
Abstract
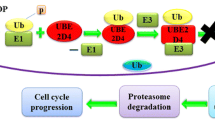
Cancer is characterized by abnormal growth of cells. Targeting ubiquitin proteins in the discovery of new anticancer therapeutics is an attractive strategy. The present study uses the structure-based drug discovery methods to identify new lead structures, which are selective to the putative ubiquitin-conjugating enzyme E2N-like (UBE2NL). The 3D structure of the UBE2NL was evaluated using homology modeling techniques. The model was validated using standard in silico methods. The hydrophobic pocket of UBE2NL that aids in binding with its natural receptor ubiquitin-conjugating enzyme E2 variant (UBE2V) was identified through protein-protein docking study. The binding site region of the UBE2NL was identified using active site prediction tools. The binding site of UBE2NL which is responsible for cancer cell progression is considered for docking study. Virtual screening study with the small molecular structural database was carried out against the active site of UBE2NL. The ligand molecules that have shown affinity towards UBE2NL were considered for ADME prediction studies. The ligand molecules that obey the Lipinski’s rule of five and Jorgensen’s rule of three pharmacokinetic properties like human oral absorption etc. are prioritized. The resultant ligand molecules can be considered for the development of potent UBE2NL enzyme inhibitors for cancer therapy.














Similar content being viewed by others

References
Roos, W. P., Thomas, A. D., & Kaina, B. (2016). DNA damage and the balance between survival and death in cancer biology. Nature Reviews. Cancer, 16, 20–33.
Zhang, A., Yan, G., Han, Y., & Wang, X. (2014). Metabolomics approaches and applications in prostate cancer research. Applied Biochemistry and Biotechnology, 174, 6–12.
Morvan, G. M., & Lanier, L. L. (2016). NK cells and cancer: you can teach innate cells new tricks. Nature Reviews. Cancer, 16, 7–19.
Chen, D., & Dou, Q. P. (2010). The ubiquitin-proteasome system as a prospective molecular target for cancer treatment and prevention. Current Protein & Peptide Science, 11, 459–470.
Nalepa, G., Rofle, M., & Harper, W. J. (2006). Drug discovery in the ubiquitin-proteasome system. Nature Reviews. Drug Discovery, 5, 596–613.
Hamilton, S. K., Ellison, J. M., Barber, R. K., Williams, S. R., Huzil, T. J., Mckenna, S., Ptak, C., Glover, M., & Shaw, S. G. (2001). Structure of a conjugating enzyme-ubiquitin thiol ester intermediate reveals a novel role for the ubiquitin tail. Structure, 9, 897–904.
Ciechnover, A. (2015). The unravelling of the ubiquitin system. Nature Reviews. Molecular Cell Biology, 16, 322–324.
Li, W., & Ye, Y. (2009). Polyubiquitin chains: functions, structures, and mechanisms. Cellular and Molecular Life Sciences, 65, 2397–2406.
Fezza, M., Schmitt, S., & Dou, S. P. (2011). Targeting the ubiquitin-proteasome pathway: an emerging concept in cancer therapy. Current Topics in Medicinal Chemistry, 11, 2888–2905.
Moraes, F. T., Edwards, R. A., McKenna, S., Pastushok, L., Xiao, W., Glover, J. N. M., & Ellison, J. M. (2001). Crystal structure of the human ubiquitin conjugating enzyme complex, hMms2-hUbc13. Nature Structural Biology, 8, 669–673.
Hashizume, R., Fukuda, M., Maeda, I., Nishikawa, H., Oyake, D., Yabuki, Y., Ogata, H., & Ohta, T. (2001). The RING heterodimer BRACA1-BARD1 is ubiquitin ligase inactivated by a breast cancer-derived mutation. The Journal of Biological Chemistry, 276, 14537–14540.
Argentini, M., Barboule, N., & Wasylyk, B. (2000). The contribution of the RING finger domain of MDM2 to cell cycle progression. Oncogene, 19, 3849–3857.
Sali, A., Glaeser, R., Earnest, T., & Baumeister, W. (2003). From words to literature in structural proteomics. Nature, 422, 216–225.
Apweiler, R., Bairoch, A., Wu, C. H., Barker, W. C., Boeckmann, B., Ferro, S., Gasteiger, E., Huang, H., Lopez, R., Magrane, M., Martin, M. J., Natale, D. A., O’Donovan, C., Redaschi, N., & Yeh, L. S. (2004). UniProt: the universal protein knowledge base. Nucleic Acids Research, 32, 115–119.
Gasteiger, E., Gattiker, A., Hoogland, C., Ivanyi, I., Appel, R. D., & Bairoch, A. (2003). ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Research, 31, 3784–3788.
Jhonson, M., Zaretskaya, I., Raytselis, Y., Merezhuk, Y., McGinnis, S., & Madden, T. L. (2008). NCBI BLAST: a better web interface. Nucleic Acids Research, 36, 5–9.
Christian, C., Jonathan, D. B., & Geoffrey, J. B. (2008). The Jpred 3 secondary structure prediction server. Nucleic Acids Research, 36, 197–201.
Kelley, L. A., & Sternberg, M. J. E. (2009). Protein structure prediction on the web: a case study using the Phyre server. Nature Protocols, 4, 363–371.
Ye, J., McGinnis, S., & Madden, T. L. (2006). BLAST: improvements for better sequence analysis. Nucleic Acids Research, 34, 6–9.
Fiser, A. (2010). Template-based protein structure modeling. Methods in Molecular Biology, 673, 73–94.
Thompson, J. D., Higgins, D. G., & Gibson, T. J. (1994). Clustal W: improving the sensitivity of sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research, 22, 4673–4680.
Gonnet, G. H., Cohen, M. A., & Benner, S. A. (1992). Exhaustive matching of the entire protein sequence database. Science, 256, 1443–1445.
Webb, B., & Sali, A. (2014). Comparative protein structure modeling using MODELLER. Current Protocols in Bioinformatics, 54, 5.6.1–5.6.37.
Jacobson, M., & Sali, A. (2004). Comparative protein structure modeling and its applications to drug discovery. Annual Reports in Medicinal Chemistry, 39, 259–276.
Marti-Renom, M. A., Stuart, A. C., Fiser, A., Sanchez, R., Melo, F., & Sali, A. (2000). Comparative protein structure modeling of genes and genomes. Annuals Review Biophysics Bimol Structural, 29, 291–325.
Guex, N., Peitsch, M. C., & Schwede, T. (2009). Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis, 1, 162–173.
Jorgensen, W. L., Maxwell, D. S., & Tirado-Rives, J. (1996). Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. Journal of the American Chemical Society, 118, 11225–11236.
Jorgensen, W. L., & Tirado-Rives, J. (1996). The OPLS (optimized potentials for liquid simulations) potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. Journal of the American Chemical Society, 110, 1657–1666.
Laskowski, R. A., MacArthur, M. W., Moss, D. S., & Thornton, J. M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography, 26, 283–291.
Sheik, S. S., Sundararajan, P., Hussain, A. S., & Sekar, K. (2002). Ramachandran plot on the web. Bioinformatics, 18, 1548–1549.
Wiederstein, M., & Sippl, M. J. (2007). ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research, 35, 407–441.
Sippl, M. J. (1995). Knowledge-based potentials for proteins. Current Opinion in Structural Biology, 5, 229–235.
Luthy, R., Bowie, J. U., & Eisenberg, D. (1992). Assessment of protein models with three dimensional profiles. Nature, 356, 83–85.
Kalman, M., & Ben-Tal, N. (2010). Quality assessment of protein model-structures using evolutionary conservation. Bioinformatics, 26, 1299–1307.
Dundas, J., Ouyang, Z., Seng, T. J., Binkowski, A., Trupaz, Y., & Liang, J. (2006). CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Research, 34, 116–118.
Laurie, A. T., & Jackson, R. M. (2005). Q-site finder: an energy-based method for the prediction of protein-ligand sites. Bioinformatics, 21, 1908–1916.
Halgren, T. (2009). Identifying and characterizing binding sites and assessing druggability. Journal of Chemical Information and Modeling, 49, 377–389.
Binkowski, T. A., Naghibzadeh, S., & Liang, J. (2003). CASTp: computed atlas of surface topography of proteins. Nucleic Acids Research, 31, 3352–3355.
Liang, J., Edelsbrunner, H., & Woodward, C. (1998). Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Science, 7, 1884–1897.
Halgren, T. A. (2007). New method for fast and accurate binding site identification and analysis. Chemical Biology & Drug Design, 69, 146–148.
Schneidman-Duhovny, D., Inbar, Y., Nussinov, R., & Wolfson, H. J. (2005). PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Research, 33, 363–367.
Konc, J., Miller, B. T., Stular, T., Lesnik, S., Woodcock, H. L., Brooks, B. R., & Janezic, D. (2015). ProBiS-CHARMMing: web interface for prediction and optimization of ligands in protein binding sites. Journal of Chemical Information and Modeling, 55, 2308–2314.
Konc, J., & Janezic, D. (2010). ProBIS: a web server for detection of structurally similar protein binding sites. Nucleic Acids Research, 38, 436–440.
Kitchen, D. B., Decornez, H., Furr, J. R., & Bajorath, J. (2004). Docking and scoring in virtual screening for drug discovery: methods and applications. Nature Reviews. Drug Discovery, 3, 935–949.
Schnedier, G., & Bohm, H. J. (2002). Virtual screening and fast automated docking methods. Drug Discovery Today, 7, 64–70.
Chen, I. J., & Folopee, N. (2010). Drug-like bioactive structures and conformational coverage with the LigPrep/ConfGen suite: comparison to programs MOE and catalyst. Journal of Chemical Information and Modeling, 50, 822–839.
Friesner, R. A., Murphy, R. B., Repasky, M. P., Frye, L. L., Greenwood, J. R., Halgren, T. A., Sanschagrin, P. C., & Mainz, D. T. (2006). Extra precision glide: docking and scoring incorporating a model of hydrophobic encloser for protein-ligand complexes. Journal of Medicinal Chemistry, 49, 6177–6196.
Lill, M. A., & Danielson, M. L. (2011). Computer-aided drug design platform using PyMoL. Journal of Computer-Aided Molecular Design, 25, 13–19.
Connolly, M. (1983). Solvent accessible surfaces of proteins and nucleic acids. Science, 221, 709–713.
Marsh, J. A., & Teichmann, S. A. (2011). Relative solvent accessible surface area predicts protein conformational changes upon binding. Structure, 19, 859–867.
Pulaganti, M., Banaganapalli, B., Mulakayala, C., Chitta, S. K., & C. M. A. (2014). Molecular modeling and docking studies of o-succinylbenzoate synthase of M. Tuberculosis - - a potential target for antituberculosis drug design. Applied Biochemistry and Biotechnology, 172, 1407–1432.
Ramatenki, V., Dumpati, R., Vadija, R., Vellanki, S., Potlapally, S. R., Rondla, R. and Vuruputuri, U. (2016). Targeting the ubiquitin-conjugating enzyme E2D4 for cancer drug discovery – a structure- based approach. doi: 10.1007/s12154-016-0164-6. 1-17.
Morya, V. K., Dewaker, V., & Kim, E. K. (2012). In silico study and validation of phosphotransacetylase (PTA) as a putative drug target for Staphylococcus aureus by homology-based modelling and virtual screening. Applied Biochemistry and Biotechnology, 168, 1792–1805.
Vadija, R., Mustyala, K. K., Nambigari, N., Dulapalli, R., Dumpati, R. K., Ramatenki, V., Vellanki, S. P., & Vuruputuri, U. (2016). Homology modeling and virtual screening studies of FGF-7 protein- a structure-based approach to design new molecules against tumor angiogenesis. Journal of Chemical Biology, 9, 69–78.
Sasikala, D., Jeyakanthan, J., & Srinivasan, P. (2016). Structural insights on identification of potential lead compounds targeting WbpP in Vibrio Vulnificus through structure-based approaches. Journal of Receptor and Signal Transduction Research, 36, 515–530.
Mustyala, K. K., Malkhed, V., Potlapally, S. R., Chittireddy, V. R., & Vuruputuri, U. (2016). Identification of small molecular inhibitors for efflux protein: DrrA of Mycobacterium tuberculosis. Cellular and Molecular Bioengineering, 9, 190–202.
Dumpati, R., Dulapalli, R., Kondagari, B., Ramatenki, V., Vellanki, S., Vadija, R., & Vuruputuri, U. (2016). Suppressor of cytokine signalling-3 as a drug target for type 2 diabetes mellitus: a structure-guided approach. Chemistry Select, 1, 2502–2514.
Laskowski, R. A. (2001). PDBsum: summaries and analyses of PDB structures. Nucleic Acids Research, 29, 221–222.
Singh, T., Biswas, D., & Jayaram, B. (2011). AADS- an automated active site identification, docking, and scoring protocol for protein targets based on physicochemical descriptors. Journal of Chemical Information and Modeling, 51, 2515–2527.
Ramatenki, V., Potlapally, S. R., Dumpati, R. K., Vadija, R., & Vuruputuri, U. (2015). Homology modeling and virtual screening of ubiquitin conjugation enzyme E2A for designing a novel selective antagonist against cancer. Journal of Receptor and Signal Transduction Research, 35, 536–549.
Malkhed, V., Mustyala, K. K., Potlapally, S. R., & Vuruputuri, U. (2014). Identification of novel leads applying in silico studies for mycobacterium multidrug resistant (MMR) protein. Journal of Biomolecular Structure and Dynamics, 12, 1889–1906.
Kaur, N., Khokhar, M., Jain, V., Bharatam, P. V., Sandhir, R., & Tewari, R. (2013). Identification of druggable targets for acinetobacter baumannii va substrative genomics and plausible inhibitors for MurA and Murb. Applied Biochemistry and Biotechnology, 171, 417–436.
Sarita, R. P., Malkhed, V., & Uma, V. (2011). Identification of novel selective antagonists for cyclin C by homology modeling and virtual screening. International Journal of Biological Macromolecules, 48, 292–300.
Ioakimids, L., Thoukydidis, L., Mirza, A., Naeem, S., & Reynisson, J. (2008). Benchmarketing the reliability of QikProp. Correlation between experimental and predicted values. QSAR and Combinatorial Science, 27, 445–456.
Norinder, U., & Bergstrom, C. A. S. (2006). Prediction of ADMET properties. Chem. Med. Chem., 9, 920–937.
Chikan, N. A., Bhavaniprasad, V., Anbarasu, K., Shabir, N., & Patel, T. N. (2013). From natural products to drugs for epimutation computer-aided drug design. Applied Biochemistry and Biotechnology, 170, 164–175.
Lipinski, C. A. (2004). Lead- and drug-like compounds: the rule-of-five revolution. Drug Discovery Today: Technologies, 1, 337–341.
Congreve, M., Carr, R., Murray, C., & Jhoti, H. (2003). A ‘rule of three’ for fragment-based lead discovery? Drug Discovery Today, 8, 876–877.
Acknowledgements
The author VR acknowledges Council of Scientific and Industrial Research (CSIR)-INDIA for the financial support (File No: 09/132(0821)/2012-EMR-I). The author RV acknowledges Council of Scientific and Industrial Research (CSIR)-INDIA for SRF. The authors RKD and SPV acknowledge UGC for SRF under RFSMS scheme. The author RR acknowledges UGC for PDF. The authors also acknowledge Principal and Head, Department of Chemistry, University College Science, Osmania University, Hyderabad for providing facilities to carry out this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors state that no human studies and no animal studies were carried out for this article.
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Ramatenki, V., Dumpati, R., Vadija, R. et al. Identification of New Lead Molecules Against UBE2NL Enzyme for Cancer Therapy. Appl Biochem Biotechnol 182, 1497–1517 (2017). https://doi.org/10.1007/s12010-017-2414-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-017-2414-7