
Abstract
Chlorine (Cl) in the terrestrial environment is of interest from multiple perspectives, including the use of chloride as a tracer for water flow and contaminant transport, organochlorine pollutants, Cl cycling, radioactive waste (radioecology; 36Cl is of large concern) and plant science (Cl as essential element for living plants). During the past decades, there has been a rapid development towards improved understanding of the terrestrial Cl cycle. There is a ubiquitous and extensive natural chlorination of organic matter in terrestrial ecosystems where naturally formed chlorinated organic compounds (Clorg) in soil frequently exceed the abundance of chloride. Chloride dominates import and export from terrestrial ecosystems while soil Clorg and biomass Cl can dominate the standing stock Cl. This has important implications for Cl transport, as chloride will enter the Cl pools resulting in prolonged residence times. Clearly, these pools must be considered separately in future monitoring programs addressing Cl cycling. Moreover, there are indications that (1) large amounts of Cl can accumulate in biomass, in some cases representing the main Cl pool; (2) emissions of volatile organic chlorines could be a significant export pathway of Cl and (3) that there is a production of Clorg in tissues of, e.g. plants and animals and that Cl can accumulate as, e.g. chlorinated fatty acids in organisms. Yet, data focusing on ecosystem perspectives and combined spatiotemporal variability regarding various Cl pools are still scarce, and the processes and ecological roles of the extensive biological Cl cycling are still poorly understood.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chlorine (Cl) is one of the 20 most abundant elements on earth and has various essential functions for living organisms. Chloride, the only stable ionic form of Cl, is the major anion in blood and is present at concentrations of approximately 100 mmol L−1 in plasma and interstitial fluid (Yunos et al. 2010). Chloride participates in osmoregulation of cells (White and Broadley 2001) and is as an important electrolyte for regulation of muscle function and synaptic transmission in the neural system. It is also an essential co-factor in enzymes involved in photosynthesis, e.g. PSII photosystem oxidation of water (Winterton 2000). Hence, Cl is a critical plant nutrient and a minimum requirement of Cl for crops of 1 g kg−1 dry mass (d.m.) is indicated (White and Broadley 2001).
Many of the most debated organic contaminants, including the well-known persistent organic pollutants (POPs), are chlorinated (Godduhn and Duffy 2003). Although natural halogenated organic compounds have been known since the late nineteenth century (Gribble 2010), this was not widely recognised by environmental chemists, and the dominating view until recently was that chlorinated organic compounds (Clorg) in the environment were primarily anthropogenic and often toxic. It is now evident that there is a large production of natural Clorg. Nearly 5000 natural Clorg have been identified; they are produced by fungi, lichens, plants, marine organisms, insects and vertebrates including humans (Gribble 2003, 2010, 2015; Öberg 2002). Some specific Clorg have well known physiological functions, e.g. several important antibiotics such as vancomycin. Others have important environment effects, e.g. volatile Clorg (VOCls) that contribute to atmospheric ozone destruction (Winterton 2000). However, the ecological functions of most Clorg and the reasons for their production are largely unknown.
Cl is central in hydrological research as Cl−, the globally dominating chlorine species, is highly soluble in water, and oceanic water has a high enrichment factor compared with riverine water; seawater concentrations are in the order of 2500 times larger than freshwater concentrations (Winterton 2000). At a first glance, this has been interpreted to indicate that Cl− is unreactive in the environment, which until recently has been the prevailing view (White and Broadley 2001). Accordingly, Cl− has been seen as a suitable and inexpensive tracer of soil and ground water movements (Herczeg and Leaney 2011; Hruska et al. 2012; Cartwright et al. 2017), and studies using Cl− as a tracer have been the foundation for contaminant transport models (Kirchner et al. 2000). However, as discussed below, there is now clear evidence that in some environments, Cl− is more active than previously thought.
Chlorine-36, 36Cl, is a radioactive isotope with a half-life of 3.01 × 105 years and has attracted interest because of its presence in waste from nuclear facilities (Tanaka and Thiry 2020; van den Hoof and Thiry 2012). The long half-life, high mobility in the pedosphere and the potential for substantial biological uptake creates a need for long-term risk assessments related to handling and storage of radioactive waste (Limer et al. 2009). A growing awareness of the complex cycling of Cl in terrestrial environments necessitates a re-evaluation of risk assessments based on the previous assumptions that 36Cl in soils primarily occur as 36Cl− and is highly soluble and unreactive. This has been highlighted in for instance agricultural soil-plant systems (Le Dizès and Gonze 2019).
During the past decades, there have been several unexpected discoveries regarding the terrestrial chlorine (Cl) cycle. Experiments with 36Cl as tracer have confirmed natural chlorination rates corresponding to as much as 50–300% of the annual wet deposition of Cl in several types of soils (Bastviken et al. 2009). Substantial chlorination of organic matter occurs in all studied types of temperate and boreal soils in agricultural and forest areas (Gustavsson et al. 2012; Redon et al. 2013). Moreover, extensive accumulation of Clorg over 30 years in forest soils was recently demonstrated at the ecosystem level (Montelius et al. 2015). In addition, simultaneous and rapid dechlorination of Clorg in soils was also confirmed (Montelius et al. 2016). Enzymatic control of chlorination processes has been described (van Pee and Unversucht 2003; Bastviken et al. 2009; Wever and Barnett 2017), and the genetic capacity to carry out chlorination is widespread among prokaryotes and eukaryotes alike (Bengtson et al. 2009; Bengtson et al. 2013; Weigold et al. 2016). The extensive natural chlorination processes in soil suggest that the Cl turnover likely is linked to common ecosystem processes. Indeed, chlorination rates were recently linked to microbial activity (Svensson et al. 2017), but the fundamental reasons for the extensive soil Cl-cycling are still unclear. A recent review discusses the microbial metabolism of Clorg and the possible links between chlorinating and dechlorinating microbes (Atashgahi et al. 2018).
Several aspects of Cl have been summarised previously, e.g., physiological roles (White and Broadley 2001; Yunos et al. 2010), and the POP perspectives (Winterton 2000; Bidleman et al. 2019) and will not be the primary focus here. Rather, this review represents an update and supplement to previous reviews focusing on the terrestrial Cl cycling (Clarke et al. 2009; Graedel and Keene 1996; Öberg 2002; Leri and Myneni 2010; Öberg and Bastviken 2012; Vodyanitskii and Makarov 2017) called upon by the emerging interest in the behaviour of 36Cl in soils and terrestrial ecosystems (Limer et al. 2009; Sheppard et al. 1996; van den Hoof and Thiry 2012; Le Dizès and Gonze 2019; Tanaka and Thiry 2020). Here, we provide an overview of current knowledge on the Cl cycle and the fate of Cl in terrestrial environments. We highlight the latest findings on the occurrence of chloride and Clorg and suggested translocations and transformation processes related with suggested ecosystem Cl budgets. Finally, we discuss knowledge gaps and possible future research directions towards improved understanding of Cl-dynamics in terrestrial environments.
Fundamental chemical aspects of Cl
Cl, having the atomic number 17, belongs to the halogen group in the periodic table. Molecular Cl is a strong oxidant due to the high electron affinity and electronegativity of Cl. Consequently, molecular Cl is rare in nature, and Cl−, which is highly soluble in water, typically dominates in the hydrosphere and in minerals, but, as discussed below, not necessarily in soil.
Cl isotopes and sources of 36Cl
In nature, Cl occurs primarily as the two stable isotopes 35Cl (ca. 76%) and 37Cl (ca. 24%). In addition, seven radioactive isotopes exist of which six have half-lives of less than 1 h and are of less interest with respect to Cl cycling in the environment. In contrast, 36Cl has a half-life of 3.01 105 years and decays with a maximum energy of 709.6 keV either by emitting a beta particle (98.1%) or by electron capture (1.9%) resulting in the end products argon-36 (36Ar) and sulphur-36 (36S), respectively (Peterson et al. 2007; Rodriguez et al. 2006).
In the environment, 36Cl is produced by natural nuclear reactions; in the atmosphere by the spallation of argon with cosmic ray protons and in soil and rock by neutron activation of potassium (K), calcium (Ca) and Cl (White and Broadley 2001). The resulting radiological dose to individuals can be estimated from the ratio of 36Cl to stable chlorine (36Cl/Cl) in the surface environment and varies with geographical location. The natural 36Cl/Cl ratio is between 10−15 and 10−12 (Campbell et al. 2003). The dose can thereby differ by several orders of magnitude between coastal and inland areas due to the difference in concentration of stable Cl. 36Cl/Cl ratios exceeding 10−12 (up to 2·× 10−11) were found in a 100-km2 area in the Tokaimura region, Japan, where four nuclear power reactors and one nuclear fuel reprocessing plant were once operated (Seki et al. 2007).
36Cl was produced in large amounts by neutron activation of seawater upon nuclear weapons testing between 1952 and 1958 (Peterson et al. 2007). These peaks in 36Cl have been used for dating ground water (Campbell et al. 2003; White and Broadley 2001). 36Cl is also produced during nuclear power reactor operation due to neutron capture of stable 35Cl that may be present at trace levels in core materials, graphite, coolant water and construction materials such as steel and concrete (Frechou and Degros 2005; Hou et al. 2007). In addition, 36Cl can be produced in considerable amounts via spallation reactions of other concrete components, such as K and Ca, primarily in fast reactors where high-energy particles such as fast neutrons are present (Aze et al. 2007). Although ambient 36Cl levels typically are low, the active uptake of Cl by organisms leads to higher concentrations in plants than in the soil in which they grow (Kashparov et al. 2007a, 2007b; White and Broadley 2001). Therefore, information about Cl cycling in soils, sediments and vegetation, including bioavailability and residence end exposure times, is necessary for relevant risk assessments (Limer et al. 2009).
Major Cl reservoirs and large-scale cycling
The largest Cl reservoirs on the earth’s surface are the crust and the ocean (Graedel and Keene 1996) (Table 1). Inorganic Cl by far dominates these reservoirs. Estimates for the other reservoirs are also largely based on Cl− concentration measurements. This assumption of a general dominance of Cl− in the pedosphere is problematic as Clorg have been shown to range from 11 to near 100% of the total Cl pool in a large range of soil types (Gustavsson et al. 2012; Johansson et al. 2003a; Redon et al. 2011; Redon et al. 2013); the pedosphere Cl pool may be at least twice as large if Clorg is included.
Biosphere estimates of Cl pool have not been included in prior studies. If we assume a plant biomass concentration of Cl− of 1 mg g−1 d.m. (dry matter), which the plant does not show deficiency symptoms (Marschner 2012) and a global plant biomass of 900 Gt (Bar-On et al. 2018), the plant biomass reservoir of Cl (0.9 × 1015 g) is considered smaller than the pedosphere Cl pool (48 × 1015 g).
Rapidly growing plants take up large amounts of Cl−. In common crops, the ratios of Cl− concentrations in fresh plant tissue to Cl− concentrations per dry mass in the top 20 cm of soil were 1.5–305 (Kashparov et al. 2007a, 2007b). This is in line with the high proportion, 10%, of total catchment Cl found in biomass from a Danish forest (Öberg et al. 1998). In coniferous forests, a much higher root uptake of Cl− than the plant Cl− demand was found resulting in large leaching and throughfall from the trees (Montelius et al. 2015; Redon et al. 2011; van den Hoof and Thiry 2012). The reasons for the excess uptake and the extensive internal ecosystem Cl− cycling in some environments are presently unknown but can dramatically increase Cl− residence times and soil concentrations (Montelius et al. 2015; Tanaka and Thiry 2020).
In the large-scale inorganic Cl cycle, mineral weathering contributes with Cl− to freshwaters that subsequently reach the oceans (Graedel and Keene 1996). The largest contribution of Cl− to the atmosphere is sea salt aerosols while minor contributions include HCl from volcanic activity and biomass burning, mineral aerosols and VOCls of natural or anthropogenic origin. Cl− is transported to oceans and soils by wet and dry deposition. Fluxes between the reservoirs have been proposed (Graedel and Keene 1996), but these values are poorly constrained. For example, to balance the overall budget, a yearly Cl loss of 30 Tg year−1 from the pedosphere (equivalent to 1.25‰ of the total pedosphere reservoir) had to be assumed. Given the other budget elements, such a flux would lead to a rapid depletion of the pedosphere stock which is unrealistic. Such budget calculations illustrate the lack of relevant information regarding the large-scale fluxes and a possible bias caused by not including Clorg in the budget.
Cl in terrestrial ecosystems
During the past decades, there has been a rapid development towards improved qualitative understanding of the terrestrial Cl cycle. Below, we summarise ecosystem inputs, transformation, translocation and export processes, reservoirs and finally ecosystem budgets regarding Cl.
Deposition
Prior to the mid-1950s, the chemical composition of surface waters was considered a result of physical land use in combination with the geochemical, hydrological processes and features of the surrounding area. In the mid-1950s, it was suggested that the chemical composition of rivers mirrors the chemical composition of precipitation (Eriksson 1955). The arguments were based on extensive data on Cl− and sulphate in precipitation and rivers and implied that these ions originated from oceans (Eriksson 1960). The aerosols are carried with the winds and deposited either back to the sea or on land by precipitation that washes out Cl− from the atmosphere. Gases and particles can also contain Cl−. These can undergo ‘dry deposition’, i.e. be deposited directly on the ground or vegetation, or ‘wet deposition’, i.e. washed out of the air by precipitation. The total deposition is generally higher in forested areas than over open land because atmospheric particles are intercepted by vegetation; leaching from the leaves is also possible.
The quantification of wet deposition of Cl− can be done with high precision and is relatively well constrained but variable with higher depositions close to the sea and influenced by the prevailing winds (Clarke et al. 2009). For instance, in Europe, the average Cl wet deposition is approximately less than 20 kg ha−1 year−1 but varies from 0.5 to 220 kg ha−1 year−1 (Clarke et al. 2009). In contrast to wet deposition, dry deposition is difficult to measure as it, besides distance to the sea, also includes inputs via gases, aerosols and particles and is affected by interception by surfaces, e.g., tree canopies. Therefore, quantitative figures of dry deposition are considerably more uncertain than those of wet deposition but have been estimated to be 15–73% (average 43%) of total deposition based on data from North America and Europe (Svensson et al. 2012).
It is well known that precipitation, in addition to Cl−, also contains Clorg (Enell and Wennberg 1991; Grimvall et al. 1991; Laniewski et al. 1995). Measurements of individual halogens in organic matter derived from precipitation show that most of the organically bound halogens detected as adsorbable organic halogens (AOX) are chlorinated compounds (Laniewski et al. 1999). Brominated compounds are widespread but less common, and organically bound iodine has only been detected at sites close to the sea (Laniewski et al. 1999).
A major part of Clorg present in precipitation and snow consists of relatively polar semi- or non-volatile compounds, particularly organic bases and acids (Laniewski et al. 1999). Chloroacetic acids can occasionally explain up to 6% of the Clorg in precipitation (von Sydow 1999) while the relative contribution from VOCls usually is smaller, often at ng L−1 concentrations (Schleyer 1996).
Little is known about the origin of the Clorg in precipitation. Known industrial pollutants, such as flame retardants (e.g., chlorinated alkyl phosphates) and pesticides (e.g., lindane), are typically present at ng L−1 levels (Stringer and Johnston 2001), i.e. in concentrations about three orders of magnitude less than the total Clorg concentrations. Chloroacetic acids (CCAs) have been detected in rain (Frank 1991; Reimann et al. 1996), but the origin is under debate (Berg et al. 2000; Cape et al. 2006; Laturnus et al. 2005). In addition, a study conducted at Klosterhede in northwest Denmark suggests that Clorg in throughfall mainly originates from plant internal sources rather than from dry deposition (and thus external) sources (Öberg et al. 1998). Although there is important and extensive work on the formation and emissions of volatile organic halogens, their low concentrations in rain (ng L−1; Laniewski et al. 1999; Svensson et al. 2007a) cannot explain the total rain Clorg levels (μg L−1; Öberg et al. 1998; Montelius et al. 2015).
Weathering
As mentioned above, it has long been believed that Cl− primarily participates in geochemical processes and is only negligibly affected by biological processes or interactions with organic matter. Riverine Cl− has likewise often been considered to originate from the atmosphere only, despite possible weathering processes during the pathway through the soil (Eriksson 1960; Schlesinger and Bernhardt 2020). There are limited analyses of Cl− in rocks, but felsic bedrocks such as granite contain low amounts of Cl−, and the highest amounts are found in mafic bedrocks (Melkerud et al. 1992) and, obviously, in halide-rich evaporites. Felsic minerals can be considered to have a lower chemical weathering rate than mafic minerals. The weathering rate has been estimated for a small stream at Hubbard Brook, New Hampshire, USA, with bedrock consisting mainly of granite, and approximately 4–8% of the Cl− stream output originated from weathering (Lovett et al. 2005).
Land rise in previously glaciated regions can result in soils originating from marine sediments and therefore are rich in Cl−. Release of Cl− from such marine deposits constitutes a special case with potentially significant subsurface contribution of Cl− to soils, water and organisms.
Input from irrigation, fertilisation and road de-icing
Anthropogenic Cl− input from irrigation and fertilisation can represent substantial inputs to terrestrial environments. Irrigation with low to medium level salinity water can contribute in the order of 500–1000 kg ha−1 year−1, i.e. anthropogenic contributions can be the major Cl− input (Xu et al. 2000).
Since the start of de-icing of roads in mid-twentieth century, studies have shown increased Cl− concentrations in both surface water and groundwater in the vicinity of roads (Dugan et al. 2017; Kaushal et al. 2005; Kelly et al. 2008). Road salt effects can be chemical, e.g. induce ion exchange affecting acidification and metal and nutrient leaching (Bäckström et al. 2004; Löfgren 2001), or biological, i.e. affect aquatic food webs (Hintz and Relyea 2019; Todd and Kaltenecker 2012; Van Meter et al. 2011). The application of road salt has been estimated to be substantial compared with deposition (Forczek et al. 2011), and there are indications of associated extra Cl retention in soil (Robinson et al. 2017; Kincaid and Findlay 2009; Perera and Gharabaghi 2013).
Processes driving terrestrial Cl cycling
Most of the soil chlorination is driven by enzymes, i.e. organisms, but abiotic chlorination seems also to occur at significant rates (Bastviken et al. 2009; Rohlenova et al. 2009; Matucha et al. 2006; Weigold et al. 2016; Atashgahi et al. 2018). Chlorination rates have been experimentally investigated at temperatures ranging from 4 to 50 °C, where low rates were found at 4 and 50 °C, possibly representing primarily abiotic rates near the temperature extremes, while there was an enzymatic response pattern with a maximum at 20 °C indicating a dominance of biotic chlorination at many common temperatures. There are indications that abiotic processes related to iron cycling in soils contribute to Cl− retention (Fahimi et al. 2003; Keppler et al. 2000). However, since the redox cycling of iron is usually a consequence of microbial activity, the proposed abiotic processes may be linked to biological processes, albeit indirectly. Under experimental conditions, formation of volatile Clorg in hypersaline lake sediments from Western Australia is higher under biotic conditions than in sterilised samples but were not stimulated via Fe redox transformations or the formation of reactive Fe species (Ruecker et al 2014).
Chlorination of organic matter can occur both inside and outside cells. The intracellular chlorination seems strictly regulated by enzymatic processes (van Pee and Unversucht 2003). Enzymes known to mediate intracellular chlorination include FADH2-dependent halogenases and perhydrolases. In addition, biotic chlorination has been suggested to involve an enzymatic reaction, where methyl halide transferase catalyses the formation via the reaction of S-adenosyl-l-methionine (SAM) with chloride (Wuosmaa and Hager 1990; Atashgahi et al. 2018). The underlying process for the extracellular chlorination seems to be a formation of reactive chlorine (e.g. hypochlorous acid, HOCl) formed by reactions between hydrogen peroxide and Cl− (Neidleman and Geigert 1986). Reactive Cl is a strong oxidant and reacts with surrounding organic matter rendering unspecific chlorination of various organic compounds in the large and complex pool of soil organic matter (Hoekstra et al. 1998a; van Pee and Unversucht 2003). Extracellular chlorination also depends on the production of reactive chlorine by enzymes, such as heme and vanadium containing haloperoxidases, but the enzymatic control is less rigorous compared with intracellular chlorination (van Pee 2001).
Given the rapid chlorination rates and consequent Cl− retention in soil, ubiquity of Clorg and the widespread capacity among organisms to perform/promote chlorination, a fundamental, albeit unknown, ecological/evolutionary explanation for organic matter chlorination is likely. Intracellular chlorination processes have been explained as ways of detoxification or are believed to be produced as chemical defence compounds (e.g. antibiotics), hormones or pheromones (Hoekstra et al. 1998b). However, direct verification of these hypotheses is limited. Extracellular chlorination represents a different process, although it is well documented that reactive chlorine species such as hypochlorous acid are potent bactericides used by phagocytes to kill invading microorganisms (Apel and Hirt 2004). Several microorganisms and plants produce chlorinated allomones, i.e. substances that deter or kill competing or pathogenic organisms. The ability to use reactive chlorine in the chemical defence against competing microorganisms could provide a substantial advantage and be evolutionarily favoured. Indeed, screening genetic databases for identified haloperoxidases from terrestrial environments indicates that many originate from organisms associated with plants or decomposing plant material; the ability to produce reactive chlorine may be especially common in environments that are known for antibiotic-mediated competition for resources (Bengtson et al. 2009).
Another hypothesis relates to microbial processing of organic material representing their substrates. There is a general perception that chlorinated organic matter is less bioavailable than non-chlorinated organic compounds. However, chloroperoxidases, like many other oxidases, catalyse production of small reactive molecules (hypochlorous acid in the case of chloroperoxidase) that can break C–C bonds in complex, refractory organic compounds (Hoekstra et al. 1998b; van Pee and Unversucht 2003) whereby smaller, more bioavailable fragments of the refractory compounds may be formed. The exposure of lignin to reactive chlorine enhances its biodegradability, (Johansson et al. 2000), and fungal chloroperoxidase activity results in depolymerisation and breakdown of synthetic lignin supports this hypothesis (Ortiz-Bermúdez et al. 2003). Similarly, biodegradation of lignin in the effluent of chlorine bleached pulp mills is higher than the degradation of corresponding chlorine-free lignin (Bergbauer and Eggert 1994). Hence, promoting formation of Clorg could be a way of increasing the organic substrate supply for microorganisms as these compounds could be preferred as substrates by microorganisms after dechlorination.
A third potential reason for microbial chlorination is defence against oxygen radicals. Formation of reactive chlorine is related to consumption and detoxification of reactive oxygen species including hydrogen peroxide and oxygen radicals; by formation of, e.g., extracellular hypochlorous acid, reactive oxygen species may be prevented from entering the cell. Interestingly, repeated oxidative stress exposure induces the expression of chloroperoxidase genes and increases the production of reactive chlorine in some algae and bacteria (Bengtson et al. 2013).
Establishing how various environmental factors regulate chlorination and influence chlorination rates is important for understanding Cl cycling. Several hypotheses have been proposed, but these need further clarification. Tests with different nitrogen levels are ambiguous and local variability large (Bastviken et al. 2006; Rodstedth et al. 2003). Chlorination rates are slower under anoxic conditions than oxic (Bastviken et al. 2009), which is reasonable given that chlorination of organic matter is an oxidative process. This indicates an indirect regulation by soil moisture, but the overall regulation of natural chlorination of organic matter is still unclear. Varying the nitrogen levels has yielded ambiguous results (Bastviken et al. 2006, Rodhstedt 2000), and local variability seems large. A factorial study showed that total chlorination was hampered by addition of nitrate or by nitrate in combination with water but enhanced by addition of chloride as well as labile organic matter (glucose and maltose) (Svensson et al. 2017). These estimates were based on studies of bulk soil excluding roots, which likely has underestimated the chlorination potential as it was later suggested that most of the chlorination takes place in the rhizosphere (Montelius et al. 2019).
Dechlorination processes (transformation from Clorg to Cl− by either organic matter decomposition or by selective removal of Cl atoms from organic molecules) have been extensively studied in relation to Clorg pollution and bioremediation (van Pee and Unversucht 2003). Chlorinated compounds can be used as terminal electron acceptors in microbial metabolism. Interestingly, the Gibbs-free energy yield of this process is similar to the energy yield with nitrate as the electron acceptor and only slightly lower than the energy yield of oxic respiration (Smidt and de Vos 2004). Hence, chlorinated organic compounds can be very potent as electron acceptors. Dechlorination could therefore be the result of either degradation of the chlorinated organic matter or the microbial use of chlorinated organic molecules as electron acceptors, i.e. organohalide respiration; there is a wide literature regarding dehalogenation processes in terms of biochemistry of specific compounds (Dolfing 2000; Fetzner 1998; Olivas et al. 2002; Pries et al. 1994; Smidt and de Vos 2004; van Pee and Unversucht 2003). There are also studies that show net-uptake of chloromethane without evidence of organohalide respiration (Peng et al. 2020), and a cometabolic dechlorination has been suggested (Atashgahi et al. 2018; Peng et al. 2020). However, rates and regulation of dechlorination of bulk Clorg in nature are still rarely quantified although the limited available information suggests rates are high (Montelius et al. 2016). Studies have shown a pH dependence of inhibition of dechlorination and alternative electron acceptors (Paul and Smolders 2014; Aulenta et al. 2007; Yang et al. 2017). Further studies on the activities are needed to confirm dechlorination in non-contaminated soils, as organohalide respiring organisms, using Clorg as electron acceptors for growth, are likely present and active in different types of uncontaminated soils (Krzmarzick et al 2011; Montelius et al. 2016; Zlamal et al. 2017). There are already results on that the potential of dechlorination in terrestrial environments is both common and widespread with a variety of genes encoding for enzymes capable of dehalogenation (Temme et al. 2019).
Directly measured dechlorination rates of bulk Clorg in terrestrial environments indicate that soil Clorg levels result from a dynamic equilibrium between the chlorination and rapid dechlorination of some Clorg compounds, whereas another Clorg pool is dechlorinated more slowly (Montelius et al. 2016). Hence both chlorination and dechlorination processes are important for understanding the behaviour of Cl− in soils, and it has even been suggested that the balance between chlorination and dechlorination is more important for soil Cl− levels than Cl− deposition (Gustavsson et al. 2012; Montelius et al. 2015).
Soil microorganisms seem capable of rapidly taking up large quantities of Cl− during growth phase adding to ecosystem Cl dynamics (Bastviken et al. 2007). After addition of 36Cl to experiment soil, 20% was incorporated into microbial biomass within 5 days (Bastviken et al. 2007), suggesting that rapid microbial growth following a system disturbance (a rain event, leaf fall in autumn etc.) could lead to rapid microbial uptake of Cl− based on physiological need. It is unclear if this can affect Clorg formation rates.
Terrestrial reservoirs of chlorine
Soil
Total Cl typically range from 20 to > 1000 mg kg−1 d.m. in non-saline soils (Table 2). The concentrations of Clorg in surface soil are in most cases higher than Cl− concentrations (Table 2). The dry mass fraction of Clorg in surface soils (0.01–0.5%) is in parity with that of phosphorous (0.03–0.2%) and only slightly lower than nitrogen (1–5%) and sulphur (0.1–1.5%). Bulk density, horizon thicknesses and stoniness affect total storage of Cl more than concentration differences. Unfortunately, these parameters make the calculation of total Cl storage capacity difficult. Total storage is usually largest in the mineral soil layer because of its greater thickness compared with the organic surface layer, although Clorg concentrations are typically 2–5 times higher in the organic surface soil layer (Redon et al. 2011). Available data only separates organic and mineral soil layers, and more detailed discussions about specific soil profiles are therefore not possible at this point. Clorg levels are higher in soils with more organic matter (Redon et al. 2011), while the percentage Clorg is frequently higher (> 80%) in mineral soils (Table 2). Hence, higher soil total Cl levels may relate to soil organic matter content, probably because soil chlorination processes retain and accumulate Cl as Clorg (Gustavsson et al. 2012, Montelius et al. 2015). However, available information is scarce and non-conclusive, but high levels of Clorg (average 245 μg g−1) have been found in coastal Arctic wet tundra soils (Zlamal et al. 2017).
Despite the large number of identified chlorinated organic compounds, the molecular composition of the bulk Clorg in soils is largely unknown. The soil organic matter seems mainly composed of high molecular mass substances, usually > 1000 Dalton (Hjelm and Asplund 1995). However, Clorg content in different types of soil organic matter has rarely been determined. Of the Clorg in coniferous soil, 1–10% was associated with water-leachable fractions of the organic matter (Bastviken et al. 2009, 2007). Further, in lysimeter soil, Clorg is associated with organic matter with a molecular mass < 10 000 Dalton, while most organic matter had molecular mass, i.e., > 10,000 Dalton (Lee et al. 2001).
The concentration of Clorg is usually found higher in coniferous forest than deciduous forest soils (Johansson et al. 2003a). This pattern was confirmed by Redon et al. (2011) in a study of more than 50 forested sites in France. In addition, soil humus in plots with Norway spruce (Picea abies) had higher net accumulation of Cl− (7 times) and Clorg (9 times) than soil humus of plots with Sessile oak (Quercus sessiliflora) over an experimental period of 30 years (Montelius et al. 2015). Thus, it seems that vegetation characteristics can explain local soil Cl− and Clorg levels, which may explain why these are independent of atmospheric deposition.
Water
In contrast to soils, Cl− concentrations generally exceed Clorg concentrations in water. For example, the Cl− concentration in various waters is measured in mg L−1, while Clorg is typically measured in μg L−1 and VOCls are in the range of ng L−1 (Table 3) (Asplund and Grimvall 1991; Enell and Wennberg 1991; Eriksson 1960, McCulloch 2003). Hence, the atmospheric deposition of Clorg is three orders of magnitude lower than deposition of Cl− and thereby often assumed to be negligible from a total Cl perspective. While ground water has higher Cl− concentrations than precipitation and surface waters, Clorg and VOCl concentrations can be higher in surface waters than in precipitation (Table 3).
Sediment
Analysis of sediment Clorg has usually focused on contamination from industrial activities (Jonsson 1992; Poykio et al. 2008). There is a large body of literature on specific chlorinated pollutants (e.g. polychlorinated biphenyls [PCB] and dichlorodiphenyltrichloroethane and its degradation products [DDTs]). Among the bulk Clorg measurements, AOX has been used to study sediment pore waters, but such efforts in non-contaminated sediments are rare. The AOX method has been adapted for other types of bulk Clorg analyses including extractable organic halogens (EOX) and volatile organic halogens (VOX). There has also been a suggestion to avoid AOX analyses for sediment pore waters as it does not discriminate between natural and anthropogenic Clorg (Müller 2003). Extractable organic halogens (EOX; extraction of sediments with cyclohexane–isopropanol under sonication) yielded concentrations of 5–70 μg g−1 (lipid mass) in the upper 2 cm of Bay of Bothnia sediments in an area with contamination from pulp and paper mills to evaluate the effects of chlorine bleaching (Poykio et al. 2008). Another study reported Cl concentrations of < 10 to 840 μg g−1 organic matter in seven non-polluted inland water sediments (Suominen et al. 1997). The analysis methods differed (AOX and EOX after various extractions) making comparisons difficult. Analyses using similar methodology as for soils, such as total organic halogens (TOX), appear to be largely missing, and therefore, Cl and Clorg levels in sediments are presently unclear.
Biomass
The Cl− content of plant biomass varies between species. For plant growth, a general Cl− requirement of 1 mg g−1 d.m. has been suggested; deficiency symptoms have been observed at 0.1–5.7 mg g−1 d.m., while toxicity has been reported at 4–50 mg g−1 d.m. (White and Broadley 2001). Thus, extrapolations across species are uncertain. Plant Clorg content has been estimated to 0.01–0.1 mg g−1 d.m. (Öberg et al. 2005), but this is based on scattered measurements from beech (Fagus sylvatica) leaves, spruce needles, Sphagnum moss and bulk samples of grass, and the variability between species and plant parts is unknown at present. Table 4 shows a large variation in different types of vegetation.
Based on measurements of different plant parts, the standing stock Cl in trees in a pine forest in Belgium was estimated to be 4.7 and 5.5 kg ha−1 for wood plus leaves, and roots, respectively (van den Hoof and Thiry 2012). Fresh leaves had the highest Cl concentration (0.59 mg g−1 d.m.) corresponding to 35% of the total Cl in the trees. Clorg accounted for less than 10% of the Cl in the leaves and the bark but constituted 20% of the total biomass Cl of the whole tree. Cl is an essential element, but the potentially high enrichment levels indicate that the role of Cl for trees is not fully understood, and that processes regulating Cl uptake in vegetation affect the residence times of chlorine in terrestrial ecosystems (Epp et al. 2020; Tanaka and Thiry 2020).
Investigation of total Cl in various landscape compartments, including soil, sediment, water and biomass, indicates that the terrestrial biomass Cl pool dominates over other pools and account for in the order of 60% of the total catchment Cl (Tröjbom and Grolander 2010). Cl was substantially enriched in biomass compared with other comparable elements (e.g., bromine and iodine) and nutrients (nitrogen, phosphorus, potassium, calcium). Cl is an essential element, but this level of enrichment indicates that the roles of Cl for organisms may not be fully understood, and that a large part of potential contaminant 36Cl reaching terrestrial parts of the landscape will be taken up by biota.
In terrestrial vascular plants alone, a couple of hundred chlorinated compounds have been identified (Gribble 2010), but many of these are relatively short lived and have a specific function, e.g. as auxins (Engvild 1994; Walter et al. 2020). Of interest for risk assessment are compounds that are toxic, and which may be transferred between organisms in the food chain. Several natural chlorinated compounds have structural similarities with persistent organic pollutants (POPs) that are believed to be produced at low trophic levels in algae and sponges (Gribble 2010). Many clearly accumulate through the food chain; very high concentrations may occur in, e.g. cetaceans (Alonso et al. 2012; Mwevura et al. 2010).
Several types of natural halogenated carboxylic acids are known (Dembitsky and Srebnik 2002). Most of these are structurally complex, often containing reactive functional groups that shorten their environmental half-lives. However, chlorinated fatty acids (ClFAs) have both a long half-life in the environment and undergo food-chain transfer (Björn 1999; Mu et al. 2004). ClFAs constituted the bulk of extractable organically bound chlorine (EOCl; 70% or more) in environmental samples in the 1990s (Håkansson et al. 1991; Mu et al. 2004). To date, the origin of ClFAs is not understood. There are clearly anthropogenic influences as EOCl concentrations are particularly high close to point sources of Clorg. An important type of point source was production of chlorine-bleached pulp and paper (Håkansson et al. 1991), but there are also reports indicating that the ClFA-profile depends on the type of anthropogenic pollutants present (Vereskuns 1999). Research is lacking, but as far as known, ClFAs are ubiquitous and present also in areas where the concentrations of anthropogenic POPs are low. Thus, de novo synthesis of ClFAs in organisms is possible, and formation of CLFAs via chloroperoxidase action has been shown in some marine organisms (Mu et al. 2004).
ClFAs are particularly interesting from a risk assessment point of view as they are not recognised by biota as xenobiotics although they may exert toxicity (Ewald 1998). In contrast to legacy POPs, ClFAs do not primarily accumulate in fatty tissues but are incorporated in membrane lipids; the concentrations of ClFAs relative other fatty acids (FAs) are higher in muscle tissues than in depot fat, at least in mammals (Björn 1999; Åkesson Nilsson 2004). Of particular interest for this review, a de novo synthesis of ClFAs incorporating 36Cl released into the environment could potentially introduce a long-lived source of radioactivity in areas where critical membrane functions take place.
A possible complication in assessing the environmental problems is that toxicity, detoxification pathways and uptake or emission of halogenated compound in terrestrial or aquatic environments may change upon dehalogenation (Skladanka et al. 2012). For full understanding of possible ecotoxic problems, the toxicity of individual compounds as well as their degradation products may have to be considered (Weissflog et al. 2007). Some compounds may also react directly with compounds that are key for cell function. For example, as halogens are leaving groups, halogenated aliphatics my react with endogenous or exogenous nucleophiles, including DNA and proteins (Motwani et al. 2011), affecting the efficiency of biological processes.
Litter
Simultaneous leaching of Cl− and formation of Clorg has been shown in litter (detached dead or dying plant biomass) (Myneni 2002). A study of senescent leaves from white oak (Quercus alba) showed Cl− and Clorg contents of 335 and 165 mg kg−1 (Leri and Myneni 2010). Cl fractions were quantified using X-ray absorption near-edge structure (XANES) spectroscopy. The results were that (1) total Clorg content in the leaves increased during the senescence and gradual degradation of the organic matter, (2) aliphatic Clorg was present at stable levels over time and seems contributed by plant processes and resistant to degradation and (3) that water-soluble aromatic Clorg was first leached from the leaves followed by later accumulation of non-soluble aromatic Clorg during senescence. There are however few studies that have quantified chlorine content in litter. Clorg in spruce needle litter in Denmark was 51–196 mg kg−1 d.m. (median 101) (Öberg et al. 1998). In a study of 51 different forest sites in France with both coniferous and broad leaf tree species, total Cl content in the litter was 46–528 mg kg−1 d.m. (median 147), and the percentage Clorg was 11–100% (median 40%) (Redon et al. 2011). Again, available data suggest substantial variability within and between species and locations.
Translocation within systems
There are scattered indications of extensive internal cycling of Cl in terrestrial ecosystems. For example, the annual root uptake of Cl by Scots pine (Pinus silvestris) was nine times larger than the Cl demand by the tree (van den Hoof and Thiry 2012). The excess Cl was returned to soil primarily as Cl− in throughfall Cl deposition and to some extent by litterfall. Similarly, a study integrating data from 27 forests of different types in France show that throughfall was variable, but on an average twice as high as the total atmospheric Cl deposition, 41 and 20 kg ha−1 year−1, respectively, while the average Cl in litterfall ranged from 0.1 to 2.5 kg ha−1 year−1 (Redon et al. 2011). This cycling within trees and soil, if a general phenomenon, will prolong Cl residence times in the forest ecosystem. The reasons for excess uptake of Cl relative to needs are unknown but could be related with evapotranspiration if Cl− enters the plant with the water without discrimination. Therefore, vegetation seems to have a prominent role in the production and distribution of organohalogens.
The plant root-soil interface, the rhizosphere, is dynamic with numerous biogeochemical processes taking place that are important for terrestrial carbon cycling and other element cycling that sustain plant growth. Through root exudation of various chemical compounds, roots may regulate the nearby soil microbial community, plant defence, attracting beneficial microbes, change the chemical and physical properties of the soil or inhibit the growth of competing plant species (Philippot et al. 2013). The majority of the studies on Cl biogeochemical cycling have hitherto focused on bulk soil in which plant roots were removed by sieving, despite the knowledge that vegetation and vegetation-associated organisms have a strong influence on element turnover (Clemmensen et al. 2013; Pausch and Kuzyakov 2018). In a recent experimental study, we observed that increased availability of labile organic matter increased the Clorg formation rates (Svensson et al. 2017). Hence, it seems that the most labile organic matter was rapidly degraded and fuelled a greater chlorination rate before being depleted. This indicates that increased chlorination rates are to be expected whenever more labile organic matter is present, e.g. in zones with more root exudate. These findings agree with the idea that chlorination is driven by biotic activity, which is highest in surface soil layers with higher root density and more input of organic material. Hence, root exudates in combination with plant-specific microbial interactions in the root zone may be important. Recent results from a radiotracer (36Cl) soil-plant (wheat, Triticum vulgare), the experiment shows that most halogenation took place in the rhizosphere. The specific halogenation rates (day−1) in soil with plants was at least two orders of magnitude higher (0.01 d−1) than without plants (0.0007 day−1), suggesting that plants play an active role in halogenation processes (Montelius et al. 2019).
There is also an extensive cycling of Cl in soils. Microbial activity in the topsoil results in formation of Clorg from Cl− (Bastviken et al. 2009; Öberg et al. 2005). In incubated soil samples from 14 sites, the amount of Cl converted to Clorg in soil dry matter via microbial chlorination was 1.4–90 ng g−1 day−1 or 0.2–3‰ of the Cl− pool being transformed to Clorg per day (Gustavsson et al. 2012). Estimates based on field data are rare, but one study estimated the net chlorination to be 2 kg ha−1 year−1 from mass balance calculations, while laboratory estimates using the same soil yielded gross chlorination rates of 2–13 kg ha−1 year−1 corresponding to 50–300% of the wet deposition to this catchment (Bastviken et al. 2009; Bastviken et al. 2007). This discovery challenges the use of chloride as a tracer of water movement in the soil, in turn undermining many current hydrological and contaminant transport models. Substantial chlorination of organic matter occurs in agricultural, forest and peat soils (Gustavsson et al. 2012; Redon et al. 2013; Silk et al. 1997), implying that natural chlorination is a general and widespread phenomenon across many biomes and not limited to certain types of environments. The extensive natural chlorination of organic matter in soil suggests that the Cl turnover likely is linked to common ecosystem processes. Indeed, the chlorination rates were linked with microbial activity (Svensson et al. 2017), but the fundamental reasons for the extensive soil Cl-cycling are still unclear.
Available data (Table 5) indicates higher chlorination rates on a mass basis in litter compared with deeper soil layers. Extensive chlorination has been shown upon litter degradation (Myneni 2002), and seasonal patterns, with an increased aromatic Clorg concentration during summer months, have been suggested (Leri and Myneni 2010).
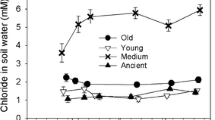
Migration through the profile may be important for internal Cl cycling in soils. Intensive chlorination has been observed in surface soil and litter layers, while Clorg levels decrease with soil depth, and the form of Cl dominating in the hydrological export from catchments is Cl− suggesting that Clorg is leached from surface upland soils and either retained and preserved or transformed to Cl− in deeper soil layers (Fig. 1) (Rodstedth et al. 2003; Svensson et al. 2007b; Öberg et al. 2005). Wetland soils also appear to retain Cl−, as a significant retention of road salt, 4–41%, has been indicated (McGuire and Judd 2020).

Estimated organic chlorine transport in forest ecosystem soil. Concentration data and flux estimations for topsoil are based on data from Rodhstedt (2000) and Svensson et al. (2007) leached from the topsoil and gradually lost from the soil water by precipitation or adsorption to the solid phase or by organic matter degradation while the water moves downward through the soil column. Cl− shows an opposite pattern with lower relative concentrations in surface soils and increasing concentrations downward partly due to the release of Cl− from Clorg. This model can explain why the water released from soils has higher concentrations of Cl− than Clorg, while Clorg dominates in surface soils
VOCls are produced in a wide variety of ecosystems such as wetlands, salt marshes and forests (Albers et al. 2010; Dimmer et al. 2001; Rhew et al. 2002), and in different climatic regions including arctic, temperate and tropical areas (Albers et al. 2017; Rhew et al. 2008; Jiao and Ruecker 2018; Yokouchi et al. 2002). Despite the growing knowledge of the field, data on VOCl emission rates are scattered and inconsistent. Budget and transport estimates on various scales are uncertain, partly because low concentrations of each specific VOCl make sampling and analyses challenging (Pickering et al. 2013). Furthermore, VOCl sources and sinks in terrestrial environments are not well understood, and continuous observations over time are scarce.
VOCls have been found in several terrestrial biomes such as tropical forest, grasslands, deciduous angiosperm forests, taiga, tundra and rice fields (Dimmer et al. 2001; Frank et al. 1989; Haselmann et al. 2002; Hoekstra et al. 2001; Khalil et al. 1998; Laturnus et al. 2000; Redeker et al. 2000; Rhew et al. 2008; Wang et al. 2007; Varner et al. 1999). Most of the available information has been gathered in the northern hemisphere. Previous studies on terrestrial ecosystems have primarily considered seven different VOCls. The most studied compounds are chloromethane (CH3Cl) and chloroform (CHCl3). Other VOCl compounds reported include CCl4 (tetrachlorometane), C2H3Cl (chloroethylene), CH2Cl2 (dichloromethane), CH3CCl3 (methyl chloroform) and C2H3Cl3 (trichloroethane) (Haselmann et al. 2002; Hoekstra et al. 2001; Rhew et al. 2008; Wang et al. 2007). In addition, other halogenated compounds such as bromomethane, iodomethane, trichlorofluoromethane (Freon-11) and dichlorodifluoromethane (Freon-12) are released from terrestrial sources (Khalil and Rasmussen 2000; Rhew et al. 2000; Varner et al. 2003).
Emissions of VOCl are likely small compared with wet and dry deposition of Cl. However, a 36Cl radiotracer study indirectly indicated substantial VOCl-associated release of Cl in soils corresponding to 0.18 g m−2 year−1 or 44% of the annual wet deposition (Bastviken et al. 2009). This number needs validation, but, interestingly, it includes all possible VOCls in contrast to other estimates that measure specific VOCl compounds only. Previous studies showed average Cl emission from a coniferous forest soil of 0.13 and 0.04 g m−2 year−1 released as chloroform and chloromethane, respectively (Dimmer et al. 2001), while a Cl emission of < 0.01 g m−2 year−1 was released as chloroform from a Scots pine forest soil (Hellen et al. 2006). Coastal areas and wetlands seem to be significant sources with large emissions of chloromethane at, on average 0.6 g m−2 year−1 and 0.2 g m−2 year−1 respectively (Svensson 2019). Given this, the formation of VOCl would not only represent a substantial proportion of the emission to the atmosphere but also a significant part of the chlorine cycle.
Ecosystem Cl budgets
Ecosystem budgets are typically based on concentration measurements in combination with information about carbon and water cycling that supports estimates of Cl transport and transformation. Different attempts to estimate Cl budgets (Montelius et al. 2015; Redon et al. 2011; van den Hoof and Thiry 2012; Öberg et al. 2005) all show that the order of magnitude of Cl pools is similar, though there are less data on vegetation. Figure 2 shows the current most solid Cl ecosystem budget. Further, the studies only cover temperate forest ecosystems in Northern Europe.

A terrestrial Cl budget of a Norway spruce forested ecosystems in France (at the experimental forest site at Breuil-Chenue, Eastern France). Pool (kg ha−1) and fluxes (kg ha−1 year−1). Volatilisation and soil leaching were not measured in the study (data obtained from Montelius et al. 2015)
Net ecosystem budgets of Cl−, i.e. comparison of atmospheric deposition and stream export, are common because Cl− is often included in monitoring efforts. A data summary indicates imbalances in many catchments (Svensson et al. 2012). This is not surprising given the new understanding of processes that either retain (plant uptake, formation of Clorg) or release (decay of biomass, dechlorination) Cl−. Imbalances were most striking in areas with a wet Cl− deposition below 6 kg ha−1 year−1 suggesting unknown dechlorination/degradation processes (Svensson et al. 2012).
Lysimeter studies have specifically addressed Cl− balances by irrigating soil cores with artificial precipitation with known Cl− concentrations and monitoring the efflux (Bastviken et al. 2006; Rodstedth et al. 2003). These studies showed imbalances that the soil in some cases had acted as a sink and in others as a source of Cl− indicating Cl transformation in topsoil. Changes in the large storage of Clorg in soil were suggested to explain the pattern, and substantial leaching of Clorg was observed. The soil in the experiment was, however, too heterogeneous to determine whether a change such as net loss or accumulation in the soil had taken place or not.
Cl transport through soils was previously assumed to reflect water transport in catchments. The application of Cl− as a tracer is probably sufficient in areas with a large Cl− deposition such as in the study by Kirchner et al. (2000), whereas studies of soils with low Cl− deposition indicate retention of chloride, which may lead to a change in residence time (Bastviken et al. 2006). Thus, Cl− may occasionally be a poor indicator of water retention. Few studies have addressed this issue. In a field tracer study in a small catchment, simultaneously injecting 36Cl and radioactive water (3H2O) into soil over a 30-day period, the recoveries in runoff of 36Cl and 3H2O were 47% and 78% (of the injected amount), respectively, when the tracers were injected into surface soil, while they were 83% and 98% (of the injected amount), respectively, after injections into deeper soils (Nyberg et al. 1999). Clearly, Cl− is preferentially retained in topsoil. Further, overall Cl residence times, i.e. considering both Cl− and Clorg pools and fluxes, were five times higher than residence times based on Cl− only (Redon et al. 2011). However, data have primarily been collected in forests, and to some extent also arable land, and peat bogs, while wetlands, sediments and discharge areas, where potential 36Cl contamination will leave underground aquifers, are poorly studied.
Future challenges
Previous findings, primarily based on studies of bulk chlorine pools, reveal that (1) Cl− is more reactive than previously thought, (2) many organisms actively produce enzymes that are involved in an extensive and rapid chlorine cycling, (3) that this cycling is linked to carbon and possibly other elements and (4) that Cl is probably much more important to, e.g. vegetation, microorganisms and carbon cycling than hitherto understood. However, several fundamental questions remain. Some of these are discussed below based on the interest to understand transport, uptake and/or exposure and residence times of Cl and 36Cl in terrestrial and limnic ecosystems. Cl− and Clorg pools will behave differently, e.g. with respect to solubility, bioavailability and residence time, and inorganic and organic Cl pools have to be considered separately in order to understand and model the dynamics of Cl in the environment over time.
What is the ecosystem variability of Cl− and Clorg?
This fundamental question includes inputs, standing stocks and outputs from various types of landscape units. Previous studies, generally non-repeated snapshots, have revealed differences between soil types, as well as local variations within sample plots, but only include studies in temperate regions and terrestrial ecosystems within a few different ecosystems (Johansson et al. 2003b). Currently, there are no systematic assessments of the temporal variability at different spatial scales. Such measurements are key for further studies, and for very fundamental aspects such as (1) the design of sampling programs, (2) understanding data comparability over time (seasonality) and space and (3) assessing model uncertainties. Although seemingly simple and basic, studies of ecosystems variability are fundamental to build appropriate environmental models. Soil and ground water discharge areas, e.g. wetlands, streams and lakes, are likely recipients of sub-surface sources of Cl. At present, while Cl− measurements are common in limnic monitoring, there is, to our knowledge, no systematic data regarding Cl cycling of the separate aquatic Cl− and Clorg pools, and information on Cl cycling in aquatic systems is largely missing (Dugat-Bony et al. (2016).
What are the main drivers of Cl− and Clorg accumulation and transport?
Long-term modelling with ambitions to include varying environmental conditions requires an understanding of how levels of Cl− and Clorg are regulated. Fundamental environmental variables such as water content, organic matter content, primary productivity, dominating vegetation, nutrient levels and temperature may have large impact on the relative distribution of Cl− and Clorg and affect bioavailability, transport pathways and residence times in ecosystems. In addition, a better understanding of biological chlorination would greatly facilitate modelling of chlorine cycles and for instance management of Cl accumulation in soils affected by salinisation.
Dechlorination rates, i.e. release of organically bound Cl from the organic matter, of bulk soil Clorg are a poorly understood key factor for understanding terrestrial Cl cycling (Montelius et al. 2016). It is likely that some of the Clorg we observe in soils represents a dynamic pool undergoing rapid turnover while some of the measured soil Clorg pool may consist of very stable compounds, but the balance between these Clorg pools is not clear. Therefore, rates and regulation of dechlorination are important to understand and predict the fate of Clorg, such as upon of land use change causing leakage or accumulation of Cl (Kauffman et al. 2003; Lovett et al. 2005; Mannerkoski et al. 2005) and potential effects due to land management such as deforestation.
In terms of transport, circumstantial evidence presented above highlight that the emissions of the total VOCl from various landscape compartments need to be quantified. Available data indicate that total VOCl emissions (in contrast to individual VOCl compounds) could be a substantial part of the export of chlorine from terrestrial ecosystems, but this remains to be confirmed.
Most questions regarding the regulation of Cl− and Clorg levels and transport have hitherto not been addressed and will require experimental assessments. However, other approaches may also prove valuable. For example, better understanding of the chemical composition and structure of Clorg would greatly facilitate interpretations regarding chemical prerequisites for chlorination and dechlorination, what enzymes are involved and under what conditions biotic/abiotic processes may occur.
Reaching a more comprehensive understanding of Cl cycling in terrestrial environments
As explained above, Cl cycling models need information about both Cl− and Clorg. Presently available data for both Cl− and Clorg are typically based on single “snap-shot” measurement with information from a limited number of environments. These data support static mass balance models, models that are restricted by several assumptions, including the steady-state assumption, i.e. that Clorg behaves as organic matter in general. Further, model uncertainties are difficult to assess given the poor knowledge of spatial and temporal variability. Also, the relatively large importance of biomass part of the total catchment Cl can be found in biomass show that many previous beliefs influencing model assumptions need to be re-evaluated.
Thus, without data covering the temporal dimension for both Cl− and Clorg separately, along with relevant environmental variables in sediment, water, soil and biomass pools, and without information of how important processes (e.g. chlorination and dechlorination) are regulated, long-term dynamic models of Cl-cycling largely remain guesswork. Hence, we still depend on very fundamental and partly descriptive studies of Cl in multiple environments to reach a deeper understanding about the terrestrial Cl cycling. Such studies need to consider Cl− and Clorg separately in as much detail as possible and should preferably be linked to simultaneous studies of water and other element cycles and biological activity at the same locations.
Data availability
All data generated or analysed during this study are included in this published article.
References
Åkesson Nilsson G (2004). Determination of chlorinated fatty acids using SPE, XSD and GC/MS with particular regard to cultured human cells, PhD Dissertation, Swedish University of Agricultural Sciences. PhD
Albers CN, OS Jacobsen, et al. (2010). Spatial variation in natural formation of chloroform in the soils of four coniferous forests Biogeochemistry
Albers CN, Jacobsen OS, Flores EMM, Johnsen AR (2017) Arctic and subarctic natural soils emit chloroform and brominated analogues by alkaline hydrolysis of trihaloacetyl compounds. Environ Sci Technol 51:6131–6138
Alonso MB, Eljarrat E, Gorga M, Secchi ER, Bassoi M, Barbosa L, Bertozzi CP, Marigo J, Cremer M, Domit C, Azevedo AF, Dorneles PR, Torres JPM, Lailson-Brito J, Malm O, Barceló D (2012) Natural and anthropogenically-produced brominated compounds in endemic dolphins from Western South Atlantic: another risk to a vulnerable species. Environ Pollut 170:152–160
Apel K, Hirt H (2004) Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol 55:373–399
Asplund G, Grimvall A (1991) Organohalogens in nature. More widespread than prevoiusly assumed. Environ Sci Technol 25:1347–1350
Atashgahi S, Liebensteiner M et al (2018) Microbial synthesis and transformation of inorganic and organic chlorine compounds. Front Microbiol 9:3079
Aulenta F, Pera A, Rossetti S, Petrangeli Papini M, Majone M (2007) Relevance of side reactions in anaerobic reductive dechlorination microcosms amended with different electron donors. Water Res 41:27–38
Aze T, Fujimura M, Matsumura H, Masumoto K, Nakao N, Matsuzaki H, Nagai H, Kawai M (2007) Measurement of the production rates of 36Cl from Cl, K, and Ca in concrete at the 500 MeV neutron irradiation facility at KENS. J Radioanal Nucl Chem 272:491–494
Bäckström M, Karlsson S, Bäckman L, Folkeson L, Lind B (2004) Mobilisation of heavy metals by deicing salts in a roadside environment. Water Res 38:720–732
Bar-On YM, Phillips R, Milo R (2018) The biomass distribution on Earth. Proc Natl Acad Sci U S A 115:6506–6511
Bastviken D, Sandén P, Svensson T, Ståhlberg C, Magounakis M, Öberg G (2006) Chloride retention and release in a boreal forest soil – effects of soil water residence time and nitrogen and chloride loads. Environ Sci Technol 40:2977–2982
Bastviken D, Thomsen F, Svensson T, Karlsson S, Sandén P, Shaw G, Matucha M, Öberg G (2007) Chloride retention in forest soil by microbial uptake and by natural chlorination of organic matter. Geochim Cosmochim Acta 71:3182–3192
Bastviken D, Svensson T, Karlsson S, Sandén P, Öberg G (2009) Temperature sensitivity indicates that chlorination of organic matter in forest soil is primarily biotic. Environ Sci Technol 43:3569–3573
Bengtson P, Bastviken D, de Boer W, Öberg G (2009) Possible role of reactive chlorine in microbial antagonism and organic matter chlorination in terrestrial environments. Environ Microbiol 11:1330–1339
Bengtson P, Bastviken D, Öberg G (2013) Possible roles of reactive chlorine II: assessing biotic chlorination as a way for organisms to handle oxygen stress. Environ Microbiol 15:991–1000
Berg M, Müller S et al (2000) Concentrations and mass fluxes of chloroacteci acids and trifluoroacetic acid in rain and natural waters in Switzerland. Environ Sci Technol 34:2675–2683
Bergbauer M, Eggert C (1994) Degradability of chlorine-free bleachery effluent lignins by two fungi - effects on lignin subunit type and on polymer molecular-weight. Can J Microbiol 40:192–197
Bertills U (1995) Grundvattnets kemi i Sverige. Solna, Naturvårdsverket
Bidleman T, Andersson A et al (2019) A review of halogenated natural products in Arctic, subarctic and Nordic ecosystems. Emerging Contaminants 5:89–115
Biester H, Keppler F, Putschew A, Martinez-Cortizas A, Petri M (2004) Halogen retention, organohalogens, and the role of organic matter decomposition on halogen enrichment in two Chilean peat bogs. Environ Sci Technol 38:1984–1991
Björn H (1999). Uptake, turnover and distribution of chlorinated fatty acids in aquatic biota. PhD Dissertation, PhD Dissertation, Lund University. PhD.
Campbell K, Wolfsberg A et al (2003) Chlorine-36 data at Yucca Mountain: statistical tests of conceptual models for unsaturated-zone flow. J Contam Hydrol 62-3:43–61
Cape JN, Forczek ST et al (2006) Progress in understanding the sources, deposition and above-ground fate of trichloroacetic acid. Environ Sci Pollut Res 13:276–286
Cartwright I, Cendón D, Currell M, Meredith K (2017) A review of radioactive isotopes and other residence time tracers in understanding groundwater recharge: possibilities, challenges, and limitations. J Hydrol 555:797–811
Clarke N, Fuksova K et al (2009) The formation and fate of chlorinated organic substances in temperate and boreal forest soils. Environ Sci Pollut Res 16:127–143
Clemmensen KE, Bahr A, Ovaskainen O, Dahlberg A, Ekblad A, Wallander H, Stenlid J, Finlay RD, Wardle DA, Lindahl BD (2013) Roots and associated fungi drive long-term carbon sequestration in boreal forest. Science 339:1615–1618
Dembitsky VM, Srebnik M (2002) Natural halogenated fatty acids: their analogues and derivatives. Prog Lipid Res 41:315–367
Dimmer CH, Simmonds PG, Nickless G, Bassford MR (2001) Biogenic fluxes of halomethanes from Irish peatland ecosystems. Atmos Environ 35:321–330
Dolfing J (2000) Energetics of anaerobic degradation pathways of chlorinated aliphatic compounds. Microb Ecol 40:2–7
Dugan HA, Bartlett SL, Burke SM, Doubek JP, Krivak-Tetley FE, Skaff NK, Summers JC, Farrell KJ, McCullough IM, Morales-Williams AM, Roberts DC, Ouyang Z, Scordo F, Hanson PC, Weathers KC (2017) Salting our freshwater lakes. Proc Natl Acad Sci U S A 114:4453–4458
Dugat-Bony E, Peyret P, Biderre-Petit C (2016) New insights into the microbial contribution to the chlorine cycle in aquatic ecosystems. In: Sime-Ngando T, Boivin P, Chapron E, Jezequel D, Meybeck M (eds) Lake Pavin. Springer, Cham
Edwards I, Kalra Y, Radford F (1981) Chloride determination and levels in the soil-plant environment. Environ Pollut B Chem Phys. 2:109–117
Enell M, Wennberg L (1991) Distribution of halogenated organic-compounds (AOX) - Swedish transport to surrounding sea areas and mass balance studies in 5 drainage systems. Water Sci Technol 24:385–395
Engvild KC (1994) The chloroindole auxins of pea, strong plant growth hormones or endogenous herbicides? Risø National Laboratory
Epp T, Neidhardt H et al (2020) Vegetation canopy effects on total and dissolved Cl, Br, F and I concentrations in soil and their fate along the hydrological flow path. Sci Total Environ 712:135473
Eriksson E (1955) Air borne salts and the chemical composition of river waters. Tellus 7:243–250
Eriksson E (1960) The yearly circulation of chloride and sulfur in nature; metoerological, geochemical and pedological implications. Part II Tellus 12:63–109
Ewald G (1998) Chlorinated fatty acids - environmental pollutants with intriguing properties. Chemosphere 37:2833–2837
Fahimi IJ, Keppler F, Schöler HF (2003) Formation of chloroacetic acids from soil, humic acid and phenolic moieties. Chemosphere 52:513–520
Fetzner S (1998) Bacterial dehalogenation. Appl Microbiol Biotechnol 50:633–657
Flodin C, Johansson E, Borén H, Grimvall A, Dahlman O, Mörck R (1997) Chlorinated structures in high molecular weight organic matter isolated from fresh and decaying plant material and soil. Environ Sci Technol 31:2464–2468
Forczek S, Benada O et al (2011) Influence of road salting on the adjacent Norway spruce (Picea abies) forest. Plant Soil Environ 57:344–350
Frank H (1991) Chlorocarbons, photooxidants, and forest decline. Ambio 20:13–18
Frank H, Frank W, Thiel D (1989) C1-halocarbons and C2-halocarbons in soil-air of forests. Atmos Environ 23:1333–1335
Frechou C, Degros JP (2005) Measurement of Cl-36 in nuclear wastes and effluents: validation of a radiochemical protocol with an in-house reference sample. J Radioanal Nucl Chem 263:333–339
Gielen S, Batlle J et al (2016) Concentrations and distributions of Al, Ca, Cl, K, Mg and Mn in a Scots pine forest in Belgium. Ecol Model 324:1–10
Godduhn A, Duffy LK (2003) Multi-generation health risks of persistent organic pollution in the far north: use of the precautionary approach in the Stockholm Convention. Environ Sci Pol 6:341–353
Graedel TE, Keene WC (1996) The budget and cycle of earth´s natural chlorine. Pure Appl Chem 68:1689–1697
Gribble G (2003) The diversity of naturally produced organohalogens. Chemosphere 52:289–297
Gribble G (2010) Naturally occurring organohalogen compounds - a comprehensive update. Springer, Wien
Gribble G (2015) A recent survey of naturally occurring organohalogen compounds. Environ Chem 12:396–405
Grimvall A, Borén H, Jonsson S, Karlsson S, Sävenhed R (1991) Organohalogens of natural and industrial origin in large recipients of bleach-plant effluents. Water Sci Technol 24:373–383
Grön C (1995). AOX in groundwater. Naturally-produced organohalogens. A. Grimvall and E. de Leer. Dordrecht, Kluwer academic publishers
Gustavsson M, Karlsson S, Öberg G, Sandén P, Svensson T, Valinia S, Thiry Y, Bastviken D (2012) Organic matter chlorination rates in different boreal soils: the role of soil organic matter content. Environ Sci Technol 46:1504–1510
Håkansson H, Sundin P, Andersson T, Brunström B, Dencker L, Engwall M, Ewald G, Gilek M, Holm G, Honkasalo S, Idestam-Almquist J, Jonsson P, Kautsky N, Lundberg G, Lund-Kvernheim A, Martinsen K, Norrgren L, Personen M, Rundgren M, Stålberg M, Tarkpea M, Wesén C (1991) In vivo and in vitro toxicity of fractionated fish lipids, with particular regard to their content of chlorinated organic compounds. Pharmacol Toxicol 69:459–471
Haselmann KF, Laturnus F, Grøn C (2002) Formation of chloroform in soil -a year-round study at a Danish spruce forest site. Water Air Soil Pollut 139:35–41
Hellen H, Hakola H et al (2006) C-2-C-10 hydrocarbon emissions from a boreal wetland and forest floor. Biogeosciences 3:167–174
Herczeg AL, Leaney FW (2011) Review: environmental tracers in arid-zone hydrology. Hydrogeol J 19:17–29
Hintz WD, Relyea RA (2019) A review of the species, community, and ecosystem impacts of road salt salinisation in fresh waters. Freshw Biol 64:1081–1097
Hjelm O, G Asplund (1995) Chemical characterization of organohalogens in a coniferous forest soil. Naturally-produced organohalogens. A. Grimvall and E. W. B. d. Leer, Kluwer Academic Publishers: 105-111.
Hoekstra E, de Leer E et al (1998a) Natural formation of chloroform and brominated trihalomethanes in soil. Environ Sci Technol 32:3724–3729
Hoekstra E, Verhagen F et al (1998b) Natural production of chloroform by fungi. Phytochemistry 49:91–97
Hoekstra EJ, Duyzer JH, de Leer EWB, Brinkman UAT (2001) Chloroform -concentration gradients in soil air and atmospheric air, and emission fluxes from soil. Atmos Environ 35:61–70
Hou XL, Ostergaard LF et al (2007) Determination of Cl-36 in nuclear waste from reactor decommissioning. Anal Chem 79:3126–3134
Hruska J, Oulehle F et al (2012) Long-term forest soil acidification, nutrient leaching and vegetation development: linking modelling and surveys of a primeval spruce forest in the Ukrainian Transcarpathian Mts. Ecol Model 244:28–37
Jiao Y, Ruecker A (2018) Halocarbon emissions from a degraded forested wetland in coastal South Carolina impacted by sea level rise. ACS Earth Space Chem 2:955–967
Johansson E, Krantz-Rulcker C et al (2000) Chlorination and biodegradation of lignin. Soil Biol Biochem 32:1029–1032
Johansson E, Ebenå G, Sandén P, Svensson T, Öberg G (2001) Organic and inorganic chlorine in Swedish spruce forest soil: influence of nitrogen. Geoderma 101:1–13
Johansson E, Sandén P, Öberg G (2003a) Organic chlorine in deciduous and coniferous forest soil in southern Sweden. Soil Sci 168:347–355
Johansson E, S P, et al. (2003b) Spatial patterns of organic chlorine and chloride in Swedish forest soil. Chemosphere 52:391–397
Jonsson P (1992). Large-scale changes of contaminants in Baltic Sea sediments during the twentieth century. Acta Univ. Ups., , Uppsala University. PhD
Kashparov V, Colle C, Levchuk S, Yoschenko V, Svydynuk N (2007a) Transfer of chlorine from the environment to agricultural foodstuffs. J Environ Radioact 94:1–15
Kashparov V, Colle C, Levchuk S, Yoschenko V, Zvarich S (2007b) Radiochlorine concentration ratios for agricultural plants in various soil conditions. J Environ Radioact 95:10–22
Kauffman S, Royer D et al (2003) Export of chloride after clear-cutting in the Hubbard Brook sandbox experiment. Biogeochemistry 63:23–33
Kaushal SS, Groffman PM, Likens GE, Belt KT, Stack WP, Kelly VR, Band LE, Fisher GT (2005) Increased salinization of fresh water in the Northeastern United States. Proc Natl Acad Sci U S A 102:13517–13520
Kelly VR, Lovett GM, Weathers KC, Findlay SEG, Strayer DL, Burns DJ, Likens GE (2008) Long-term sodium chloride retention in a rural watershed: legacy effects of road salt on streamwater concentration. Environ Sci Technol 42:410–415
Keppler F, Eiden R, Niedan V, Pracht J, Schöler HF (2000) Halocarbons produced by natural oxidation processes during degradation of organic matter. Nature 403:298–301
Khalil MAK, Rasmussen RA (2000) Soil-atmosphere exchange of radiatively and chemically active gases. Environ Sci Pollut Res 7:79–82
Khalil MAK, Rasmussen RA, Shearer MJ, Chen ZL, Yao H, Yang J (1998) Emissions of methane, nitroux oxide, and other trace gases from rice fields in China. J Geophys Res 103:25241–25250
Kincaid D, Findlay S (2009) Sources of elevated chloride in local streams: groundwater and soils as potential reservoirs. Water Air Soil Pollut 203:335–342
Kindbohm K, Svensson A et al. (2001). Trends in air concentration and deposition at background monitoring sites in Sweden -major inorganic compounds, heavy metals and ozone. Göteborg, IVL report B1429
Kirchner JW, Feng XH et al. (2000). Fractal stream chemistry and its implications for contaminant transport in catchments. 403: 524-527
Laniewski K, Borén H et al. (1995). Chemical characterization of adsorbable organic halogens (AOX) in precipitation. Naturally-produced organohalogens. A. Grimvall and E. de Leer. Dordrecht, Kluwer Academic Publishers: 113-130
Laniewski K, Borén H, Grimvall A (1999) Fractionation of halogenated organic matter present in rain and snow. Chemosphere 38:393–409
Laturnus F, Lauritsen FR, Grøn C (2000) Chloroform in a pristine aquifer system: toward an evidence of biogenic origin. Water Resour Res 36:2999–3009
Laturnus F, Fahimi I, Gryndler M, Hartmann A, Heal M, Matucha M, Schöler HF, Schroll R, Svensson T (2005) Natural formation and degradation of chloroacetic acids and volatile organochlorines in forest soil - challenges to understanding. Environ Sci Pollut Res 12:233–244
Le Dizès S, Gonze MA (2019) Behavior of 36Cl in agricultural soil-plant systems: a review of transfer processes and modelling approaches. J Environ Radioact 196:82–90
Lee R, Shaw G et al (2001) Specific association of 36Cl with low molecular with humic substances in soil. Chemosphere 43:1063–1070
Leri AC, Myneni SCB (2010) Organochlorine turnover in forest ecosystems: the missing link in the terrestrial chlorine cycle. Glob Biogeochem Cycles 24(4). https://doi.org/10.1029/2010GB003882
Limer L, Albrecht A et al. (2009). Cl-36 dose assessment uncertainties and variability Rep. www.bioprota.com
Löfgren S (2001) The chemical effects of deicing salt on soil and stream water of five catchments in southeast Sweden. Water Air Soil Pollut 130:863–868
Lovett GM, Likens GE, Buso DC, Driscoll CT, Bailey SW (2005) The biogeochemistry of chlorine at Hubbard Brook, New Hampshire, USA. Biogeochemistry 72:191–232
Mannerkoski H, Finér L, Piirainen S, Starr M (2005) Effect of clear-cutting and site preparation on the level and quality of groundwater in some headwater catchments in eastern Finland. For Ecol Manag 220:107–117
Marschner P (2012). Marschner’s mineral nutrition of higher plants, Academic Press.
Matucha M, Rohlenova J et al (2006) Determination of trichloroacetic acid in environmental studies using carbon 14 and chlorine 36. Chemosphere. 63:1924–1932
McCulloch A (2003) Chloroform in the environment: occurrence, sources, sinks and effects. Chemosphere 50:1291–1308
McGuire KM, Judd KE (2020) Road salt chloride retention in wetland soils and effects on dissolved organic carbon export. Chem Ecol 36:342–359
Melkerud P-A, Olsson M et al. (1992). Geochemical atlas of Swedish forest soils, Rapporter i skogsekologi och skoglig marklära nr 65. SLU, Uppsala
Montelius M, Thiry Y, Marang L, Ranger J, Cornelis JT, Svensson T, Bastviken D (2015) Experimental evidence of large changes in terrestrial chlorine cycling following altered tree species composition. Environ Sci Technol 49:4921–4928
Montelius M, Svensson T, Lourino-Cabana B, Thiry Y, Bastviken D (2016) Chlorination and dechlorination rates in a forest soil - a combined modelling and experimental approach. Sci Total Environ 554–555:203–210
Montelius M, Svensson T, Lourino-Cabana B, Thiry Y, Bastviken D (2019) Radiotracer evidence that the rhizosphere is a hot-spot for chlorination of soil organic matter. Plant Soil 443:245–257
Motwani HV, Qui S et al (2011) Cob(I)alamin reacts with sucralose to an alkycobalamin: relevance to in vivo cobalamin and sucralose interaction. Food Chem Toxicol 49:750–757
Mu HL, Ewald G, Nilsson E, Sundin P, Wesén C (2004) Fate of chlorinated fatty acids in migrating sockeye salmon and their transfer to arctic grayling. Environ Sci Technol 38:5548–5554
Müller G (2003) Sense or no-sense of the sum parameter for water soluble “adsorbabale oorganic halogens” (AOX) and “adsorbed organic halogens” (AOX-S18) for the assessment of organohalogens in sludges and sediments. Chemosphere. 52:371–379
Mwevura H, Amir OA, Kishimba M, Berggren P, Kylin H (2010) Organohalogen compounds in blubber of Indo-Pacific bottlenose dolphin (Tursiops aduncus) and spinner dolphin (Stenella longirostris) from Zanzibar, Tanzania. Environ Pollut 158:2200–2207
Myneni SCB (2002) Formation of stable chlorinated hydrocarbons in weathering plant material. Science 295:1039–1041
Neidleman SL, Geigert J (1986) Biohalogenation. John Wiley and Sons, Chichester
Nkusi G, Müller G (1995) Naturally produced organohalogens: AOX-monitoring in plants and sediments. In: A. Grimvall and E. W. B. De Leer (eds.) Naturally-Produced Organohalogens
Nyberg L, Rodhe A, Bishop K (1999) Water transit times and flow paths from two line injections of 3H and 36Cl in a microcatchment at Gårdsjön, Sweden. Hydrol Process 13:1557–1575
Öberg G (2002) The natural chlorine cycle - fitting the scattered pieces. Appl Microbiol Biotechnol 58:565–581
Öberg G, Bastviken D (2012) Transformation of chloride to organic chlorine in terrestrial environments: variability, extent, and implications. Crit Rev Environ Sci Technol 42:2526–2545
Öberg G, Nordlund E, Berg B (1996) In situ formation of organically bound halogens during decomposition of Norway spruce needles: effect of fertilization. Can J For Res 26:1040–1048
Öberg G, Johansen C, Grøn C (1998) Organic halogens in spruce forest throughfall. Chemosphere 36:1689–1701
Öberg G, Holm M, Sandén P, Svensson T, Parikka M (2005) The role of organic-matter-bound chlorine in the chlorine cycle: a case study of the Stubbetorp catchment, Sweden. Biogeochemistry 75:241–269
Olivas Y, Dolfing J, Smith GB (2002) The influence of redox potential on the degradation of halogenated methanes. Environ Toxicol Chem 21:493–499
Ortiz-Bermúdez P, Srebotnik E et al (2003) Chlorination and cleavage of lignin structures by fungal chloroperoxidases. Appl Environ Microbiol 69:5015–5018
Paul L, Smolders E (2014) Inhibition of iron (III) minerals and acidification on the reductive dechlorination of trichloroethylene. Chemosphere. 111:471–477
Pausch J, Kuzyakov Y (2018) Carbon input by roots into the soil: quantification of rhizodeposition from root to ecosystem scale. Glob Chang Biol 24:1–12
Peng P, Lu Y et al (2020) Metagenomic- and cultivation-based exploration of anaerobic chloroform biotransformation in hypersaline sediments as natural source of chloromethanes. Microorganisms 8:665
Perera N, Gharabaghi B (2013) Creek watershed due to road salt application: a re-assessment after 20 years. J Hydrol 479:159–168
Peterson J, M MacDonell, et al. (2007). Radiological and chemical fact sheets to support health risk analyses for contaminated areas, Argonne National Laboratory Environmental Science Division
Philippot L, Raaijmakers JM, Lemanceau P, van der Putten WH (2013) Going back to the roots: the microbial ecology of the rhizosphere. Nat Rev Microbiol 11:789–799
Pickering L, Black TA, Gilbert C, Jeronimo M, Nesic Z, Pilz J, Svensson T, Öberg G (2013) Portable chamber system for measuring chloroform fluxes from terrestrial environments - methodological challenges. Environ Sci Technol 47:14298–14305
Poykio R, Nurmesniemi H et al (2008) EOX concentrations in sediment in the part of the Bothnian Bay affected by effluents from the pulp and paper mills at Kemi, Northern Finland. Environ Monit Assess 139:183–194
Pries F, Vanderploeg JR et al (1994) Degradation of halogenated aliphatic compounds - the role of adaption. FEMS Microbiol Rev 15:279–295
Redeker KR, Wang NY, Low JC, McMillan A, Tyler SC, Cicerone RJ (2000) Emissions of methyl halides and methane from rice paddies. Science 290:966–969
Redon PO, Abdelouas A, Bastviken D, Cecchini S, Nicolas M, Thiry Y (2011) Chloride and organic chlorine in forest soils: storage, residence times, and influence of ecological conditions. Environ Sci Technol 45:7202–7208
Redon PO, Jolivet C, Saby NPA, Abdelouas A, Thiry Y (2013) Occurrence of natural organic chlorine in soils for different land uses. Biogeochemistry 114:413–419
Reimann S, Grob K, Frank H (1996) Chloroacetic acids in rainwater. Environ Sci Technol 30:2340–2344
Rhew RC, Miller BR, Weiss RF (2000) Natural methyl bromide and methyl chloride emissions from coastal salt marshes. Nature 403:292–295
Rhew RC, Miller BR et al (2002) Environmental and biological controls on methyl halide emissions from southern California coastal salt marshes. Biogeochemistry 60:141–161
Rhew RC, Teh YA et al (2008) Chloroform emissions from the Alaskan Arctic tundra. Geophys Res Lett 35:5
Robinson H, Hasenmuller E, Chambers L (2017) Soil as a reservoir for road salt retention leading to its gradual release to groundwater. Appl Geochem 83:72–85
Rodriguez M, Pina G et al (2006) Radiochemical analysis of chlorine-36. Czechoslov J Phys 56:D211–D217
Rodstedth M, Ståhlberg C, Sandén P, Öberg G (2003) Chloride imbalances in soil lysimeters. Chemosphere 52:381–389
Rohlenova J, Gryndler M et al (2009) Microbial chlorination of organic matter in forest soil: investigation using 36Cl-chloride and its methodology. Environ Sci Technol 43:3652–3655
Schlesinger WH, Bernhardt E (2020) Biogeochemistry: an analysis of global change, 4th edn. Academic Press, San Diego
Schleyer R (1996) Beeinflussing der Grundwasserqualitaet durch Deposition anthropogener organischer Stoffe aus der Atmosphaere. Schriftenr Ver Wasser Boden Lufthyg 10:247–265
Schleyer R, Renner I et al (1991) Beeinflussung der Grundwasserqualitaet durch luftgetragene organische Schadstoffe. Schriftenr Ver Wasser Boden Lufthyg 5:1–96
Seki R, Matsuhiro T et al (2007) Isotopic ratios of Cl-36/Cl in Japanese surface soil. Nuclear Instruments & Methods in Physics Research Section B-Beam Interactions with Materials and Atoms 259:486–490
Sheppard SC, Johnson LH, Goodwin BW, Tait JC, Wuschke DM, Davison CC (1996) Chlorine-36 in nuclear waste disposal .1. Assessment results for used fuel with comparison to I-129 and C-14. Waste Manag 16:607–614
Silk P, Lonergan G et al (1997) Evidence of natural organochlorine formation in peat bogs. Chemosphere 35:2865–2880
Skladanka J, Adam V, Zitka O, Krystofova O, Beklova M, Kizek R, Havlicek Z, Slama P, Nawrath A (2012) Investigation into the effect of molds in grasses on their content of low molecular mass thiols. Int J Environ Res Public Health 9:3789–3805
Smidt H, de Vos WM (2004) Anaerobic microbial dehalogenation. 58: 43-73.
Stringer R, Johnston P (2001) Chlorine and the environment. Kluwer Academic Publishers, Dordrecht
Suominen KP, Jaakkola T, Elomaa E, Hakulinen R, Salkinoja-Salonen MS (1997) Sediment accumulation of organic halogens in pristine forest lakes. Environ Sci Pollut Res 4:21–30
Svensson T (2019) Measurements and fluxes of volatile chlorinated organic compounds (VOCl) from natural terrestrial sources. Measurement techniques and spatio-temporal variability of flux estimates. Swedish Nuclear Fuel and Waste Management Co. SKB TR-18-09
Svensson T, Laturnus F, Sandén P, Öberg G (2007a) Chloroform in runoff water—a two-year study in a small catchment in Southeast Sweden. Biogeochemistry 82:139–151
Svensson T, Sandén P, Bastviken D, Öberg G (2007b) Chlorine transport in a small catchment in southeast Sweden during two years. Biogeochemistry 82:181–199
Svensson T, Lovett GM et al. (2012). Is chloride a conservative ion in forested ecosystems? Biogeochemistry DOI: 10.1007/s10533-010-9538-y: 125-134
Svensson T, Högbom L et al (2013) Effects of previous nitrogen addition on chlorine in forest soil, soil solution and biomass. Biogeochemistry 116:3–13
Svensson T, Montelius M, Andersson M, Lindberg C, Reyier H, Rietz K, Danielsson Å, Bastviken D (2017) Influence of multiple environmental factors on organic matter chlorination in podsol soil. Environ Sci Technol 51:14114–14123
Tanaka T, Thiry Y (2020) Assessing the recycling of chlorine and its long-lived 36Cl isotope in terrestrial ecosystems through dynamic modeling. Science of the Total Environment 700
Temme H, Carlson A, Novak P (2019) Presence, diversity, and enrichment of respiratory reductive dehalogenase and non-respiratory hydrolytic and oxidative dehalogenase genes in terrestrial environments. Front Microbiol 10:1258
Todd AK, Kaltenecker MG (2012) Warm season chloride concentrations in stream habitats of freshwater mussel species at risk. Environ Pollut 171:199–206
Tröjbom M, Grolander S (2010) Chemical conditions in present and future ecosystems in Forsmark – implications for selected radionuclides in the safety assessment SR-Site, SKB Report SKB R-10-27
van den Hoof C, Thiry Y (2012) Modelling of the natural chlorine cycling in a coniferous stand: implications for chlorine-36 behaviour in a contaminated forest environment. J Environ Radioact 107:56–67
Van Meter RJ, Swan CM et al (2011) Road salt stress induces novel food web structure and interactions. Wetlands 31:843–851
van Pee KH (2001) Microbial synthesis of halometabolites. Arch Microbiol 175:250–258
van Pee KH, Unversucht S (2003) Biological dehalogenation and halogenation reactions. Chemosphere 52:299–312
Varner RK, Crill PM, Talbot RW (1999) Wetlands: a potentially significant source of atmospheric methyl bromide and methyl chloride. Geophys Res Lett 26:2433–2435
Varner RK, White ML et al (2003) Production of methyl bromide in a temperate forest soil. Geophys Res Lett 30:1521
Vereskuns G (1999). Chlorinated fatty acids in freshwater fish and some biological effects of dichloristearic acid, PhD Dissertation, Swedish University of Agricultural Sciences. PhD
Vodyanitskii Y, Makarov M (2017) Organochlorine compounds and the biogeochemical cycle of chlorine in soils: a review. Eurasian Soil Science 50:1025–1032
von Sydow L (1999). Haloacetates in precipitation Linköping, Linköping University. PhD Thesis
Walter A, Caputi L, O’Connor S, van Pée KH, Ludwig-Müller J (2020) Chlorinated auxins—how does Arabidopsis Thaliana deal with them? Int J Mol Sci 21:2567
Wang JX, Qin P, Sun S (2007) The flux of chloroform and tetrachloromethane along an elevational gradient of a coastal salt marsh, East China. Environ Pollut 148:10–20
Weigold P, El-Hadidi M et al (2016) A metagenomic-based survey of microbial (de)halogenation potential in a German forest soil. Sci Rep 6:28958
Weissflog L, Krüger GHJ, Forczek ST, Lange CA, Kotte K, Pfennigsdorff A, Rohlenová J, Fuksová K, Uhlířová H, Matucha M, Schröder P (2007) Oxidative biodegradation of tetrachloroethene in needle of Norway spruce (Picea abies L.). S Afr J Bot 73:89–96
Wever R, Barnett P (2017) Vanadium chloroperoxidases: the missing link in the formation of chlorinated compounds and chloroform in the terrestrial environment? Chemistry and Asian journal 12:1997–2007
White P, Broadley M (2001) Chloride in soils and its uptake and movement within the plant: a review. Ann Bot 88:967–988
Wilander A (1997) Referenssjöarnas vattenkemi under 12 år: tillstånd och trender Stockholm. Naturvårdsverket.
Winterton N (2000) Chlorine: the only green element - towards a wider acceptance of its role in natural cycles. Green Chem 2:173–225
Wuosmaa A, Hager L (1990) Methyl chloride transferase: a carbocation route for biosynthesis of halometabolites. Science 249:160–162
Xu GH, Magen H et al. (2000). Advances in chloride nutrition of plants. Advances in Agronomy, Vol 68. D. L. Sparks. San Diego, Elsevier Academic Press Inc. 68: 97-150
Yang Y, Cápiro N, et al. (2017) Organohalide respiration with chlorinated ethenes under low pH conditions
Yokouchi Y, Toom-Sauntry D, Yazawa K, Inagaki T, Tamaru T (2002) Recent decline of methyl bromide in the troposphere. Atmos Environ 36:4985–4989
Yunos NM, Bellomo R et al (2010) Bench-to-bedside review: chloride in critical illness. Crit Care 14:226
Zlamal J, Raab T et al (2017) Biological chlorine cycling in the Arctic Coastal Plain. Biogeochemistry 134:243–260
Acknowledgements
We would also like to thank Yves Thiry at the Research and Development Division of ANDRA, France, for the useful and appreciated input to the manuscript.
Funding
Open Access funding provided by Linköping University. Financial support was received from the Swedish Nuclear Fuel and Waste Management Co (SKB).
Author information
Authors and Affiliations
Contributions
TS, HK, MM, PS and DB have contributed to the text development and have given approval to the final version of the manuscript. TS led the work and DB shaped an early version of the text.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Responsible Editor: Kitae Baek
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Svensson, T., Kylin, H., Montelius, M. et al. Chlorine cycling and the fate of Cl in terrestrial environments. Environ Sci Pollut Res 28, 7691–7709 (2021). https://doi.org/10.1007/s11356-020-12144-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-020-12144-6