., Sotiriou, C., Brantley, K. R., Boyd, J., & Liu, E. T. (2002). Gene expression profiles of BRCA1-linked, BRCA2-linked, and sporadic ovarian cancers. Journal of the National Cancer Institute, 94, 990–1000.
Berman, D. B., Costalas, J., Schultz, D. C., Grana, G., Daly, M., & Godwin, A. K. (1996). A common mutation in BRCA2 that predisposes to a variety of cancers is found in both Jewish Ashkenazi and non-Jewish individuals. Cancer Research, 56, 3409–3414.
Friedenson, B. (2005). BRCA1 and BRCA2 pathways and the risk of cancers other than breast or ovarian. Med Gen Med, 7, 60.
D’Andrea, A. D., & Grompe, M. (2003). The Fanconi anaemia/BRCA pathway. Nature Reviews Cancer, 3, 23–34.
Hucl, T., & Gallmeier, E. (2011). DNA repair: Exploiting the Fanconi anemia pathway as a potential therapeutic target. Physiological Research, 60, 453–465.
d’Adda di Fagagna, F., Reaper, P. M., Clay-Farrace, L., Fiegler, H., Carr, P., Von Zglinicki, T., et al. (2003). A DNA damage checkpoint response in telomere-initiated senescence. Nature, 426, 194–198.
Jones, R. M., Mortusewicz, O., Afzal, I., Lorvellec, M., García, P., Helleday, T., & Petermann, E. (2012). Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene. doi:10.1038/onc.2012.387.
Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N., Johnson, D. A., Richardson, T. B., et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature, 434, 917–921.
Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E., et al. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature, 434, 913–917.
Harper, J. W., & Elledge, S. J. (2007). The DNA damage response: Ten years after. Molecular Cell, 28, 739–745.
Polo, S. E., & Jackson, S. P. (2011). Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes & Development, 25, 409–433.
Huen, M. S., & Chen, J. (2008). The DNA damage response pathways: At the crossroad of protein modifications. Cell Research, 18, 8–16.
Alpi, A. F., & Patel, K. J. (2009). Monoubiquitylation in the Fanconi anemia DNA damage response pathway. DNA Repair (Amsterdam), 8, 430–435.
Panier, S., & Durocher, D. (2009). Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair (Amsterdam), 8, 436–443.
Messick, T. E., & Greenberg, R. A. (2009). The ubiquitin landscape at DNA double-strand breaks. Journal of Cell Biology, 187, 319–326.
Fraile, J. M., Quesada, V., Rodríguez, D., Freije, J. M., & López-Otín, C. (2012). Deubiquitinases in cancer: New functions and therapeutic options. Oncogene, 31, 2373–2388.
Jackson, S. P., & Durocher, D. (2013). Regulation of DNA responses by ubiquitin and SUMO. Molecular Cell (in press).
Hershko, A., & Ciechanover, A. (1998). The ubiquitin system. Annual Review of Biochemistry, 67, 425–479.
Wilkinson, K. D. (2000). Ubiquitination and deubiquitination: Targeting of proteins for degradation by the proteasome. Seminars in Cell & Developmental Biology, 11, 141–148.
Varshavsky, A. (2012). The ubiquitin system, an immense realm. Annual Review of Biochemistry, 81, 167–176.
Hicke, L. (2001). Protein regulation by monoubiquitin. Nature Reviews Molecular Cell Biology, 2, 195–201.
Ikeda, F., & Dikic, I. (2008). Atypical ubiquitin chains: New molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Reports, 9, 536–542.
Iwai, K. (2012). Diverse ubiquitin signaling in NF-κB activation. Trends in Cell Biology, 22, 355–364.
Komander, D., & Rape, M. (2012). The ubiquitin code. Annual Review of Biochemistry, 81, 203–229.
Hay, R. T. (2007). SUMO-specific proteases: A twist in the tail. Trends in Cell Biology, 17, 370–376.
Hochstrasser, M. (2009). Origin and function of ubiquitin-like proteins. Nature, 2009(458), 422–429.
van der Veen, A. G., & Ploegh, H. L. (2012). Ubiquitin-like proteins. Annual Review of Biochemistry, 81, 323–357.
Reyes-Turcu, F. E., Ventii, K. H., & Wilkinson, K. D. (2009). Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annual Review of Biochemistry, 78, 363–397.
Reyes-Turcu, F. E., & Wilkinson, K. D. (2009). Polyubiquitin binding and disassembly by deubiquitinating enzymes. Chemical Reviews, 109, 1495–1508.
Sacco, J. J., Coulson, J. M., Clague, M. J., & Urbé, S. (2010). Emerging roles of deubiquitinases in cancer-associated pathways. IUBMB Life, 62, 140–157.
Zhang, D., & Zhang, D. E. (2011). Interferon-stimulated gene 15 and the protein ISGylation system. Journal of Interferon and Cytokine Research, 31, 119–130.
Dou, H., Huang, C., Singh, M., Carpenter, P. B., & Yeh, E. T. (2010). Regulation of DNA repair through deSUMOylation and SUMOylation of replication protein A complex. Molecular Cell, 39, 333–345.
Komander, D., Clague, M. J., & Urbé, S. (2009). Breaking the chains: Structure and function of the deubiquitinases. Nature Reviews Molecular Cell Biology, 10, 550–563.
Lindner, H. A. (2007). Deubiquitination in virus infection. Virology, 362, 245–256.
Sun, S. C. (2008). Deubiquitylation and regulation of the immune response. Nature Reviews Immunology, 8(7), 501–511.
Hussain, S., Zhang, Y., & Galardy, P. J. (2009). DUBs and cancer: The role of deubiquitinating enzymes as oncogenes, non-oncogenes and tumor suppressors. Cell Cycle, 8, 1688–1697.
Ramakrishna, S., Suresh, B., & Baek, K. H. (2011). The role of deubiquitinating enzymes in apoptosis. Cellular and Molecular Life Sciences, 68, 15–26.
Hibbert, R. G., Mattiroli, F., & Sixma, T. K. (2009). Structural aspects of multi-domain RING/Ubox E3 ligases in DNA repair. DNA Repair (Amsterdam), 8, 525–535.
Hofmann, K. (2009). Ubiquitin-binding domains and their role in the DNA damage response. DNA Repair (Amsterdam), 8, 544–556.
Al-Hakim, A., Escribano-Diaz, C., Landry, M. C., O’Donnell, L., Panier, S., Szilard, R. K., et al. (2010). The ubiquitous role of ubiquitin in the DNA damage response. DNA Repair (Amsterdam), 9, 1229–1240.
Nijman, S. M., Huang, T. T., Dirac, A. M., Brummelkamp, T. R., Kerkhoven, R. M., D’Andrea, A. D., et al. (2005). The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Molecular Cell, 17, 331–339.
Huang, T. T., Nijman, S. M., Mirchandani, K. D., Galardy, P. J., Cohn, M. A., Haas, W., et al. (2006). Regulation of monoubiquitinated PCNA by DUB autocleavage. Nature Cell Biology, 8, 339–347.
Taniguchi, T., Garcia-Higuera, I., Andreassen, P. R., Gregory, R. C., Grompe, M., & D’Andrea, A. D. (2002). S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood, 100, 2414–2420.
Wang, X., Andreassen, P. R., & D’Andrea, A. D. (2004). Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Molecular and Cellular Biology, 24, 5850–5862.
Montes de Oca, R., Andreassen, P. R., Margossian, S. P., Gregory, R. C., Taniguchi, T., Wang, X., et al. (2005). Regulated interaction of the Fanconi anemia protein, FANCD2, with chromatin. Blood, 105, 1003–1009.
Cohn, M. A., Kowal, P., Yang, K., Haas, W., Huang, T. T., Gygi, S. P., et al. (2007). A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Molecular Cell, 28, 786–797.
Lee, S. W., Lee, M. H., Park, J. H., Kang, S. H., Yoo, H. M., Ka, S. H., Oh, Y. M., Jeon, Y. J., & Chung, C. H. (2012, October 23). SUMOylation of hnRNP-K is required for p53-mediated cell-cycle arrest in response to DNA damage. EMBO Journal.
Sikdar, N., Banerjee, S., Lee, K. Y., Wincovitch, S., Pak, E., Nakanishi, K., et al. (2009). DNA damage responses by human ELG1 in S phase are important to maintain genomic integrity. Cell Cycle, 8, 3199–3207.
Kim, J. M., Parmar, K., Huang, M., Weinstock, D. M., Ruit, C. A., Kutok, J. L., et al. (2009). Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Developmental Cell, 16, 314–320.
Oestergaard, V. H., Langevin, F., Kuiken, H. J., Pace, P., Niedzwiedz, W., Simpson, L. J., et al. (2007). Deubiquitylation of FANCD2 is required for DNA crosslink repair. Molecular Cell, 28, 798–809.
Murai, J., Yang, K., Dejsuphong, D., Hirota, K., Takeda, S., & D’Andrea, A. D. (2011). The USP1/UAF1 complex promotes double-strand break repair through homologous recombination. Molecular and Cellular Biology, 31, 2462–2469.
Guervilly, J. H., Renaud, E., Takata, M., & Rosselli, F. (2011). USP1 deubiquitinase maintains phosphorylated CHK1 by limiting its DDB1-dependent degradation. Human Molecular Genetics, 20, 2171–2181.
Jones, M. J., Colnaghi, L., & Huang, T. T. (2011). Dysregulation of DNA polymerase κ recruitment to replication forks results in genomic instability. EMBO Journal, 31, 908–918.
Ogi, T., Limsirichaikul, S., Overmeer, R. M., Volker, M., Takenaka, K., Cloney, R., et al. (2010). Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Molecular Cell, 37, 714–727.
Cotto-Rios, X. M., Jones, M. J., Busino, L., Pagano, M., & Huang, T. T. (2011). APC/CCdh1-dependent proteolysis of USP1 regulates the response to UV-mediated DNA damage. Journal of Cell Biology, 194, 177–186.
Williams, S. A., Maecker, H. L., French, D. M., Liu, J., Gregg, A., Silverstein, L. B., et al. (2011). USP1 deubiquitylates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell, 146, 918–930.
Lasorella, A., Uo, T., & Iavarone, A. (2001). Id proteins at the cross-road of development and cancer. Oncogene, 20, 8326–8333.
Lasorella, A., Stegmuller, J., Guardavaccaro, D., Liu, G., Carro, M. S., Rothschild, G., et al. (2006). Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature, 442, 471–474.
Priolo, C., Tang, D., Brahamandan, M., Benassi, B., Sicinska, E., Ogino, S., et al. (2006). The isopeptidase USP2a protects human prostate cancer from apoptosis. Cancer Research, 66, 8625–8632.
Stevenson, L. F., Sparks, A., Allende-Vega, N., Xirodimas, D. P., Lane, D. P., & Saville, M. K. (2007). The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO Journal, 26, 976–986.
Allende-Vega, N., Sparks, A., Lane, D. P., & Saville, M. K. (2010). MdmX is a substrate for the deubiquitinating enzyme USP2a. Oncogene, 29, 432–441.
Nicassio, F., Corrado, N., Vissers, J. H., Areces, L. B., Bergink, S., Marteijn, J. A., et al. (2007). Human USP3 is a chromatin modifier required for S phase progression and genome stability. Current Biology, 17, 1972–1977.
Doil, C., Mailand, N., Bekker-Jensen, S., Menard, P., Larsen, D. H., Pepperkok, R., et al. (2009). RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell, 136, 435–446.
Buus, R., Faronato, M., Hammond, D. E., Urbé, S., & Clague, M. J. (2009). Deubiquitinase activities required for hepatocyte growth factor-induced scattering of epithelial cells. Current Biology, 19, 1463–1466.
Zhang, X., Berger, F. G., Yang, J., & Lu, X. (2011). USP4 inhibits p53 through deubiquitinating and stabilizing ARF-BP1. EMBO Journal, 30, 2177–2189.
Liang, L., Fan, Y., Cheng, J., Cheng, D., Zhao, Y., Cao, B., et al. (2012). TAK1 ubiquitylation regulates doxorubicin-induced NF-κB activation. Cellular Signalling, 25, 247–254.
Zhou, F., Zhang, X., van Dam, H., Ten Dijke, P., Huang, H., & Zhang, L. (2012). Ubiquitin-specific protease 4 mitigates Toll-like/interleukin-1 receptor signaling and regulates innate immune activation. Journal of Biological Chemistry, 287, 11002–11010.
Zhang, L., Zhou, F., Drabsch, Y., Gao, R., Snaar-Jagalska, B. E., Mickanin, C., et al. (2012). USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-β type I receptor. Nature Cell Biology, 14, 717–726.
Aggarwal, K., & Massagué, J. (2012). Ubiquitin removal in the TGF-β pathway. Nature Cell Biology, 14, 656–657.
Xiao, N., Li, H., Luo, J., Wang, R., Chen, H., Chen, J., et al. (2012). Ubiquitin-specific protease 4 (USP4) targets TRAF2 and TRAF6 for deubiquitylation and inhibits TNFα-induced cancer cell migration. Biochemistry Journal, 441, 979–986.
Uras, I. Z., List, T., Nijman, S. M. (2012). Ubiquitin-specific protease 4 inhibits mono-ubiquitylation of the master growth factor signaling kinase PDK1. PLoS One, 7, 31003.
Zhao, B., Schlesiger, C., Masucci, M. G., & Lindsten, K. (2009). The ubiquitin specific protease 4 (USP4) is a new player in the Wnt signalling pathway. Journal of Cellular and Molecular Medicine, 8B, 1886–1895.
Dayal, S., Sparks, A., Jacob, J., Allende-Vega, N., Lane, D. P., & Saville, M. K. (2009). Suppression of the deubiquitinating enzyme USP5 causes the accumulation of unanchored polyubiquitin and the activation of p53. Journal of Biological Chemistry, 284, 5030–5041.
Meulmeester, E., Maurice, M. M., Boutell, C., Teunisse, A. F., Ovaa, H., Abraham, T. E., et al. (2005). Loss of HAUSP-mediated deubiquitylation contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Molecular Cell, 18, 565–576.
Faustrup, H., Bekker-Jensen, S., Bartek, J., Lukas, J., & Mailand, N. (2009). USP7 counteracts SCFbetaTrCP- but not APCCdh1-mediated proteolysis of Claspin. Journal of Cell Biology, 18, 13–19.
Khoronenkova, S. V., Dianova, I. I., Ternette, N., Kessler, B. M., Parsons, J. L., & Dianov, G. L. (2012). ATM-dependent downregulation of USP7/HAUSP by PPM1G activates p53 response to DNA damage. Molecular Cell, 45, 801–813.
van der Horst, A., de Vries-Smits, A. M., Brenkman, A. B., van Triest, M. H., van den Broek, N., Colland, F., et al. (2006). FOXO4 transcriptional activity is regulated by monoubiquitylation and USP7/HAUSP. Nature Cell Biology, 10, 1064–1073.
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell, 144, 646–674.
Khoronenkova, S. V., Dianova, I. I., Parsons, J. L., & Dianov, G. L. (2011). USP7/HAUSP stimulates repair of oxidative DNA lesions. Nucleic Acids Research, 7, 2604–2609.
Hanawalt, P. C., & Spivak, G. (2008). Transcription-coupled DNA repair: Two decades of progress and surprises. Nature Reviews Molecular Cell Biology, 12, 958–970.
Schwertman, P., Lagarou, A., Dekkers, D. H., Raams, A., van der Hoek, A. C., Laffeber, C., et al. (2012). UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nature Genetics, 5, 598–602.
Li, M., Chen, D., Shiloh, A., Luo, J., Nikolaev, A. Y., Qin, J., et al. (2002). Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature, 416, 648–653.
Youle, R. J., & Strasser, A. (2008). The BCL-2 protein family: Opposing activities that mediate cell death. Nature Reviews Molecular Cell Biology, 9, 47–59.
Schwickart, M., Huang, X., Lill, J. R., Liu, J., Ferrando, R., French, D. M., et al. (2010). Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature, 463, 103–107.
Trivigno, D., Essmann, F., Huber, S. M., & Rudner, J. (2012). Deubiquitinase USP9x confers radioresistance through stabilization of Mcl-1. Neoplasia, 14, 893–904.
Harris, D. R., Mims, A., & Bunz, F. (2012). Genetic disruption of USP9X sensitizes colorectal cancer cells to 5-fluorouracil. Cancer Biology & Therapy, 13, 1319–1324.
Yuan, J., Luo, K., Zhang, L., Cheville, J. C., & Lou, Z. (2010). USP10 regulates p53 localization and stability by deubiquitinating p53. Cell, 140, 384–396.
Liu, J., Xia, H., Kim, M., Xu, L., Li, Y., Zhang, L., et al. (2011). Beclin1 controls the levels of p53 by regulating the deubiquitylation activity of USP10 and USP13. Cell, 147, 223–234.
Maertens, G. N., El Messaoudi-Aubert, S., Elderkin, S., Hiom, K., & Peters, G. (2010). Ubiquitin-specific proteases 7 and 11 modulate Polycomb regulation of the INK4a tumour suppressor. EMBO Journal, 29, 2553–2565.
Wiltshire, T. D., Lovejoy, C. A., Wang, T., Xia, F., O’Connor, M. J., & Cortez, D. (2010). Sensitivity to poly(ADP-ribose) polymerase (PARP) inhibition identifies ubiquitin-specific peptidase 11 (USP11) as a regulator of DNA double-strand break repair. The Journal of Biological Chemistry, 285, 14565–14571.
Patel, K. J., Yu, V. P., Lee, H., Corcoran, A., Thistlethwaite, F. C., Evans, M. J., et al. (1998). Involvement of Brca2 in DNA repair. Molecular Cell, 1, 347–357.
Tutt, A., Gabriel, A., Bertwistle, D., Connor, F., Paterson, H., Peacock, J., et al. (1999). Absence of BRCA2 causes genome instability by chromosome breakage and loss associated with centrosome amplification. Current Biology, 9, 1107–1110.
Yu, V. P., Koehler, M., Steinlein, C., Schmid, M., Hanakahi, L. A., van Gool, A. J., et al. (2000). Gross chromosomal rearrangements and genetic exchange between nonhomologous chromosomes following BRCA2 inactivation. Genes & Development, 14, 1400–1406.
Moynahan, M. E., Pierce, A. J., & Jasin, M. (2001). BRCA2 is required for homology-directed repair of chromosomal breaks. Molecular Cell, 7, 263–272.
Tutt, A., Bertwistle, D., Valentine, J., Gabriel, A., Swift, S., Ross, G., et al. (2001). Mutation in BRCA2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO Journal, 20, 4704–4716.
Schoenfeld, A. R., Apgar, S., Dolios, G., Wang, R., & Aaronson, S. A. (2004). BRCA2 is ubiquitinated in vivo and interacts with USP11, a deubiquitinating enzyme that exhibits prosurvival function in the cellular response to DNA damage. Molecular and Cellular Biology, 24, 7444–7455.
Joo, H. Y., Zhai, L., Yang, C., Nie, S., Erdjument-Bromage, H., Tempst, P., et al. (2007). Regulation of cell cycle progression and gene expression by H2A deubiquitylation. Nature, 449, 1068–1072.
Shanbhag, N. M., Rafalska-Metcalf, I. U., Balane-Bolivar, C., Janicki, S. M., & Greenberg, R. A. (2010). ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell, 141, 970–981.
Mattiroli, F., Vissers, J. H., van Dijk, W. J., Ikpa, P., Citterio, E., Vermeulen, W., et al. (2012). RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell, 150, 1182–1195.
Zhang, L., Lubin, A., Chen, H., Sun, Z., & Gong, F. (2012). The deubiquitinating protein USP24 interacts with DDB2 and regulates DDB2 stability. Cell Cycle, 1, 23.
Bakkenist, C. J., & Kastan, M. B. (2004). Initiating cellular stress responses. Cell, 118, 9–17.
Wang, B., Matsuoka, S., Carpenter, P. B., & Elledge, S. J. (2002). 53BP1, a mediator of the DNA damage checkpoint. Science, 298, 1435–1438.
Ward, I. M., Minn, K., van Deursen, J., & Chen, J. (2003). p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Molecular and Cellular Biology, 7, 2556–2563.
Zhang, D., Zaugg, K., Mak, T. W., & Elledge, S. J. (2006). A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell, 126, 529–542.
Popov, N., Wanzel, M., Madiredjo, M., Zhang, D., Beijersbergen, R., Bernards, R., et al. (2007). The ubiquitin-specific protease USP28 is required for MYC stability. Nature Cell Biology, 7, 765–774.
Jiang, M. R., Li, Y. C., Yang, Y., & Wu, J. R. (2003). C-Myc degradation induced by DNA damage results in apoptosis of CHO cells. Oncogene, 2, 2325–2359.
Herbst, A., Hemann, M. T., Tworkowski, K. A., Salghetti, S. E., Lowe, S. W., & Tansey, W. P. (2005). A conserved element in Myc that negatively regulates its proapoptotic activity. EMBO Reports, 2, 177–183.
Popov, N., Herold, S., Llamazares, M., Schülein, C., & Eilers, M. (2007). Fbw7 and Usp28 regulate myc protein stability in response to DNA damage. Cell Cycle, 6, 2327–2331.
Liu, J., Chung, H. J., Vogt, M., Jin, Y., Malide, D., He, L., et al. (2011). JTV1 co-activates FBP to induce USP29 transcription and stabilize p53 in response to oxidative stress. EMBO Journal, 30, 846–858.
Musacchio, A., & Salmon, E. D. (2007). The spindle-assembly checkpoint in space and time. Nature Reviews Molecular Cell Biology, 8, 379–393.
Stegmeier, F., Rape, M., Draviam, V. M., Nalepa, G., Sowa, M. E., Ang, X. L., et al. (2007). Anaphase initiation is regulated by antagonistic ubiquitylation and deubiquitylation activities. Nature, 446, 876–881.
Song, L., & Rape, M. (2010). Regulated degradation of spindle assembly factors by the anaphase-promoting complex. Molecular Cell, 38, 369–382.
Zhang, Y., van Deursen, J., & Galardy, P. J. (2011). Overexpression of ubiquitin specific protease 44 (USP44) induces chromosomal instability and is frequently observed in human T-cell leukemia. PLoS ONE, 6, e23389.
Zhang, Y., Foreman, O., Wigle, D. A., Kosari, F., Vasmatzis, G., Salisbury, J. L., et al. (2012). USP44 regulates centrosome positioning to prevent aneuploidy and suppress tumorigenesis. The Journal of Clinical Investigation, 122, 4362–4374.
Lindahl, T. (1993). Instability and decay of the primary structure of DNA. Nature, 362, 709–715.
Dianov, G. L., & Parsons, J. L. (2007). Co-ordination of DNA single strand break repair. DNA Repair (Amsterdam), 6, 454–460.
Parsons, J. L., Dianova, I. I., Khoronenkova, S. V., Edelmann, M. J., Kessler, B. M., & Dianov, G. L. (2011). USP47 is a deubiquitylating enzyme that regulates base excision repair by controlling steady-state levels of DNA polymerase beta. Molecular Cell, 41, 609–615.
Peschiaroli, A., Skaar, J. R., Pagano, M., & Melino, G. (2010). The ubiquitin-specific protease USP47 is a novel beta-TRCP interactor regulating cell survival. Oncogene, 29, 1384–1393.
Xiang, T., Li, L., Yin, X., Yuan, C., Tan, C., Su, X., et al. (2012). The ubiquitin peptidase UCHL1 induces G0/G1 cell cycle arrest and apoptosis through stabilizing p53 and is frequently silenced in breast cancer. PLoS ONE, 7, e29783.
Li, L., Tao, Q., Jin, H., van Hasselt, A., Poon, F. F., Wang, X., et al. (2010). The tumor suppressor UCHL1 forms a complex with p53/MDM2/ARF to promote p53 signaling and is frequently silenced in nasopharyngeal carcinoma. Clinical Cancer Research, 16, 2949–2958.
Ummanni, R., Jost, E., Braig, M., Lohmann, F., Mundt, F., Barett, C., et al. (2011). Ubiquitin carboxyl-terminal hydrolase 1 (UCHL1) is a potential tumour suppressor in prostate cancer and is frequently silenced by promoter methylation. Molecular Cancer, 10, 129.
Hussain, S., Foreman, O., Perkins, S. L., Witzig, T. E., Miles, R. R., van Deursen, J., et al. (2010). The de-ubiquitinase UCH-L1 is an oncogene that drives the development of lymphoma in vivo by deregulating PHLPP1 and Akt signaling. Leukemia, 24, 1641–1655.
Zhong, J., Zhao, M., Ma, Y., Luo, Q., Liu, J., Wang, J., et al. (2012). UCHL1 acts as a colorectal cancer oncogene via activation of the β-catenin/TCF pathway through its deubiquitinating activity. International Journal of Molecular Medicine, 30, 430–436.
Dong, Y., Hakimi, M. A., Chen, X., Kumaraswamy, E., Cooch, N. S., Godwin, A. K., et al. (2003). Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair. Molecular Cell, 12, 1087–1099.
Chen, X., Arciero, C. A., Wang, C., Broccoli, D., & Godwin, A. K. (2006). BRCC36 is essential for ionizing radiation-induced BRCA1 phosphorylation and nuclear foci formation. Cancer Research, 66, 5039–5046.
Sobhian, B., Shao, G., Lilli, D. R., Culhane, A. C., Moreau, L. A., Xia, B., et al. (2007). RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science, 316, 1198–1202.
Shao, G., Patterson-Fortin, J., Messick, T. E., Feng, D., Shanbhag, N., Wang, Y., et al. (2009). MERIT40 controls BRCA1–RAP80 complex integrity and recruitment to DNA double-strand breaks. Genes & Development, 23, 740–754.
Shao, G., Lilli, D. R., Patterson-Fortin, J., Coleman, K. A., Morrissey, D. E., & Greenberg, R. A. (2009). The RAP80–BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitylation events at DNA double strand breaks. Proceedings of the National Academy of Sciences of the United States of America, 106, 3166–3171.
Coleman, K. A., & Greenberg, R. A. (2011). The BRCA1–RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. Journal of Biological Chemistry, 286, 13669–13680.
Fisch, P., Forster, A., Sherrington, P. D., Dyer, M. J., & Rabbitts, T. H. (1993). The chromosomal translocation t(X;14)(q28;q11) in T-cell pro-lymphocytic leukaemia breaks within one gene and activates another. Oncogene, 8, 3271–3276.
Zhu, P., Zhou, W., Wang, J., Puc, J., Ohgi, K. A., Erdjument-Bromage, H., et al. (2007). A histone H2A deubiquitinase complex coordinating histone acetylation and H1 dissociation in transcriptional regulation. Molecular Cell, 27, 609–621.
Jiang, X. X., Nguyen, Q., Chou, Y., Wang, T., Nandakumar, V., Yates, P., et al. (2011). Control of B cell development by the histone H2A deubiquitinase MYSM1. Immunity, 35, 883–896.
Nijnik, A., Clare, S., Hale, C., Raisen, C., McIntyre, R. E., Yusa, K., et al. (2012). The critical role of histone H2A-deubiquitinase Mysm1 in hematopoiesis and lymphocyte differentiation. Blood, 119, 1370–1379.
Panier, S., Ichijima, Y., Fradet-Turcotte, A., Leung, C. C., Kaustov, L., Arrowsmith, C. H., et al. (2012). Tandem protein interaction modules organize the ubiquitin-dependent response to DNA double-strand breaks. Molecular Cell, 47, 383–395.
Bekker-Jensen, S., & Mailand, N. (2011). The ubiquitin- and SUMO-dependent signaling response to DNA double-strand breaks. FEBS Letters, 585, 2914–2919.
Butler, L. R., Densham, R. M., Jia, J., Garvin, A. J., Stone, H. R., Shah, V., et al. (2012). The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. EMBO Journal, 31, 3918–3934.
Morris, J. R. (2012). Attenuation of the ubiquitin conjugate DNA damage signal by the proteasomal DUB POH1. Cell Cycle, 11, 22.
Galanty, Y., Belotserkovskaya, R., Coates, J., & Jackson, S. P. (2012). RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes & Development, 26, 1179–1195.
Nakada, S., Tai, I., Panier, S., Al-Hakim, A., Iemura, S., Juang, Y. C., et al. (2010). Non-canonical inhibition of DNA damage-dependent ubiquitylation by OTUB1. Nature, 466, 941–946.
Juang, Y. C., Landry, M. C., Sanches, M., Vittal, V., Leung, C. C., Ceccarelli, D. F., et al. (2012). OTUB1 co-opts Lys48-linked ubiquitin recognition to suppress E2 enzyme function. Molecular Cell, 45, 384–397.
Sato, Y., Yamagata, A., Goto-Ito, S., Kubota, K., Miyamoto, R., Nakada, S., et al. (2012). Molecular basis of Lys-63-linked polyubiquitylation inhibition by the interaction between human deubiquitinating enzyme OTUB1 and ubiquitin-conjugating enzyme UBC13. Journal of Biological Chemistry, 287, 25860–25868.
Wiener, R., Zhang, X., Wang, T., & Wolberger, C. (2012). The mechanism of OTUB1-mediated inhibition of ubiquitylation. Nature, 483, 618–622.
Sun, X. X., Challagundla, K. B., & Dai, M. S. (2011). Positive regulation of p53 stability and activity by the deubiquitinating enzyme Otubain 1. EMBO Journal, 31, 576–592.
Yang, Y., Fu, W., Chen, J., Olashaw, N., Zhang, X., Nicosia, S. V., et al. (2007). SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nature Cell Biology, 9, 1253–1262.
Yates, K. E., Korbel, G. A., Shtutman, M., Roninson, I. B., & DiMaio, D. (2008). Repression of the SUMO-specific protease Senp1 induces p53-dependent premature senescence in normal human fibroblasts. Aging Cell, 7, 609–621.
Bawa-Khalfe, T., Cheng, J., Lin, S. H., Ittmann, M. M., & Yeh, E. T. (2010). SENP1 induces prostatic intraepithelial neoplasia through multiple mechanisms. Journal of Biological Chemistry, 285, 25859–25866.
Li, X., Lee, Y. K., Jeng, J. C., Yen, Y., Schultz, D. C., Shih, H. M., et al. (2007). Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1-mediated transcriptional repression. Journal of Biological Chemistry, 282, 36177–36189.
Lee, M. H., Mabb, A. M., Gill, G. B., Yeh, E. T., & Miyamoto, S. (2011). NF-κB induction of the SUMO protease SENP2: A negative feedback loop to attenuate cell survival response to genotoxic stress. Molecular Cell, 43, 180–191.
Moumen, A., Masterson, P., O’Connor, M. J., & Jackson, S. P. (2005). hnRNP K: An HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell, 123, 1065–1078.
Jiang, M., Chiu, S. Y., & Hsu, W. (2011). SUMO-specific protease 2 in Mdm2-mediated regulation of p53. Cell Death and Differentiation, 18, 1005–1015.
Dou, H., Huang, C., Van Nguyen, T., Lu, L. S., & Yeh, E. T. (2011). SUMOylation and de-SUMOylation in response to DNA damage. FEBS Letters, 585, 2891–2896.
Cheng, Z., Ke, Y., Ding, X., Wang, F., Wang, H., Wang, W., et al. (2008). Functional characterization of TIP60 sumoylation in UV-irradiated DNA damage response. Oncogene, 27, 931–941.
Isaacson, M. K., & Ploegh, H. L. (2009). Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host & Microbe, 5, 559–570.
Whitehurst, C. B., Vaziri, C., Shackelford, J., & Pagano, J. S. (2012). Epstein-Barr virus BPLF1 deubiquitylates PCNA and attenuates polymerase η recruitment to DNA damage sites. Journal of Virology, 86, 8097–8106.
Adams, J. (2004). The development of proteasome inhibitors as anticancer drugs. Cancer Cell, 5, 417–421.
Vassilev, L. T. (2007). MDM2 inhibitors for cancer therapy. Trends in Molecular Medicine, 13, 23–31.
Soucy, T. A., Smith, P. G., Milhollen, M. A., Berger, A. J., Gavin, J. M., Adhikari, S., et al. (2009). An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature, 458, 732–736.
Ceccarelli, D. F., Tang, X., Pelletier, B., Orlicky, S., Xie, W., Plantevin, V., et al. (2011). An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell, 145, 1075–1087.
Bedford, L., Lowe, J., Dick, L. R., Mayer, R. J., & Brownell, J. E. (2011). Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nature Reviews Drug Discovery, 10, 29–46.
Nicholson, B., Marblestone, J. G., Butt, T. R., & Mattern, M. R. (2007). Deubiquitinating enzymes as novel anticancer targets. Future Oncology, 3, 191–199.
Cohen, P., & Tcherpakov, M. (2010). Will the ubiquitin system furnish as many drug targets as protein kinases? Cell, 143, 686–693.
Edelmann, M. J., Nicholson, B., & Kessler, B. M. (2011). Pharmacological targets in the ubiquitin system offer new ways of treating cancer, neurodegenerative disorders and infectious diseases. Expert Reviews in Molecular Medicine, 13, e35.
Brough, R., Frankum, J. R., Costa-Cabral, S., Lord, C. J., & Ashworth, A. (2011). Searching for synthetic lethality in cancer. Current Opinion in Genetics & Development, 21, 34–41.
Yap, T. A., & Workman, P. (2012). Exploiting the cancer genome: Strategies for the discovery and clinical development of targeted molecular therapeutics. Annual Review of Pharmacology and Toxicology, 52, 549–573.
Morandell, S., & Yaffe, M. B. (2012). Exploiting synthetic lethal interactions between DNA damage signaling, checkpoint control, and p53 for targeted cancer therapy. Progress in Molecular Biology and Translational Science, 110, 289–314.
Bouwman, P., & Jonkers, J. (2012). The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nature Reviews Cancer, 12, 587–598.
Chen, J., Dexheimer, T. S., Ai, Y., Liang, Q., Villamil, M. A., Inglese, J., et al. (2011). Selective and cell-active inhibitors of the USP1/UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chemistry & Biology, 18, 1390–1400.
Colland, F., Formstecher, E., Jacq, X., Reverdy, C., Planquette, C., Conrath, S., et al. (2009). Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Molecular Cancer Therapeutics, 8, 2286–2295.
Colombo, M., Vallese, S., Peretto, I., Jacq, X., Rain, J. C., Colland, F., et al. (2010). Synthesis and biological evaluation of 9-oxo-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile analogues as potential inhibitors of deubiquitinating enzymes. Chem Med Chem, 5, 552–558.
Fernández-Montalván, A., Bouwmeester, T., Joberty, G., Mader, R., Mahnke, M., Pierrat, B., et al. (2007). Biochemical characterization of USP7 reveals post-translational modification sites and structural requirements for substrate processing and subcellular localization. FEBS Journal, 274, 4256–4270.
Wrigley, J. D., Eckersley, K., Hardern, I. M., Millard, L., Walters, M., Peters, S. W., et al. (2011). Enzymatic characterisation of USP7 deubiquitinating activity and inhibition. Cell Biochemistry and Biophysics, 60, 99–111.
Chauhan, D., Tian, Z., Nicholson, B., Kumar, K. G., Zhou, B., Carrasco, R., et al. (2012). A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell, 22, 345–358.
Weinstock, J., Wu, J., Cao, P., Kingsbury, W., McDermott, J., Kodrasov, M., et al. (2012). Selective dual inhibitors of the cancer-related deubiquitylating proteases USP7 and USP47. ACS Medicinal Chemistry Letters, 3, 789–792.
Reverdy, C., Conrath, S., Lopez, R., Planquette, C., Atmanene, C., Collura, V., et al. (2012). Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chemistry & Biology, 19, 467–477.
Zhang, Y., Zhou, L., Rouge, L., Phillips, A. H., Lam, C., Liu, P., et al. (2013). Conformational stabilization of ubiquitin yields potent and selective inhibitors of USP7. Nature Chemical Biology, 9, 51–58.
Liu, Y., Lashuel, H. A., Choi, S., Xing, X., Case, A., Ni, J., et al. (2003). Discovery of inhibitors that elucidate the role of UCH-L1 activity in the H1299 lung cancer cell line. Chemistry & Biology, 10, 837–846.
Li, Z., Melandri, F., Berdo, I., Jansen, M., Hunter, L., Wright, S., et al. (2004). Delta12-prostaglandin J2 inhibits the ubiquitin hydrolase UCH-L1 and elicits ubiquitin-protein aggregation without proteasome inhibition. Biochemical and Biophysical Research Communications, 319, 1171–1180.
Verbitski, S. M., Mullally, J. E., Fitzpatrick, F. A., & Ireland, C. M. (2004). Punaglandins, chlorinated prostaglandins, function as potent Michael receptors to inhibit ubiquitin isopeptidase activity. Journal of Medicinal Chemistry, 47, 2062–2070.
Hirayama, K., Aoki, S., Nishikawa, K., Matsumoto, T., & Wada, K. (2007). Identification of novel chemical inhibitors for ubiquitin C-terminal hydrolase-L3 by virtual screening. Bioorganic & Medicinal Chemistry, 15, 6810–6818.
Mitsui, T., Hirayama, K., Aoki, S., Nishikawa, K., Uchida, K., Matsumoto, T., et al. (2010). Identification of a novel chemical potentiator and inhibitors of UCH-L1 by in silico drug screening. Neurochemistry International, 56, 679–686.
Kapuria, V., Peterson, L. F., Fang, D., Bornmann, W. G., Talpaz, M., & Donato, N. J. (2010). Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Research, 70, 9265–9276.
Altun, M., Kramer, H. B., Willems, L. I., McDermott, J. L., Leach, C. A., Goldenberg, S. J., et al. (2011). Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chemistry & Biology, 18, 1401–1412.
Issaenko, O. A., & Amerik, A. Y. (2012). Chalcone-based small-molecule inhibitors attenuate malignant phenotype via targeting deubiquitinating enzymes. Cell Cycle, 11, 1804–1817.
Park, J. Y., Kim, J. H., Kim, Y. M., Jeong, H. J., Kim, D. W., Park, K. H., et al. (2012). Tanshinones as selective and slow-binding inhibitors for SARS-CoV cysteine proteases. Bioorganic & Medicinal Chemistry, 20, 5928–5935.
Davies, C. W., Chaney, J., Korbel, G., Ringe, D., Petsko, G. A., Ploegh, H., et al. (2012). The co-crystal structure of ubiquitin carboxy-terminal hydrolase L1 (UCHL1) with a tripeptide fluoromethyl ketone (Z-VAE(OMe)-FMK). Bioorganic & Medicinal Chemistry Letters, 22, 3900–3940.
Albrow, V. E., Ponder, E. L., Fasci, D., Békés, M., Deu, E., Salvesen, G. S., et al. (2011). Development of small molecule inhibitors and probes of human SUMO deconjugating proteases. Chemistry & Biology, 18, 722–732.
Uno, M., Koma, Y., Ban, H. S., & Nakamura, H. (2012). Discovery of 1-[4-(N-benzylamino)phenyl]-3-phenylurea derivatives as non-peptidic selective SUMO-sentrin specific protease (SENP)1 inhibitors. Bioorganic & Medicinal Chemistry Letters, 22, 5169–5173.
Fong, P. C., Boss, D. S., Yap, T. A., Tutt, A., Wu, P., Mergui-Roelvink, M., et al. (2009). Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. New England Journal of Medicine, 361, 123–134.
Acknowledgments
We thank all our collaborators at MISSION Therapeutics for their many insightful comments on the roles played by DUBs in the DDR. We also thank the members of the SPJ academic laboratory for helpful discussions and advice. Research in the SPJ laboratory is funded by the CRUK Program Grant C6/A11224, the European Research Council and the European Community Seventh Framework Programme Grant Agreement No. HEALTH-F2-2010-259893 (DDResponse). Core infrastructure funding was provided by the CRUK (C6946/A14492) and the Welcome Trust (WT092096).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Jacq, X., Kemp, M., Martin, N.M.B. et al. Deubiquitylating Enzymes and DNA Damage Response Pathways. Cell Biochem Biophys 67, 25–43 (2013). https://doi.org/10.1007/s12013-013-9635-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-013-9635-3