
Abstract
Alzheimer’s disease (AD) is an age-related progressive neurodegenerative disease commonly found among elderly. In addition to cognitive and behavioral deficits, vision abnormalities are prevalent in AD patients. Recent studies investigating retinal changes in AD double-transgenic mice have shown altered processing of amyloid precursor protein and accumulation of β-amyloid peptides in neurons of retinal ganglion cell layer (RGCL) and inner nuclear layer (INL). Apoptotic cells were also detected in the RGCL. Thus, the pathophysiological changes of retinas in AD patients are possibly resembled by AD transgenic models. The retina is a simple model of the brain in the sense that some pathological changes and therapeutic strategies from the retina may be observed or applicable to the brain. Furthermore, it is also possible to advance our understanding of pathological mechanisms in other retinal degenerative diseases. Therefore, studying AD-related retinal degeneration is a promising way for the investigation on (1) AD pathologies and therapies that would eventually benefit the brain and (2) cellular mechanisms in other retinal degenerations such as glaucoma and age-related macular degeneration. This review will highlight the efforts on retinal degenerative research using AD transgenic mouse models.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Increasing incidences of poor vision and ocular diseases have been described in Alzheimer’s disease (AD) patients (Bayer et al. 2002). AD-related visual dysfunction can be attributed to any defects occurring in the visual pathway, including damage in visual cortical areas and degeneration of pre-cortical areas (Guo et al. 2010). Glaucoma and age-related macular degeneration (AMD) are the frequently occurring ocular diseases associated with AD (Blanks et al. 1996). Recently, many laboratories have taken efforts to investigate the link of retinal degeneration and AD. Retinal abnormalities such as extensive loss of retinal ganglion cell (RGC) (Mendez et al. 1990; Blanks et al. 1996), reduced thickness of retinal nerve fiber layer (NFL) (Hedges et al. 1996), and reduced retinal blood flow (Guo et al. 2010) are observed in AD patients.
Owing to the limitations on the investigations of AD in human postmortem studies, transgenic AD mouse models have been used to mimic the human pathological environment. AD mice are engineered to express human mutant genes found in early-onset AD patients. The majority (30–40%) of familial Alzheimer’s disease (FAD) is resulting from autosomal dominant inheritance with mutated genes encoding presenilin 1 (PS1) on the long arm of chromosome 14, presenilin 2 (PS2) on chromosome 1, and amyloid precursor protein (APP) on chromosome 21 (Price et al. 1998b). Several lines of transgenic mice bearing the abovementioned gene mutations were shown to exhibit some neuropathological features of human AD (Eriksen and Janus 2007). Many researchers have been taking these transgenic mice as experimental models to study AD-related retinal degeneration.
This review focuses on the current knowledge obtained from AD transgenic mouse models. First, it begins with their genetic backgrounds. Then, it summarizes AD-related pathologies in their brains. Finally, the review goes through the retinal changes in these mice. It is our attempt to stimulate researches in AD-related retinal degeneration, which will have a significant impact in pathologies and therapeutics in both AD and retinal degeneration such as glaucoma and AMD.
Transgenic mouse models
To study the AD pathogenesis in the brain, AD transgenic mice have been widely investigated. Different types of transgenic mice may be complementary to the disadvantages of each other as experimental models of AD. For example, transgenic mice expressing mutant genes encoding APP and PS1 can be used to evaluate the process of amyloidogenesis at the pre-dementia phase of AD as the accumulation of Aβ deposits or even plaques in the brain tissues and the retinas are observed in these transgenic lines (Zahs and Ashe 2010). The pathology of another lesion, tau, can be elucidated by studying tau mutant mice in which hyperphosphorylated tau and NFT are expressed (Frank et al. 2008). Taking these research reports together, it is possible to use different types of transgenic mouse models, which would compensate the shortcomings of each other, to study AD in a multifactorial way.
Among various models, our review focuses on mutated APP mouse models, mutated APP/PS1 mouse models, mutated tau mouse models, and triple transgenic mouse (3xTg) model (listed in Table 1). The expression of AD mutations is based on three criteria: the host strains, the choice of promoter, and the extent of gene expression.
APP mutant-related transgenic mice
APPV717I transgenic mice, also named as “London” mutation, were the first FAD mutant mice dated back to 1993 (Price et al. 1998b). The mutant APP gene expressed under Thy-1 promoter harbors the substitution of Val residue at position 717 near the γ-secretase cleavage site (Price et al. 1998b). Cells with APP mutations of Val717 were accompanied by a higher secretion of Aβ42 compared to cells with wild-type APP (Price and Sisodia 1998; Pezzini et al. 2009).
Another widely used APP mutant model known as “Swedish mutation” was subsequently developed by Karen Hsiao Ashe in 1996 (Hsiao et al. 1996). It is based on the finding that the human APP695 double mutation form (Lys670→Asn and Met671→Leu) with the Kunitz-like proteinase inhibitor domain was absent in the early-onset AD Swedish family (Dutescu et al. 2009). The construct is inserted into a hamster prion protein (PrP) cosmid vector with the PrP open reading frame replaced by variant APP. The mice carrying the gene above are named as Tg (HuAPP695.K670N-M671L) 2576 (Hsiao et al. 1996). The onset of AD is varied among studies in Tg2576 mice, ranging from the earliest at 3 months of age to the latest at 15 months of age (Hsiao et al. 1996; King et al. 1999; Morgan et al. 2000; Kawarabayashi et al. 2001). Despite the varying onset of AD, APPswe mice are often associated with age-dependent oligomeric Aβ accumulation (Duyckaerts et al. 2008).
APPswe/APPV717I co-expressed transgenic mice are also made available to recapitulate the neuropathological alteration occurring in AD (Le Cudennec et al. 2008). These two lines of transgenic mice, driven by the Thy-1 promoter, show deposition of Aβ or even plaques and hyperphosphorylated tau, depending on the extent of transgene expression (Price et al. 1998a).
PS mutant-related transgenic mice
Mutation of APP accounts for the minority cases of early-onset FAD; the majority pedigrees of AD come from the mutation of PS1 or PS2 (Spires and Hyman 2005). To date, a number of mutant PS variants were developed such as ∆E9, M146L, A246E, L286V, L392V, and H163R from PS1 mutation and N141I from PS2 mutation (Price and Sisodia 1998). Therefore, mutation of PS1 or PS2 alone is not sufficient to mimic the pathologies of FAD. Although Aβ42 was over-produced in these single mutant mice (Duyckaerts et al. 2008), the hallmark lesions of AD including either Aβ plaques or neurofibrillary tangles (NFT) were not observed in brain tissues (Price and Sisodia 1998; Spires and Hyman 2005; Duyckaerts et al. 2008).
APP/PS1 double-transgenic mice
Crossing APP mutant genes with PS1 mutant genes seems to be a reasonable alternative way to elicit amyloid pathology. Increasing opportunities of amyloidogenesis from APP can accelerate the pathogenesis of AD (Spires and Hyman 2005). Indeed various transgenic mice expressing different kinds of mutated PS1 and mutated APP genes were generated in which different levels of Aβ expressions were observed.
One example of double-transgenic mice (2xTg) is APPswe/PS1∆E9, which carries both APP Swedish mutation and human PS1 gene with an in-frame deletion of exon 9 (Price and Sisodia 1998; Price et al. 1998a). The exon 9 deletion in PS1 could elevate the secretion levels of Aβ peptides; large and homogeneous senile plaques accumulated in the human brain (Duyckaerts et al. 2008). This type of mice generated Aβ plaques at the early age of 4–5 months of age (Duyckaerts et al. 2008).
Another 2xTg mouse model, APPSL/PS1M146L, carries both Swedish and London mutation (K670N/M1671L and V717I) in human APP 751 gene together with a human mutant gene PS1M146L (Casas et al. 2004; Bayer and Wirths 2008). Aβ plaques appear at 3 months of age and the intracellular accumulation of Aβ occurred as early as 2 months (Bayer and Wirths 2008).
The APP single Tg mice or APP/PS1 2xTg mouse models have replicated the amyloidogenic events in AD. This makes them valuable in the study of amyloid pathologies in the brain. For example, APP V717I transgenic mouse model has been used to study early behavioral and cognitive deficits accompanied by over-expression of APP (Moechars et al. 1999). Tg2576 mouse line has been implicated in the investigation of accelerate amyloidogenesis such as phosphorylated elf-2α (O’Connor et al. 2008). The downstream pathological events of Aβ deposits such as inflammation have been studied by using APPSL/PS1M146L mutant mice as an experimental model (Jimenez et al. 2008). All of these transgenic models have been employed for investigating enzymatic activities of the amyloidogenic pathway, the better understanding of which is conducive to the development of drugs such as β-secretase (BACE1) inhibitors that could attenuate amyloidogenesis (Gau et al. 2002; Malamas et al. 2010). Amyloid pathology can therefore be studied in great detail with AD amyloidogenic mice.
Tau mutant-related transgenic mice
Extant amyloidogenic AD models do not express the full spectrum of neuropathology in AD but elicit only plaques without NFT (Oddo et al. 2003). To supplement that, tau mutant mice have been used to study AD, frontotemporal lobe dementia, or other tauopathies. Tau mutation occurs on chromosome 17 (Frank et al. 2008). Tau mutant mice can express hyperphosphorylated tau and even NFT in the brain (Price et al. 1998a). One type of tau-mutated gene expresses Leu instead of Pro at the position of 301 (P301L) in both the shortest (driven by mouse prion promoter, JNPL3 line) and longest (driven by mouse Thy-1 promoter) four-repeat tau isoforms, producing NFT in brain tissues and spinal cord (Spires and Hyman 2005; Eriksen and Janus 2007). This mouse line has been studied to find out the correlation between frontotemporal lobe dementia and tauopathy as well as the mediation of cognitive impairment associated with axonopathy (Terwel et al. 2005; Frank et al. 2008). Another type of tau mutant (P301S) involves the substitution of Ser instead of Pro on exon 10 of chromosome 17 driven by Thy-1.2 promoter (Frank et al. 2008). Hyperphosphorylated tau was also detected extensively in the brain and spinal cord (Allen et al. 2002).
APPswe/PS1 M146V/TauP301L triple transgenic mice (3xTg)
The transgenic mice harbor mutation of human APPswe, PS1M146V, and TauP301L. Tg2576 mice with the use of Swedish double mutation of human APP695 and four-repeat tau (4R0N) without amino terminal inserts are inserted into exon 3 with the drive of Thy-1.2 expression cassette cDNA. These constructs are then co-microinjected into mutant homozygous PS1M146V knock-in mice (Oddo et al. 2003). The manipulation of co-integration of APP and tau transgene at the same site with the PS1M146V knock-in can establish a stable source of 3xTg mice. In addition, Thy1.2 expression cassette is used to ensure transgenes to express predominantly in the CNS (Caroni 1997). This type of transgenic mice is regarded as a good model recapitulating AD pathology. The onset of AD pathology occurs at 3 months of age (Spires and Hyman 2005). Impairment of long-term potentiation was observed. Plaques and tangles were progressively developed in an age- and region-dependent manner. The pathological changes also support the amyloid cascade hypothesis in which amyloid pathology precedes tauopathy shown in an anti-Aβ antibody immunization test (Oddo et al. 2003). In addition, studies found that Aβ, especially the intracellular Aβ42, was in correlation with intracellular NFT in the AD brain via tau hyperphosphorylation pathways (Grundke-Iqbal et al. 1989; Hardy and Selkoe 2002).
Retinal degeneration in AD transgenic models
Reports of visual deficits in AD patients dated back to 20 years ago. Behavioral studies on AD transgenic mice showed that the mice suffered from visual dysfunction (Arendash et al. 2004). Emerging evidence has been attempting to reinforce the potential link between retinal degeneration and AD (Dutescu et al. 2009). Hypothetically, the over-expression of APP, the production of soluble Aβ, and Aβ deposition will lead to the formation of amyloid plaques which can induce cell death via the apoptotic pathway (Wostyn et al. 2008). Furthermore, Aβ plays a role in inducing the hyperphosphorylation of tau, which in turn induces changes of the integrity of retinal cells and their synapses in the INL (Muyllaert et al. 2008). It has been reported that the over-expression of APP, Aβ and/or tau deposition, neuronal cell loss, retinal glial cell changes, and vascular changes occur in the retina of AD transgenic mice. The retinal histopathology changes in the AD transgenic mouse models are summarized in Table 2 and demonstrated in a schematic diagram in Fig. 1.

Diagram demonstrating APP expression, Aβ deposits in retinal layers of various kinds of AD transgenic mice. The background is a resin cross-section demonstrating a layered structure of retina. Filled color shapes are positioned quantitatively based on the expression levels of deposits. Unfilled color shapes show the presence of specific deposits only but not in quantitative manner. NFL nerve fiber layer, GCL ganglion cell layer, IPL inner plexiform layer, INL inner nuclear layer, OPL outer plexiform layer, ONL outer nuclear layer, OS outer segment, RPE retinal pigment epithelium
Over-expression of APP in the retina
Compared to the wild-type mice, a significant increase in the immunoreactivity of APP in the cytoplasm of the cells was detected in RGCL and INL of various transgenic mice (APP transgenic mouse (Tg 2576), APPswe/PS1M146L and APPswe/PS1∆E9 double-transgenic mouse) (Ning et al. 2008; Dutescu et al. 2009; Liu et al. 2009; Perez et al. 2009). This over-expression of APP was age dependent as shown in APPswe/PS1M146L transgenic mice. At 27 months old, a strong immunoreactivity of APP was detected not only in the different layers of retina such as, NFL, RGCL, IPL, INL, OPL, OS, and RPE but also in the retinal vasculature (Ning et al. 2008).
Deposition of Aβ in the retina and retinal vasculature
The deposition of Aß, derived from abnormal processing of APP, was found in the retinas of AD transgenic mice. In Tg2576 mice, Aβ was found to deposit from RGCL to INL or even at the outer nuclear layer (ONL) (Dutescu et al. 2009; Liu et al. 2009). In APPswe/PS1∆E9 transgenic mice, Aβ deposits usually accumulate in NFL, RGCL, and INL (Ning et al. 2008; Dutescu et al. 2009). Aβ plaques were found in plexiform layers; the size and the number of plaques significantly increased with age (Perez et al. 2009).
Robust Aβ signal was detected around microvessels in RGCL of Tg2576 mice (Dutescu et al. 2009; Liu et al. 2009). Both the retinal and choroidal vascular Aβ deposits were reported in old-aged APPswe/PS1M146L transgenic mice and middle-aged APPswe/PS1∆E9 (Ning et al. 2008).
Deposition of hyperphosphorylated tau in the retina
Hyperphosphorylated tau was detected and found to be associated with Aβ depositions in various retinal layers including RGCL, IPL, INL, OPL, and ONL in Tg2576 mice (Liu et al. 2009). In homozygous P301S transgenic mice, hyperphosphorylated tau deposited in the NFL and aggregated into filamentous inclusions in RGCs starting from 2 months old (Gasparini et al. 2009). The hyperphosphorylated tau and tau inclusions were found in cultured retinal explants from transgenic mice at the age of 5 months (Gasparini et al. 2009).
Loss of retinal neurons in AD transgenic mice
Neuronal cell loss was reported in multiple AD transgenic lines. Apoptotic cells as shown by TUNEL staining were detected in RGCL of APPswe/PS1M146L and APPswe/PS1∆E9 transgenic mice (Ning et al. 2008). A significant reduction in the retinal thickness measured from the RGCL to ONL was detected in Tg2576 mice compared with non-transgenic control (Liu et al. 2009). This indicates that there was a loss of either the photoreceptor cells at the ONL (rod and cone cells) or neuronal cells at the inner retinal layers (RGC, horizontal cells, bipolar cells, or amacrine cells).
Glial reaction in AD retina
Glial reactions in the retina were detected in different kinds of AD transgenic mice at various ages. In APPswe/PS1M146L transgenic mice, microglia was increased in an age-dependent manner, which was in parallel with Aß deposits and TUNEL-positive RGC in the GCL. The average percentage of cells in the GCL surrounded by microglial cells increased significantly from 10% in 7.8-month-old to 50% in 27-month-old APPswe/PS1M146L transgenic mice (Ning et al. 2008). In Tg2576 transgenic mice, a significant infiltration of microglial cells in the inner retina was detected as early as 4 months of age (Liu et al. 2009). Qualitative evaluation revealed greater microglia immunoreactivity in the 12- to 19-month-old APPswe/PS1∆E9 transgenic mice when compared to age-matched non-transgenic control (Perez et al. 2009). Increased microglia reaction demonstrated by an increase of GFAP immunoreactivity was also reported in the Tg2576 transgenic mice (Liu et al. 2009).
The retina as a window of the brain
Anatomy of the retina
Being a part of the central nervous system (CNS), the retina contains a high density of neuronal cells with a laminar structure outside the brain. There are six major types of neuronal cells distributed throughout the retina. Rods and cones are photoreceptors cells restricted to the outer segment of the retina. Horizontal cells, bipolar cells, and amacrine cells are located at the INL. The latter cell type may appear at the RGCL where RGCs are accumulated. During the visual process, light is detected by photoreceptor cells. The signals will then be relayed by the cells in the INL to RGCs. The nerve fibers of RGCs converge into the optic nerve fibers where signals are further transmitted to the visual cortex for visual processing. RGCs hold the responsibility of conducting the signal to the brain. The vulnerability of RGCs to stress factors such as inflammatory factors, high intracellular calcium ions, glutamate, or free radicals, or high intraocular pressures may lead to irreversible blindness.
Importance of studying the retina in AD
As human life span is extended into old age, the prevalence of AD often increases along with the aging process. It is estimated that over 100 million people will suffer from AD by 2050 (Ron et al. 2007). Visual impairment is associated with the prevalence of AD (Rizzo et al. 2000; Berisha et al. 2007; Valenti 2010). For example, in a glaucoma study, the percentage of AD patients having glaucoma is 2.5 times higher when compared to that of normal patients (Tamura et al. 2006). A rapidly increasing list of AD cases emphasizes an urgent need for a fair and promising research into the prevention of AD as well as the associated retinal diseases.
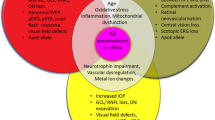
To our knowledge, it is still difficult to monitor the progressive degeneration of the brain with a fast, non-invasive, and less expensive method. The current diagnostic tools of the brain have limitations in terms of specificity of bio-markers and signal resolution (Klunk et al. 2004; Koronyo-Hamaoui et al. 2011). Thus, many attempts have been made to investigate AD with an easier approach by studying the retina. The laminar nature of retina can simplify the whole AD pathological investigation. Although there are ten layers in the retina, the retinal neurons are only located in three layers. Any abnormalities occurring in these three layers may have some pathological indications to the brain. For example, thinning of the inner retinal layer (from NFL, GCL to INL) indicates the loss of RGCs, horizontal cells, bipolar cells, or amacrine cells. A further examination of the specific neuronal cells in different layers may elucidate the pathological mechanisms induced by Aß efficiently. In addition, the retina can serve as an excellent alternative system for AD research because it can faithfully reflect the changes in the brain and offers unique advantages that can simplify the investigation of AD (Koronyo-Hamaoui et al. 2011). Multiple lines of AD transgenic mice have elicited AD-like pathological hallmarks in the retina as disease progresses. Studying retinal degeneration with the use of AD transgenic mice can be a simple gateway for investigators to look into the AD pathological changes in the more complicated brain system. Evaluating the retina thus opens up a new avenue of AD research (Fig. 2).

Schematic diagram suggests the importance of AD-related retinal degenerative research. The progressive degeneration of the brain may be associated with retinal diseases such as glaucoma and AMD. The pathological and therapeutic knowledge between the brain and the retina is transferrable. In addition, the investigation of glaucoma and AMD may be of significant therapeutic indication to AD brain. AMD age-related macular degeneration
The retina faithfully mirroring pathological events in the brain
The neuronal cells in the retina belong to the CNS. When anti-Aβ antibody was injected into a transgenic mouse model, Aβ plaques were decreased in both the retina and the brain (Ding et al. 2008). Another immunotherapy study demonstrated that APPswe/PS1∆E9 transgenic mice immunized with an altered myelin-derived peptide had a decrease of Aβ plaques in the retina and the brain to a similar extent (Koronyo-Hamaoui et al. 2011). Since the anatomical structure of the retina is far much simple than that of the brain, one can use the retina to investigate the degenerative processes, signaling mechanisms, and even neuroprotective agents. For example, the retina has been used to investigate the neuroprotective effects of autoimmune responses (Bakalash et al. 2002), which resemble the modulation of autoimmune responses to the brain so that beneficial effects can be observed. We have demonstrated that Stat3 pathway is involved in neurodegeneration after glaucoma (Ji et al. 2004), which has also been recently demonstrated to be an important signaling pathway in AD (Wan et al. 2010). Furthermore, synthesis of amyloid precursor protein and its intracellular transport have been investigated in the retina (Morin et al. 1993; Stamer et al. 2002). The retina provides a very simple model system for these authors to advance our understanding of APP synthesis and transport. All of these studies indicate that the retina can reliably represent amyloid plaque pathological events in the brain.
Simple and non-invasive imaging for the retina facilitates the monitoring of the progression of neurodegeneration
Recent advances of imaging technologies for the retina have given the opportunity for the development of a more definitive and non-invasive diagnostic tool for AD pathology (Hintersteiner et al. 2005; Nakada et al. 2008; Cordeiro et al. 2010). Koronyo-Hamaoui et al. (2011) identified amyloid plaques in the retinas from AD patients as well as those suspected as early stage cases. It was confirmed that the retina can faithfully reflect AD brain pathology. The transparent nature of the eyes allows direct tracking and visualization of the progressive changes in amyloid pathology (Cordeiro et al. 2010). The systemic injection of curcumin into live APP swe/PS1ΔE9 mice allows a high-resolution and specific non-invasive visualization of retinal Aβ plaques in situ (Koronyo-Hamaoui et al. 2011). Curcumin is a natural and safe fluorochrome that binds and labels Aβ plaques (Garcia-Alloza et al. 2007; Yang et al. 2005). In a 6-month randomized, placebo-controlled, double-blind, pilot clinical trial in AD patients, there were no significant side effects even when patients took curcumin at a dose of 4 g/day (Baum et al. 2008). Future development of high-resolution optical imaging for early AD diagnosis, prognosis assessment, and response to therapies can be achieved non-invasively through direct imaging of the retina. Progression of therapy is possible to be visualized qualitatively in the sense that one can monitor the changes of a particular neuronal cell (Hintersteiner et al. 2005; Nakada et al. 2008). Quantitative examinations of the disease stages have been performed by assessing the ratio of apoptosis to necrosis using the fluorescence counts of the respective dyes (Cordeiro et al. 2010). Even more, a high spatial resolution of images with a high signal-to-noise ratio ranging from 3:1 to 10:1 can be achieved with the imaging of the retina (Hintersteiner et al. 2005; Nakada et al. 2008). The merits of retinal imaging can provide investigators a solid support for assessing pathological status as well as developing and refining therapeutic strategies because the technique is less invasive and not sophisticated.
AD-related retinal degeneration advancing both the retinal degeneration and AD researches
Knowledge being transferable between the brain and the retina
Increasing lines of evidence highlight the commonalties shared between AD and retinal degeneration. In particular, Aβ deposits are found in the brain and the retina. As stated above, neuronal cell loss, inflammatory responses, and other pathological events that occurred in the retina are similar to those that occurred in the brain (Guo et al. 2010). However, the correlation between Aβ deposits and the retina is limited to the histopathological level. The pathological mechanisms have not been investigated in great detail. Lots of questions are waiting for answers by more researches to be conducted. What is the correlation between Aβ depositions and inflammatory responses in the retina? How do Aβ deposits trigger apoptotic pathways in RGCs? What are other mechanisms taken by Aβ and tau to cause retinal degeneration? Although the relationship for Aβ production and toxicity between the brain and the retina remains unclear, the pathological pathways are possibly shared between the two to a certain extent. For example, mitochondrial dysfunctions and activation of complement, which will be discussed below, are pathophysiological changes in both AD and retinal degeneration (Kong et al. 2009; Valenti 2010). The similarity between the brain and the retina allows the exchange of knowledge in terms of pathological mechanisms and therapeutic intervention.
The discovery of the significant involvement of double-stranded RNA-dependent protein kinase (PKR) in the apoptosis of neurons in postmortem AD brain and in experimental studies is a good example (Chang et al. 2002a, b; Suen et al. 2003). Having reported the involvement of PKR in neuronal apoptosis, it has been further found that PKR also plays important roles in the neuronal apoptosis of RGCs in endoplasmic reticulum (ER) stress-induced retinal neuronal loss (Shimazawa et al. 2007). We have recently demonstrated that the neuroprotective agents found from AD research can also be applied to eye research. Our studies on wolfberry, Lycium barbarum, an anti-aging herb, can be a good example of sharing that knowledge obtained from studying the brain can be applied into the retina (Chan et al. 2007). In AD, wolfberry can alleviate the degenerative process by promoting survival signals, suppressing ER stress, and reducing glutamate excitotoxicity (Yu et al. 2006; Chan et al. 2007; Yu et al. 2007; Ho et al. 2009; Ho et al. 2010a, 2010b). In glaucoma, wolfberry shows its beneficial effects on the retina based on suppressing the neurodestructive factors, modulating the inflammatory responses (Chiu et al. 2009), and induction of protective chaperone (Chiu et al. 2010). The neuroprotective effects of wolfberry shared between AD and glaucoma further strengthen our hypothesis that knowledge obtained from the brain and the retina are transferrable.
AD-related retinal degeneration can help us in understanding retinal degeneration in other eye diseases
Based on the findings above that Aβ deposits were observed in the retinal layers of AD transgenic mice, studying AD-related retinal degeneration in the AD models may provide some implications on how Aβ deposits are linked to retinal degeneration. Two prominent examples of retinal degeneration associated with AD are glaucoma and AMD (Blanks et al. 1996). In glaucoma, the presence of Aβ was observed in RGCs. In AMD, Aβ assemblies were accumulated in the form of drusen which are entrapped between the retinal epithelial layer and the Bruch’s membrane (McKinnon 2003; Anderson et al. 2004). Therefore, it is reasonable to speculate that the accumulation of Aβ peptides (but not plaques) may contribute to retinal degeneration in these diseases.
Aβ-mediated mitochondrial dysfunction and glaucoma
Glaucoma is characterized by the progressive degeneration of RGCs induced by increased intraocular pressure (Yin et al. 2008). However, signs of glaucoma are also elicited even after the reduction of IOP to normal level (Valenti 2010). This clearly indicates that loss of RGCs can be neurodegenerative processes and various pathological events synergistically contribute to the pathogenesis of glaucoma. One of the triggers being proposed is Aβ-induced glaucoma. Indeed recent reports have demonstrated a potential link between glaucoma and AD. Patients who suffer from AD also show changes of the retina typically found in glaucoma (Guo et al. 2010). At the molecular level, intracellular accumulation of Aβ was observed in RGCs. Accumulation of intracellular Aβ contributes to axonopathy and initiates apoptotic pathway in the brain (Ohyagi et al. 2005; Suo et al. 2007). The deleterious effects of intracellular Aβ in the brain may also be applied to the eye because intracellular Aβ exerts its toxicity to RGCs, reinforcing the degenerative process of the retina in addition to high IOP.
One of the pathological events in AD brain involves abnormal mitochondrial activities (Seo et al. 2010). Mitochondria are dynamic organelles in which the balance between fission and fusion processes is important for maintaining their healthy functions (Wang et al. 2009). Impairment of fission and fusion has been considered to be one of the pathophysiological indexes in AD (Santos et al. 2010). There are some suggestions that Aβ peptides will modulate the cytosolic Ca2+ level in mitochondria that may alter the mitochondrial morphology and physiology (Hung et al. 2010). For example, elevated cytosolic Ca2+ level may enhance the fragmentation of mitochondria and hence lead to the perturbation of fission and fusion balance which may eventually cause mitochondrial dysfunction (Saotome et al. 2008). Dysregulation of Ca2+ homeostasis may also disrupt the downstream pathways of Ca2+-dependent regulators monitoring mitochondrial dynamics (Liu and Hajnoczky 2009; Hung et al. 2010). In particular, an abnormal distribution of mitochondrial fission and fusion proteins such as OPA1 and Fis1 may occur, and thus the distribution of mitochondria may be negatively affected in neurons (Wang et al. 2009). Consequently, synaptic dysfunction may result due to the failure of meeting the energy demand in neurons, particularly in axonal and dendritic tips (Allen et al. 2002; Liu and Hajnoczky 2009; Wang et al. 2009).
The eyes are energy-demanding organs where a lot of mitochondria accumulate, particularly at the optic nerve heads, to support the visual function of the eyes (Carelli et al. 2004). Applying the same pathological mechanism to the eyes, Aβ present in RGCs may initiate similar pathways for mitochondrial dysfunctions, eventually leading to retinal degeneration. This hypothesis may be extended to one of the causes in Aβ-induced glaucoma. Intriguingly, in a glaucomatous model where cultured RGCs were subjected to elevated hydrostatic pressure, mitochondrial fission was found to be enhanced, together with morphological changes and bioenergetic dysfunction (Ju et al. 2007). The approach of studying AD-related retinal degeneration is thus valuable for evaluating the pathogenesis of glaucoma.
Aβ-mediated inflammation and AMD
AMD is characterized by the progressive degeneration of retinal pigment epithelium and photoreceptor cells (Margrain et al. 2004). Drusen as the hallmark of AMD has been reported to contain Aβ assemblies among various amounts of heterogeneous components (Luibl et al. 2006). Hypothesis has been made for the linkage between AMD and AD because the similarity between two diseases is largely based on the complement activation induced by Aβ peptides. However, there is conflicting evidence on Aβ deposits in these two diseases at the ultrastructural level (Anderson et al. 2004; Rodrigues 2007). Inflammatory factors triggered by Aβ assembly initiates exudative AMD in which vascular endothelial growth factor (VEGF), being one of the main components of blood vessel growth, may be abnormally secreted (Yoshida et al. 2005). The potential role of Aβ peptides in modulating angiogenesis may explain why AMD is related to AD because of the chronic inflammatory process. However, it should be noted that the key component of drusen contributing to AMD remains to be elucidated.
Recently, efforts have been put into the quest on how Aβ deposits take part in inflammation which is a key causative link to exudative AMD. The presence of Aβ aggregates was specific to AMD eyes (Anderson et al. 2002) and a number of evidence showed the histopathological correlation between inflammation and Aβ aggregates. Aβ assemblies, which were detected in the degenerate RPE cells, may involve the formation of amyloid vesicle-containing drusen flanking RPE cells at the sub-RPE space (Johnson et al. 2002). However, our understanding of how Aβ assemblies exactly activate complement cascade is still developing.
A hypothesis that Aβ contributes to inflammatory events along with the formation of drusen has been proposed (Anderson et al. 2004). RPE cells are vulnerable to cell debris and other accumulated substances such as lipofuscin (Anderson et al. 2004). The gradual built-up of cell debris may initiate the formation of drusen which encapsulates proteins, lipids, as well as inflammatory components (Anderson et al. 2002). Thus, there is a possibility that the amyloid vesicles found in drusen may be made up of the extracellular Aβ deposits derived from the injured RPE cells. Similar to what can be found in AD where Aβ aggregates involve the activation of complement, Aβ deposition in the extracellular RPE matrix was suggested to be a primary activator of complement cascade in which the components responsible for the alternative activation pathway of the complement system were detected in AMD (Johnson et al. 2002; Gold et al. 2006). Cleavage of C3 component into C3a and C3b and subsequently cleavage of C5 component to trigger the formation of membrane attack complex (Anderson et al. 2002) can be found in both AD and AMD.
Choroidal neovascularization is a pathological feature of exudative AMD in which VEGF-mediated angiopathy is a key pathophysiological process (Patel and Chan 2008). Aβ may be associated with abnormal VEGF production triggered by impaired cells and a series of inflammatory events. For example, Roque and Caldwell (1990) have initially proposed that VEGF was produced by the impaired Müller cells, which was later supported by Pierce et al. (1995). Recently, Yoshida et al. (2005) suggested that Aβ assembly in drusen enhanced the release of VEGF and pigment epithelium-derived factor from RPE cells and promoted angiogenesis. Accumulation of cell debris in RPE was suggested to be a pro-inflammatory stimulation activating choroidal dendritic cells which subsequently recruit macrophages to enhance the production of VEGF (Zarbin 2004). Other reports have proposed that VEGF was up-regulated by C3a and C5a (Nozaki et al. 2006). Taking all these findings into consideration, Aβ peptides can activate the complement system in which C3a and C5a are potent stimuli of angiogenesis in AMD. Studying AD-related retinal degeneration is a valuable tool in evaluating complement activation and subsequent angiopathy in AMD.
AD-related retinal degeneration shows a therapeutic significance of early AD
Early signs of AD symptoms in the brain can hardly be detected. In the most recent study of the APPswe/PS1ΔE9 mice, 5 days of systemic administration of curcumin showed that there was a qualitative age-dependent correlation between plaque deposition in the retina and the brain and increased accumulation over the course of disease progression (Koronyo-Hamaoui et al. 2011). For the very first time, they proved that Aβ plaques in the retina precede brain plaques as early as 2.5 months of age in AD-Tg mice model. The first detectable Aβ plaques in the brain were at the age of 5 months which is consistent with previous studies in this line of AD-Tg mice (Garcia-Alloza et al. 2006). Therefore, retinal degeneration is the most important site to study in early AD pathology. In APPswe/PS1ΔE9 mice, following MOG45D-loaded dendritic cells immunization, Aβ-plaque burden in the retinas was reduced as effectively as that in the brain (Koronyo-Hamaoui et al. 2011). Considering the potentiation of direct optical imaging of the retina, especially the Aβ plaques deposition in the retina labeled by curcumin, retinal degeneration in early AD is the window of monitoring disease progression as well as effectiveness of treatment.
In the study where extracellular Aβ was injected into a glaucomatous model, immunotherapy with a potential agent such as β-secretase inhibitor, Congo Red, or Aβ antibody successfully reduced Aβ-induced RGC apoptosis by suppressing further Aβ aggregation and inhibiting the enzymatic activity of amyloidogenesis (Guo et al. 2007). The effectiveness of each medication was also assessed by quantifying RGC apoptosis under the same dosage of each drug (Guo et al. 2007). This study is a good example illustrating that the retina can be a promising platform to investigate the efficacy of any potential drugs on different neuronal cells. In addition to traditional delivery system, potential neuroprotective agents can be applied in the posterior chamber by intravitreous injection (Chiu et al. 2005; Chiu et al. 2007). The bioavailability of injected compound in the vitreous can be extended (Mey and Thanos 1993). An examination of pharmacokinetic and pharmacodynamic is much easier than that in the brain. Changes in a particular layer may imply some effects of certain therapeutic agents on certain types of neuronal cells. For example, changes in INL may be an indication of some abnormalities that occurred in horizontal cells, amacrine cells, or bipolar cells. Taken together, the potent translation of treatment responses from the retina to the brain proves that studying AD-related retinal degeneration is of great therapeutic value in the early onset of AD.
Ocular disease models are beneficial to the investigation of AD pathogenesis
On one hand, studying AD-related retinal degeneration can offer chances to elucidate the pathological processes induced by Aβ in the retina of glaucoma or AMD patient. On the other hand, each of the ocular disease models mimic glaucoma and AMD can help us to understand the pathogenesis of AD.
In glaucoma, previous studies have shown that intracellular Aβ is found in the glaucomatous models (McKinnon et al. 2002). This may provide an excellent experimental model because both extra- and intracellular Aβ peptides contribute to the pathogenesis of AD. The accumulation of intracellular Aβ peptides has been shown to be neurotoxic in the brain (LaFerla et al. 2007; Li et al. 2007). This indicates that a glaucomatous model can provide hints on the pathological mechanisms of intracellular Aβ peptides.
In terms of AMD, Aβ assembly in drusen is non-fibrillar peptides or oligomers instead of fibrillar plaques observed in AD (Luibl et al. 2006). Recent studies have demonstrated that Aβ oligomers are neurotoxic because they disrupt dendritic spines and depress synaptic signaling which lead to cognitive deficits (Allen et al. 2002; Walsh et al. 2002a, b; Wang et al. 2002; Nimmrich et al. 2008). Studying AMD models can possibly provide suggestions on how Aβ oligomers lead to AD pathogenesis.
Conclusion
Brain research in AD has been widely conducted, while the retinal investigation in AD is a new arena to be explored. The interchangeable knowledge between the brain and the retina allows the concepts of Aβ pathology in the AD retina to be borrowed from those in the AD brain. Research in the retina offers many advantages over the brain in terms of cost, time, and analytical methods. Research in the AD retina resembles the pathological changes in AD brain. Therefore, studying AD-related retinal degeneration will be beneficial in assessing AD pathological changes and developing new AD therapeutic treatments in a simple and effective manner. Complementarily by AD transgenic models, the investigation of AD-related retinal degeneration may introduce a new channel to study AD in a different perspective (Fig. 2).
References
Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M (2002) Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci 22:9340–9351
Anderson DH, Mullins RF, Hageman GS, Johnson LV (2002) A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol 134:411–431
Anderson DH, Talaga KC, Rivest AJ, Barron E, Hageman GS, Johnson LV (2004) Characterization of beta amyloid assemblies in drusen: the deposits associated with aging and age-related macular degeneration. Exp Eye Res 78:243–256
Arendash GW, Lewis J, Leighty RE, McGowan E, Cracchiolo JR, Hutton M, Garcia MF (2004) Multi-metric behavioral comparison of APPsw and P301L models for Alzheimer’s disease: linkage of poorer cognitive performance to tau pathology in forebrain. Brain Res 1012:29–41
Bakalash S, Kipnis J, Yoles E, Schwartz M (2002) Resistance of retinal ganglion cells to an increase in intraocular pressure is immune-dependent. Invest Ophthalmol Vis Sci 43:2648–2653
Baum L, Lam CW, Cheung SK, Kwok T, Lui V, Tsoh J, Lam L, Leung V, Hui E, Ng C, Woo J, Chiu HF, Goggins WB, Zee BC, Cheng KF, Fong CY, Wong A, Mok H, Chow MS, Ho PC, Ip SP, Ho CS, Yu XW, Lai CY, Chan MH, Szeto S, Chan IH, Mok V (2008) Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J Clin Psychopharmacol 28:110–113
Bayer TA, Wirths O (2008) Review on the APP/PS1KI mouse model: intraneuronal Abeta accumulation triggers axonopathy, neuron loss and working memory impairment. Genes Brain Behav 7(Suppl 1):6–11
Bayer AU, Ferrari F, Erb C (2002) High occurrence rate of glaucoma among patients with Alzheimer’s disease. Eur Neurol 47:165–168
Berisha F, Feke GT, Trempe CL, McMeel JW, Schepens CL (2007) Retinal abnormalities in early Alzheimer’s disease. Invest Ophthalmol Vis Sci 48:2285–2289
Blanks JC, Torigoe Y, Hinton DR, Blanks RH (1996) Retinal pathology in Alzheimer’s disease. I. Ganglion cell loss in foveal/parafoveal retina. Neurobiol Aging 17:377–384
Carelli V, Ross-Cisneros FN, Sadun AA (2004) Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res 23:53–89
Caroni P (1997) Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods 71:3–9
Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L (2004) Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol 165:1289–1300
Chan HC, Chang RCC, Koon-Ching Ip A, Chiu K, Yuen WH, Zee SY, So KF (2007) Neuroprotective effects of Lycium barbarum Lynn on protecting retinal ganglion cells in an ocular hypertension model of glaucoma. Exp Neurol 203:269–273
Chang RCC, Suen KC, Ma CH, Elyaman W, Ng HK, Hugon J (2002a) Involvement of double-stranded RNA-dependent protein kinase and phosphorylation of eukaryotic initiation factor-2 alpha in neuronal degeneration. J Neurochem 83:1215–1225
Chang RCC, Wong AKY, Ng HK, Hugon J (2002b) Phosphorylation of eukaryotic initiation factor-2 alpha (eIF2 alpha) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 13:2429–2432
Chiu K, Lam TT, Ying Li WW, Caprioli J, Kwong JM (2005) Calpain and N-methyl-d-aspartate (NMDA)-induced excitotoxicity in rat retinas. Brain Res 1046:207–215
Chiu K, Chang RCC, So KF (2007) Intravitreous injection for establishing ocular diseases model. J Vis Exp 8:313
Chiu K, Chan HC, Yeung SC, Yuen WH, Zee SY, Chang RCC, So KF (2009) Modulation of microglia by wolfberry on the survival of retinal ganglion cells in a rat ocular hypertension model. J Ocul Biol Dis Infor 2:127–136
Chiu K, Zhou Y, Yeung SC, Lok CK, Chan OO, Chang RCC, So KF, Chiu JF (2010) Up-regulation of crystallins is involved in the neuroprotective effect of wolfberry on survival of retinal ganglion cells in rat ocular hypertension model. J Cell Biochem 110:311–320
Cordeiro MF, Guo L, Coxon KM, Duggan J, Nizari S, Normando EM, Sensi SL, Sillito AM, Fitzke FW, Salt TE, Moss SE (2010) Imaging multiple phases of neurodegeneration: a novel approach to assessing cell death in vivo. Cell Death Dis 1:e3
Ding JD, Lin J, Mace BE, Herrmann R, Sullivan P, Bowes Rickman C (2008) Targeting age-related macular degeneration with Alzheimer’s disease based immunotherapies: anti-amyloid-beta antibody attenuates pathologies in an age-related macular degeneration mouse model. Vision Res 48:339–345
Dutescu RM, Li QX, Crowston J, Masters CL, Baird PN, Culvenor JG (2009) Amyloid precursor protein processing and retinal pathology in mouse models of Alzheimer’s disease. Graefes Arch Clin Exp Ophthalmol 247:1213–1221
Duyckaerts C, Potier MC, Delatour B (2008) Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol 115:5–38
Eriksen JL, Janus CG (2007) Plaques, tangles, and memory loss in mouse models of neurodegeneration. Behav Genet 37:79–100
Frank S, Clavaguera F, Tolnay M (2008) Tauopathy models and human neuropathology: similarities and differences. Acta Neuropathol 115:39–53
Garcia MF, Gordon MN, Hutton M, Lewis J, McGowan E, Dickey CA, Morgan D, Arendash GW (2004) The retinal degeneration (rd) gene seriously impairs spatial cognitive performance in normal and Alzheimer’s transgenic mice. Neuroreport 15:73–77
Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, Purcell SM, Betensky RA, Raju S, Prada C, Greenberg SM, Bacskai BJ, Frosch MP (2006) Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis 24:516–524
Garcia-Alloza M, Borrelli LA, Rozkalne A, Hyman BT, Bacskai BJ (2007) Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J Neurochem 102:1095–1104
Gasparini L, Anthony Crowther R, Martin KR, Berg N, Coleman M, Goedert M, Spillantini MG (2009) Tau inclusions in retinal ganglion cells of human P301S tau transgenic mice: effects on axonal viability. Neurobiol Aging 32:419–433
Gau JT, Steinhilb ML, Kao TC, D’Amato CJ, Gaut JR, Frey KA, Turner RS (2002) Stable beta-secretase activity and presynaptic cholinergic markers during progressive central nervous system amyloidogenesis in Tg2576 mice. Am J Pathol 160:731–738
Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, Cramer K, Neel J, Bergeron J, Barile GR, Smith RT, Hageman GS, Dean M, Allikmets R, AMD Genetics Clinical Study Group (2006) Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet 38:458–462
Gruart A, Lopez-Ramos JC, Munoz MD, Delgado-Garcia JM (2008) Aged wild-type and APP, PS1, and APP + PS1 mice present similar deficits in associative learning and synaptic plasticity independent of amyloid load. Neurobiol Dis 30:439–450
Grundke-Iqbal I, Iqbal K, George L, Tung YC, Kim KS, Wisniewski HM (1989) Amyloid protein and neurofibrillary tangles coexist in the same neuron in Alzheimer disease. Proc Natl Acad Sci USA 86:2853–2857
Guo L, Salt TE et al (2007) Targeting amyloid-beta in glaucoma treatment. Proc Natl Acad Sci USA 104:13444–13449
Guo L, Duggan J, Cordeiro MF (2010) Alzheimer’s disease and retinal neurodegeneration. Curr Alzheimer Res 7:3–14
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356
Hedges TR 3rd, Perez Galves R, Speigelman D, Barbas NR, Peli E, Yardley CJ (1996) Retinal nerve fiber layer abnormalities in Alzheimer’s disease. Acta Ophthalmol Scand 74:271–275
Hintersteiner M, Enz A, Frey P, Jaton AL, Kinzy W, Kneuer R, Neumann U, Rudin M, Staufenbiel M, Stoeckli M, Wiederhold KH, Gremlich HU (2005) In vivo detection of amyloid-beta deposits by near-infrared imaging using an oxazine-derivative probe. Nat Biotechnol 23:577–583
Ho YS, Yu MS, Yik SY, So KF, Yuen WH, Chang RCC (2009) Polysaccharides from wolfberry antagonizes glutamate excitotoxicity in rat cortical neurons. Cell Mol Neurobiol 29:1233–1244
Ho YS, Yu MS, Yang XF, So KF, Yuen WH, Chang RCC (2010a) Neuroprotective effects of polysaccharides from wolfberry, the fruits of Lycium barbarum, against homocysteine-induced toxicity in rat cortical neurons. J Alzheimers Dis 19:813–827
Ho YS, So KF, Chang RCC (2010b) Anti-aging herbal medicine—how and why can they be used in aging-associated neurodegenerative diseases? Ageing Res Rev 9:354–362
Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274:99–102
Hung CH, Ho YS, Chang RCC (2010) Modulation of mitochondrial calcium as a pharmacological target for Alzheimer’s disease. Ageing Res Rev 9:447–456
Ji JZ, Elyaman W, Yip HK, Lee VW, Yick LW, Hugon J, So KF (2004) CNTF promotes survival of retinal ganglion cells after induction of ocular hypertension in rats: the possible involvement of STAT3 pathway. Eur J Neurosci 19:265–272
Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, Ruano D, Vizuete M, Gutierrez A, Vitorica J (2008) Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci 28:11650–11661
Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH (2002) The Alzheimer’s A beta-peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci USA 99:11830–11835
Ju WK, Liu Q, Kim KY, Crowston JG, Lindsey JD, Agarwal N, Ellisman MH, Perkins GA, Weinreb RN (2007) Elevated hydrostatic pressure triggers mitochondrial fission and decreases cellular ATP in differentiated RGC-5 cells. Invest Ophthalmol Vis Sci 48:2145–2151
Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG (2001) Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci 21:372–381
King DL, Arendash GW, Crawford F, Sterk T, Menendez J, Mullan MJ (1999) Progressive and gender-dependent cognitive impairment in the APP(SW) transgenic mouse model for Alzheimer’s disease. Behav Brain Res 103:145–162
Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B (2004) Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann Neurol 55:306–319
Kong GY, Van Bergen NJ, Trounce IA, Crowston JG (2009) Mitochondrial dysfunction and glaucoma. J Glaucoma 18:93–100
Koronyo-Hamaoui M, Koronyo Y, Ljubimov AV, Miller CA, Ko MK, Black KL, Schwartz M, Farkas DL (2011) Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 54(Suppl 1):S204–S217
LaFerla FM, Green KN, Oddo S (2007) Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci 8:499–509
Le Cudennec C, Faure A, Ly M, Delatour B (2008) One-year longitudinal evaluation of sensorimotor functions in APP751SL transgenic mice. Genes Brain Behav 7(Suppl 1):83–91
Li M, Chen L, Lee DH, Yu LC, Zhang Y (2007) The role of intracellular amyloid beta in Alzheimer’s disease. Prog Neurobiol 83:131–139
Liu X, Hajnoczky G (2009) Ca2+−dependent regulation of mitochondrial dynamics by the Miro–Milton complex. Int J Biochem Cell Biol 41:1972–1976
Liu B, Rasool S, Yang Z, Glabe CG, Schreiber SS, Ge J, Tan Z (2009) Amyloid-peptide vaccinations reduce {beta}-amyloid plaques but exacerbate vascular deposition and inflammation in the retina of Alzheimer’s transgenic mice. Am J Pathol 175:2099–2110
Luibl V, Isas JM, Kayed R, Glabe CG, Langen R, Chen J (2006) Drusen deposits associated with aging and age-related macular degeneration contain nonfibrillar amyloid oligomers. J Clin Invest 116:378–385
Malamas MS, Robichaud A, Erdei J, Quagliato D, Solvibile W, Zhou P, Morris K, Turner J, Wagner E, Fan K, Olland A, Jacobsen S, Reinhart P, Riddell D, Pangalos M (2010) Design and synthesis of aminohydantoins as potent and selective human beta-secretase (BACE1) inhibitors with enhanced brain permeability. Bioorg Med Chem Lett 20:6597–6605
Margrain TH, Boulton M, Marshall J, Sliney DH (2004) Do blue light filters confer protection against age-related macular degeneration? Prog Retin Eye Res 23:523–531
McKinnon SJ (2003) Glaucoma: ocular Alzheimer’s disease? Front Biosci 8:s1140–s1156
McKinnon SJ, Lehman DM, Kerrigan-Baumrind LA, Merges CA, Pease ME, Kerrigan DF, Ransom NL, Tahzib NG, Reitsamer HA, Levkovitch-Verbin H, Quigley HA, Zack DJ (2002) Caspase activation and amyloid precursor protein cleavage in rat ocular hypertension. Invest Ophthalmol Vis Sci 43:1077–1087
Mendez MF, Mendez MA, Martin R, Smyth KA, Whitehouse PJ (1990) Complex visual disturbances in Alzheimer’s disease. Neurology 40:439–443
Mey J, Thanos S (1993) Intravitreal injections of neurotrophic factors support the survival of axotomized retinal ganglion cells in adult rats in vivo. Brain Res 602:304–317
Moechars D, Dewachter I, Lorent K, Reversé D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F (1999) Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem 274:6483–6492
Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW (2000) A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 408:982–985
Morin PJ, Abraham CR, Amaratunga A, Johnson RJ, Huber G, Sandell JH, Fine RE (1993) Amyloid precursor protein is synthesized by retinal ganglion cells, rapidly transported to the optic nerve plasma membrane and nerve terminals, and metabolized. J Neurochem 61:464–473
Muyllaert D, Kremer A, Jaworski T, Borghgraef P, Devijver H, Croes S, Dewachter I, Van Leuven F (2008) Glycogen synthase kinase-3beta, or a link between amyloid and tau pathology? Genes Brain Behav 7(Suppl 1):57–66
Nakada T, Matsuzawa H, Igarashi H, Fujii Y, Kwee IL (2008) In vivo visualization of senile-plaque-like pathology in Alzheimer’s disease patients by MR microscopy on a 7T system. J Neuroimaging 18:125–129
Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, Hillen H, Gross G, Ebert U, Bruehl C (2008) Amyloid beta oligomers (A beta(1–42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci 28:788–797
Ning A, Cui J, To E, Ashe KH, Matsubara J (2008) Amyloid-beta deposits lead to retinal degeneration in a mouse model of Alzheimer disease. Invest Ophthalmol Vis Sci 49:5136–5143
Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, Chen Y, Zhang K, Ambati BK, Baffi JZ, Ambati J (2006) Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci USA 103:2328–2333
O’Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hébert SS, De Strooper B, Haass C, Bennett DA, Vassar R (2008) Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 60:988–1009
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39:409–421
Ohyagi Y, Asahara H, Chui DH, Tsuruta Y, Sakae N, Miyoshi K, Yamada T, Kikuchi H, Taniwaki T, Murai H, Ikezoe K, Furuya H, Kawarabayashi T, Shoji M, Checler F, Iwaki T, Makifuchi T, Takeda K, Kira J, Tabira T (2005) Intracellular Aß42 activates p53 promoter: a pathway to neurodegeneration in Alzheimer’s disease. FASEB J 19:255–257
Patel M, Chan CC (2008) Immunopathological aspects of age-related macular degeneration. Semin Immunopathol 30:97–110
Perez SE, Lumayag S, Kovacs B, Mufson EJ, Xu S (2009) Beta-amyloid deposition and functional impairment in the retina of the APPswe/PS1DeltaE9 transgenic mouse model of Alzheimer’s disease. Invest Ophthalmol Vis Sci 50:793–800
Pezzini A, Del Zotto E, Volonghi I, Giossi A, Costa P, Padovani A (2009) Cerebral amyloid angiopathy: a common cause of cerebral hemorrhage. Curr Med Chem 16:2498–2513
Pierce EA, Avery RL, Foley ED, Aiello LP, Smith LE (1995) Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci USA 92:905–909
Price DL, Sisodia SS (1998) Mutant genes in familial Alzheimer’s disease and transgenic models. Annu Rev Neurosci 21:479–505
Price DL, Sisodia SS, Borchelt DR (1998a) Genetic neurodegenerative diseases: the human illness and transgenic models. Science 282:1079–1083
Price DL, Tanzi RE, Borchelt DR, Sisodia SS (1998b) Alzheimer’s disease: genetic studies and transgenic models. Annu Rev Genet 32:461–493
Rizzo M, Anderson SW, Dawson J, Nawrot M (2000) Vision and cognition in Alzheimer’s disease. Neuropsychologia 38:1157–1169
Rodrigues EB (2007) Inflammation in dry age-related macular degeneration. Ophthalmologica 221:143–152
Ron B, Elizabeth J, Kathryn Z-G, Arrighi HM (2007) Forecasting the global burden of Alzheimer’s disease. Alz Dement 3:186–191
Roque RS, Caldwell RB (1990) Müller cell changes precede vascularization of the pigment epithelium in the dystrophic rat retina. Glia 3:464–475
Santos RX, Correia SC, Wang X, Perry G, Smith MA, Moreira PI, Zhu X (2010) A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. J Alz Dis 20(Suppl 2):S401–S412
Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnoczky G (2008) Bidirectional Ca2+−dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci USA 105:20728–20733
Scattoni ML, Gasparini L, Alleva E, Goedert M, Calamandrei G, Spillantini MG (2010) Early behavioural markers of disease in P301S tau transgenic mice. Behav Brain Res 208:250–257
Seo AY, Joseph A-M, Dutta D, Hwang JCY, Aris JP, Leeuwenburgh C (2010) New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci 123:2533–2542
Shimazawa M, Ito Y, Inokuchi Y, Hara H (2007) Involvement of Double-Stranded RNA-Dependent Protein Kinase in ER Stress-Induced Retinal Neuron Damage. Invest Ophthalmol Vis Sci 48:3729–3736
Spires TL, Hyman BT (2005) Transgenic models of Alzheimer’s disease: learning from animals. NeuroRx 2:423–437
Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM (2002) Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol 156:1051–1063
Suen KC, Yu MS, So KF, Chang RCC, Hugon J (2003) Upstream signaling pathways leading to the activation of double-stranded RNA-dependent serine/threonine protein kinase in {beta}-amyloid peptide neurotoxicity. J Biol Chem 278:49819–49827
Suo Z, Cox AA, Bartelli N, Rasul I, Festoff BW, Premont RT, Arendash GW (2007) GRK5 deficiency leads to early Alzheimer-like pathology and working memory impairment. Neurobiol Aging 28:1873–1888
Tamura H, Kawakami H, Kanamoto T, Kato T, Yokoyama T, Sasaki K, Izumi Y, Matsumoto M, Mishima HK (2006) High frequency of open-angle glaucoma in Japanese patients with Alzheimer’s disease. J Neurol Sci 246:79–83
Tanghe A, Termont A, Merchiers P, Schilling S, Demuth HU, Scrocchi L, Van Leuven F, Griffioen G, Van Dooren T (2010) Pathological Hallmarks, Clinical Parallels, and Value for Drug Testing in Alzheimer’s Disease of the APP[V717I] London Transgenic Mouse Model. Int J Alzheimers Dis doi:10.4061/2010/417314
Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, Van Leuven F (2005) Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J Biol Chem 280:3963–3973
Valenti DA (2010) Alzheimer’s disease: visual system review. Optometry 81:12–21
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002a) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416:535–539
Walsh DT, Montero RM, Bresciani LG, Jen AY, Leclercq PD, Saunders D, EL-Amir AN, Gbadamoshi L, Gentleman SM, Jen LS (2002b) Amyloid-beta peptide is toxic to neurons in vivo via indirect mechanisms. Neurobiol Dis 10:20–27
Wan J, Fu AK, Ip FC, Ng HK, Hugon J, Page G, Wang JH, Lai KO, Wu Z, Ip NY (2010) Tyk2/STAT3 signaling mediates beta-amyloid-induced neuronal cell death: implications in Alzheimer’s disease. J Neurosci 30:6873–6881
Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL (2002) Soluble oligomers of beta amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res 924:133–140
Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci 29:9090–9103
Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH (2002) The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci 22:1858–1867
Wostyn P, Audenaert K, De Deyn PP (2008) Alzheimer’s disease-related changes in diseases characterized by elevation of intracranial or intraocular pressure. Clin Neurol Neurosurg 110:101–109
Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM (2005) Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 280:5892–5901
Yin H, Chen L, Chen X, Liu X (2008) Soluble amyloid beta oligomers may contribute to apoptosis of retinal ganglion cells in glaucoma. Med Hypotheses 71:77–80
Yoshida T, Ohno-Matsui K, Ichinose S, Sato T, Iwata N, Saido TC, Hisatomi T, Mochizuki M, Morita I (2005) The potential role of amyloid beta in the pathogenesis of age-related macular degeneration. J Clin Invest 115:2793–2800
Yu MS, Ho YS, So KF, Yuen WH, Chang RCC (2006) Cytoprotective effects of Lycium barbarum against reducing stress on endoplasmic reticulum. Int J Mol Med 17:1157–1161
Yu MS, Lai CS, Ho YS, Zee SY, So KF, Yuen WH, Chang RCC (2007) Characterization of the effects of anti-aging medicine Fructus lycii on beta-amyloid peptide neurotoxicity. Int J Mol Med 20:261–268
Zahs KR, Ashe KH (2010) ‘Too much good news’—are Alzheimer mouse models trying to tell us how to prevent, not cure, Alzheimer’s disease? Trends Neurosci 33:381–389
Zarbin MA (2004) Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol 122:598–614
Acknowledgement
Research in this laboratory is partly supported by HKU Alzheimer’s Disease Research Network under Strategic Research Theme on Healthy Aging, Strategic Research Theme on Drug Discovery, Azalea (1972) Endowment Fund, and HKU Small Project Fund (20097176185).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Kin Chiu and Tin-Fung Chan contribute equally to this manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Chiu, K., Chan, TF., Wu, A. et al. Neurodegeneration of the retina in mouse models of Alzheimer’s disease: what can we learn from the retina?. AGE 34, 633–649 (2012). https://doi.org/10.1007/s11357-011-9260-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-011-9260-2