
Abstract
The crystal and molecular structures of 3-(N-methylamino)-2-nitropyridine, 5-(N-methylamino)-2-nitropyridine and 2-(N-methylamino)-5-nitropyridine have been characterized by X-ray diffraction. To perform conformational analysis, the geometries of the compounds as well as their conformers and rotamers were optimized at the B3LYP/6-311++G(3df,3pd) level. The resulting data were used to analyze the π-electron delocalization effect in relation to the methylamino group rotation in ortho-, meta- and para-substitution positions. Quantitative aromaticity indices were calculated based on which we estimated the electronic structures of the analyzed compounds. The substituent effect of the methylamino and nitro groups was also characterized by related descriptors, i.e., charge of the Substituent Active Region (cSAR(X)) calculated based on the Hirshfeld charges and Substituent Effect Stabilization Energy (SESE). It has been shown that all the used parameters were found to be mutually interrelated with much better correlations for the meta- and para- than the ortho-derivatives. The rotation of the methylamino group relative to the aromatic fragment has an effect on the change in delocalization of the π-electrons of the pyridine ring. Relations between cSAR(NHCH3) and cSAR(NO2) show almost identical sensitivity of the substituent effect in meta- and para-substituted derivatives, whereas in ortho-substituted analogs, different kinds of intramolecular interactions have been revealed. Furthermore, the analyzed dependency of SESE values on the torsional angle of the methylamino group indicates that an increase in the electron-attracting power of the substituent leads to a decrease in energy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The study of substituted nitropyridines attracts the attention of many researchers due to their wide application in organic synthesis and their use as bioactive agents in pharmaceuticals [1, 2]. It has been reported that the inhibitory properties of 3-nitropyridine derivatives and their salts can be used as therapeutic or preventive agents for hepatitis B and acquired immune deficiency syndrome (AIDS) [3]. Some non-methylated and methylated 4-nitropyridine N-oxide derivatives are particularly interesting due to their antifungal activity [4, 5] and the non-linear second-order response found in their acentric crystals [6, 7]. At the same time, N-substituted derivatives of 2-amino-5-nitropyridine exhibit promising non-linear optical properties in the crystalline state [8,9,10]. These molecules possess high molecular hyperpolarizability and a highly delocalized π-electron system bearing an electron donor, an amino group, and a nitro group as an electron acceptor. Substituents in such positions provide a pathway to intramolecular charge transfer.
Quantum chemical calculations are widely used to interpret, understand, and predict experimental data such as X-ray geometrical parameters, electronic properties, and the nature of aromatic compounds. The elucidation of the structural and electronic properties of the studied compounds is an important step towards understanding the mechanism of their biological activity. In view of the above, we present herein the characterization of three compounds, i.e., 3-(N-methylamino)-2-nitropyridine, 5-(N-methylamino)-2-nitropyridine, and 2-(N-methylamino)-5-nitropyridine. The analyzed compounds contain substituents of different electronic natures: electron acceptor (EA) and electron donor (ED). The first—nitro group—belongs to one of the most electron-accepting substituents and hence it is commonly regarded as a substituent or a functional group. A nitro group is very electronegative [11], and as a consequence, its strongly inductive effect influences the substituted molecule. This group exhibits a great range of variability of its electron-accepting properties [12] with σp+ = 0.79 and σp− = 1.27 [13], which dramatically depends on the kind of a moiety which the group is attached to [14]. The second substituent (methylamino group) acts as a π-donor, allowing for electronic stabilization of the electron-deficient carbon atom, and its resonance parameters are σp+ = − 1.81 [13]. Moreover, as showed by previous papers, higher stability of the tautomeric states could also be observed in systems with intramolecular hydrogen bonds, which contribute to the formation of six-membered quasi-rings. Therefore, we have evaluated the rotation effects of the methylamino group on electronic structures in the pyridine ring [15].
The literature contains studies reporting on the substituent effect (SE) of disubstituted pyridine derivatives. Gurzyński et al. investigated the effect of a solvent on the acidity constants in systems involving four disubstituted 4-nitropyridines. The values of acidity constants vary in non-aqueous solvents depending on the substituent effects [16]. Hamid et al. used DFT analysis to study the influence of the substituent effect on the efficacy of electron-donating in the pyridine ring. The results indicated that various substituents can exert pronounced and beneficial effects on the charge density enrichment of the pyridyl nitrogen atom in pyridine. This allows us to formulate a statement that substituent modification (in the form of the number, type or position of substitution) can be employed as a tool for controlling the donation effectiveness of the nitrogen atom in pyridines [17, 18]. In their work, Hęclik and Dobrowolski used the sEDA and pEDA substituent effect descriptors to study disubstituted pyridines. The descriptors structured based on the NBO approach express the number of electrons donated to or withdrawn from σ- and π-electron systems of the ring. As noted for all of the analyzed systems, the charge of a lone electron pair of nitrogen depends on the σ- and π-electron-withdrawing and donating properties of substituents. The electron charge of a lone pair increases as the σ-electron-donating ability of the substituent increases, and it decreases as the π-electron-donating ability increases [19]. However, to the best of our knowledge, and according to literature research, there are no results of quantum chemical calculations that would be based on aromaticity and transition state structures. Hence, this paper presents transition state activation energies and changes to π-electron delocalization of the pyridine ring based on theoretical results.
Predictions of the energy barrier of aromatic methylamino compounds around the C-N bond are of great interest to scientists, as the resulting data convey information regarding intramolecular interactions and the electronic structure of molecules. Internal rotation potentials of a substituent in different aromatic compounds constitute measures of strength of electronic conjugation (which can stabilize the planar form) relative to steric effects (which can favor the less crowded perpendicular conformation). The objective of this study is to provide theoretical predictions of the barriers and torsional potentials of substituted nitropyridines. The methylamino group substituent is in ortho-, meta-, and para- positions (Fig. 1). In general, a nitro group is coplanar with the pyridine ring. The presence of ortho-substituents may force the nitro group to rotate out of the plane. Intramolecular hydrogen bonding effects are also significant in methylaminonitropyridines. Owing to different conjugated, resonance, and intramolecular hydrogen bonding effects, the internal rotational barriers of the methylamino group ought to have different values. The torsional potentials of the methylamino group for all of the compounds were evaluated by internal rotation of the methylamino group from 0 to 180° with a 45° increment.

Chemical diagrams of the rotamers of 3-(N-methylamino)-2-nitropyridine [1(0)], 5-(N-methylamino)-2-nitropyridine [3(180)] and 2-(N-methylamino)-5-nitropyridine [6(0)]. The torsion angle values of the methylamino group are provided in brackets
Experimental
Synthesis
Compounds 1(0) and 3(180) were synthesized in line with the method that was already provided in the literature [20]. A solution of nitramine in chlorobenzene was maintained at 120 °C for 2 h. The mixture was added to a cooled solution of silica gel and hexane, and subsequently chromatographed with the use of flash chromatography. 1(0) was first eluted with the benzene-hexane mixture as the eluent. The solution was evaporated and the residue was crystallized from heptane. 3(180) was eluted with benzene.
Compound 6(0) was obtained by heating a solution containing 2-chloro-5-nitropyridine in an aqueous solution of methylamine in ethanol. After cooling, the precipitate was filtered and crystallized from methanol.
X-ray crystallography
Single crystals of 1(0), 3(180), and 6(0) were selected directly from the crystallizer. The experimental data were obtained with the use of a CCD Xcalibur diffractometer (graphite monochromatic, MoKα radiation, λ = 0.71073 Å) at 100.0(1) K. Corrections to the Lorentz and polarization factors were applied to reflection intensities. Data collection: CrysAlis CCD, cell refinement and data reduction: CrysAlis RED [21]. Crystal structures were solved by direct methods using SHELXS14 [22]. All non-hydrogen atoms were located based on the difference Fourier synthesis and refined by the least squares method in full-matrix anisotropic approximation using SHELXL14 software [23]. Hydrogen atoms were determined based on geometric concepts and treated as riding on the parent atom with methyl C-H = 0.96 Å and Uiso (H) = 1.5Ueq(C), N-H = 0.86 Å and Uiso(H) = 1.2Ueq(N), as well as CAr-H = 0.93 Å and Uiso(H) = 1.2Ueq(C). The crystallographic data for the compound and details of X-ray experiment are provided in Table 1. The structures were deposited in the Cambridge Crystallographic Data Center (CCDC), no. CCDC 1895970 1(0), 1948113 3(180), 1895969 6(0).
Theoretical calculations
All of the analyzed structures were optimized with the use of the Gaussian 09 program [24]. All calculations were made using the B3LYP/6-311++G(3df,3pd) [25,26,27,28,29] method. In all compounds, the methylamino group was gradually twisted by 45° starting from 0 up to 180°. For all rotamers (except 1(0), 3(180), and 6(0) since these stayed strictly planar and of Cs symmetry after optimization), the torsion angle was frozen. The vibrational frequencies were calculated at the same level of theory to obtain zero-point energy (ZPE) corrections.
The influence of the substituent effect on the aromaticity of transmitting moiety (pyridine ring) was described using a geometric aromaticity index called HOMA (harmonic oscillator model of aromaticity) with the following equation:
where n is the total number of bonds in the molecule and αi is the normalization constant (for C-C bonds α = 257.7, Ropt = 1.388 Å for C-N bonds α = 93.52, Ropt = 1.334 Å) fixed to give HOMA = 0 for a model of non-aromatic system and HOMA = 1 for a system with all bonds equal to the optimal value Ropt,i, assumed to be realized for fully aromatic systems [30,31,32]. The nucleus-independent chemical shift (NICS) is a descriptor of aromaticity from the magnetic point of view. An index is defined as a negative value of the absolute magnetic shielding computed at ring centers, NICS(0), and 1 Å above the center of the ring, NICS(1) [33, 34] and its zz-tensor component, NICS(1)zz, where the z-axis is normal to the plane of the molecule [35].
The substituent effect of a group with a different electronic character in relation to the pyridine ring can be explained using the idea of Substituent Effect Stabilization Energy (SESE), which is a measure of change to the stability of a system due to substituent interactions [36]. This may be accounted for by the following set of homodesmotic reactions (Fig. 2).

Homodesmotic reaction used to obtain a SESE parameter
The charge of the Substituent Active Region (cSAR(X)) [37, 38] is defined as a sum of total charges on all atoms of substituent X and the charge on the ipso-carbon atom:
All cSAR values were calculated with the use of the Hirshfeld method of the atomic charge assessment [39].
Results and discussion
Experimental vs theoretical structures
The X-ray structures of 1(0), 3(180), and 6(0) are presented in Fig. 3 showing the atom numbering scheme.

Molecular structure of the analyzed compounds (a) 1(0), (b) 3(180), (c) 6(0) with displacement ellipsoids drawn at a 50% probability level
Our discussion of the crystal structure of 1(0), 3(180), and 6(0) is limited to the most important structural parameters and hydrogen bonding geometry. The selected bond lengths, valence, and torsion angles are shown in Table S1. The title compounds consist of a pyridine ring, a nitro group, and a methylamino group. The geometry of the rings is typical and consistent with other nitropyridines [40,41,42].
Compound 1(0) crystallizes with two independent molecules, A and B, in an asymmetric unit. In all three compounds, the aromatic rings are deformed due to the resonance effect and additionally in the case of 1(0)—due to steric hindrance between the nitro and methylamino groups. For all compounds, the distortion is consistent with the position of the N-methylamino group, which is planar with a relatively short C1-N1 bond. The bond in 3(180) is slightly longer in comparison with 1(0) and 6(0), demonstrating a smaller π-donating effect of the amine nitrogen atom against the aromatic ring. The valence angles around atom N10 indicate sp2 hybridization for all molecules, therefore the methylamino group in 6(0) is coplanar with the aromatic ring, whereas in 1(0) and 3(180) the values of dihedral angles do not exceed 2°. Also, the nitro groups are twisted by only 3.9(4)° and 2.8(3)° for 1(0) and 3(180), respectively. Similar effects are observed for the geometries obtained from quantum-mechanical calculations for isolated molecules (Table S1). The largest deviation of the bond length is 0.036 Å [1(0)], 0.024 Å [3(180)] for C-NO2 bonds and 0.028 Å [6(0)] for C-NHCH3. The biggest differences between the values of torsion angles concern C2-C3-N10-C11 in 1(0) (Δ = 1.8o) which describes a twist of the C-N bond of the methylamino group. The remaining values do not differ from those in the crystal structure. The differences between experimental and calculated values may be a result of the packing effect in the crystals as the nitro groups interact with other molecules in the lattice, forming rather strong hydrogen bonds, while the optimization is limited only to monomers. Figure 4 presents crystal packing of molecules viewed along the b-axis with selected hydrogen bonds, while Table 2 shows all possible hydrogen bonds.

Crystal packing of the analyzed compounds (a) 1(0), (b) 3(180), and (c) 6(0) viewed along the b-axis
The close correlation between experimental and calculated geometries is obvious (Fig. 5). The atom positions which most differ are those for hydrogen atoms. In addition, the calculated rotamer energies correctly indicate that the most stable systems are those determined in the crystal. As a result, the level of theory B3LYP/6-311++G(3df,3pd) can be used to analyze the effect of the rotation of the methylamino group on π-electron delocalization in the pyridine ring.

Overlay diagrams of solid state and calculate molecular structures of (a1 and a2)1(0), (b) 3(180) and (c) 6(0). The RMSD values amounts a1 = 0.0231; a2 = 0.0403; b = 0.0314; c = 0.0225. Calculated structure in red, solid state structure in blue
Energy of internal rotation
To better understand the influence of the methylamino group’s internal rotation on the aromaticity of nitropyridine compounds, we analyzed the energy of systems 1–6. The calculated torsional potential of the methylamino group based on ZPE for 1–6 is presented in Table S2.
According to calculations, molecules with the methylamino group next to pyridine nitrogen have lower energy. This effect is the most noticeable in the energy difference in the para-substituted aromatic compounds with substituents possessing opposite electronic properties, due to their ability to quinonoid structure formation (6).
As expected, the highest energy values are observed for ortho- isomers when the torsional angle reaches 90° and 180° (Fig. 6). The results obtained in the present study indicate that the most stable conformations of meta- and para- isomers are those in which the methylamino group is coplanar with the pyridine ring (torsion angle of 0° and 180°). The maximum energy is observed for the structures with the methylamino group perpendicular to the pyridine ring. Indeed, the rotation barrier calculated as the energy difference between ϕ = 0° and ϕ = 90° ranges between 7.36 and 15.41 kcal/mol, depending on whether the substituent is attached to the pyridine ring. Based on the above discussion, the lowest internal rotation barrier, the C-NHCH3 bond, is in 3, while the largest barrier is observed in 4.

Energy relations between various rotamers of the analyzed compounds
Aromaticity
To analyze the effect of substituent rotation on the π-electron delocalization change in the pyridine ring, we used HOMA and NICS(1)zz (Table 3). Additionally, the two different characteristics: cSAR and SESE, were also considered. Since all of these characteristics are applied in the description of π-electron delocalization further in this paper, the relationships between them ought to be analyzed.
The results reveal the best tendency between HOMA and NICS(1)zz. For all of the considered relations (for ortho-, meta-, and para-), R2 is greater than or equal to 0.800 (Fig. 7, Table S3). Very good tendencies in other parameters are observed only for meta- and para-systems, whereas significantly worse or no correlation is found in ortho-systems.

Contour map of the R2 distributions for the obtained dependences in 1–6 compounds
The substitution in the meta-position has the smallest influence on the aromaticity index –HOMA values in 5 are greater than 0.958 in all rotamers. The largest decrease in aromaticity is observed in the ortho-position. The values amount to 0.874 for 1(0) and 0.909 for 4(0). As suggested by a bond length analysis, these effects may be influenced by the contribution of a quinonoid-like structure in the mesomeric resonance hybrids (structures) of the aromatic ring and the existence of an intramolecular Resonance-Assisted Hydrogen Bond (RAHB) [43](N1-H1…O1) in 1(0) and 4(0). Such an effect has already been described and analyzed [44], since there is a strong interrelation between π-electron delocalization and the strength of the hydrogen bonding, forming an additional quasi-aromatic ring.
The change in aromaticity results from the pyramidal conformation of the methylamino group (Fig. 8). Pyramidalization of a methylamino group is caused by a combination of two opposing interactions: the stabilizing interaction between the free electron pair of a nitrogen atom and the π-electron density over the aromatic system and the tendency of the nitrogen atom to adopt sp3 hybridization. A stronger interaction of the free electron pair of a nitrogen atom with π-electron cloud causes “flattening” of the methylamino group. In the case of systems with a 90° twist of the methylamino group, the rotation weakens interaction.

Relationships between HOMA and a torsional angle for the rotamers of methylaminonitropyridine derivatives
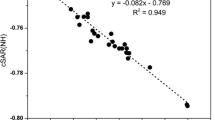
It should be noted that cSAR(X) describes a local donor/acceptor property of the chemical group X directly. As already documented [45, 46] the magnitude of cSAR(X) depends strongly on the nature of the molecular moiety to which X is attached. This characteristic represents an actual charge in the active region of a substituent and describes the amount of charge accepted from (or given back to) the moiety to which the substituent is bound. For derivatives 2, 3, 5, and 6, the tendency between cSAR(NO2) and cSAR(NHCH3) is shown in Fig. 9. The correlations range from 0.929 to 0.991 which means that the regressions are reliable. For 1 and 4, on the other hand, there is no apparent tendency. The magnitude of cSAR(NO2) denotes that the more negative its value, the higher the attraction of the electron by the nitro group. The slopes for all of the series are negative, indicating that an increase in the donor power of the substituent methylamino group is associated with an increase in the acceptor power of the nitro group, which becomes more negative. Thus, the slope value in the equation is a numerical estimation of the communication between the ED power of the methylamino group and the EA power of the nitro group. For para-substituted nitropyridines, the slopes are − 0.4593 and − 0.4964. Thus, it can be concluded that compound 3 is only slightly less sensitive to EA/ED properties of the substituents than compound 6.

Relationships between cSAR(NO2) and cSAR(NHCH3) for the rotamers of methylaminonitropyridine derivatives
SESE is an energetic characteristic of substituent effects, which takes into consideration all kinds of intramolecular interactions such as resonance, inductive (via bonds), and field effects. For cases where the substituent effect stabilizes the analyzed system, SESE values are positive. Figure 10 and Table S4 presents the dependency of SESE values on the torsional angle of the methylamino group in the analyzed compounds. The range of SESE values amounts from 1.09 kcal/mol in 2 up to 11.74 kcal/mol in 4, indicating that the values of SESE strongly depends on the position of both the functional groups. Moreover, taking into account the SESE ranges for rotamers, they can also vary significantly up to 10.91 kcal/mol for compounds with methylamino groups twisted by 180°. In general, the greatest range of SESE values is observed for ortho-compounds (8.31 kcal/mol for 1 and 11.74 kcal/mol for 4). The obtained SESE values reveal both, stabilizing (4.88 kcal/mol for 1(0)) and destabilizing (− 6.86 kcal/mol for 4(180)) substituent effects when rotating a methylamino group. The destabilization of the system results from the breaking of the intramolecular resonance-assisted hydrogen bond during the rotation methylamino group and thus the steric hindrance between substituents. Interplay between RAHB and aromaticity of the ortho-substituted compounds has been recently examined. The results showed that electron-donating substituents can enhance strength of intramolecular hydrogen bonding interactions in studied compounds [47].

Relationships between SESE and the torsional angle for the rotamers of methylaminonitropyridine derivatives
Interestingly, when looking for a direct dependency between SESE and the values of HOMA aromaticity indices, we find a close linear tendency. Thus, regularity emerges: for larger SESE values (expressing more stabilization due to an interaction between the substituent and the ring), the ring is more delocalized.
The application of SESE and comparison with cSAR allows us to determine how the electron structure in the pyridine ring in the analyzed compounds depends on the rotation of the substituent. The regressions of cSAR(NO2) plotted against SESE (Fig. 11) show that the ratio of the slopes for meta- and para- derivatives is approx. 1 and for ortho- derivatives, approx. 5. This means that the nitro group in 4 is less sensitive to the rotation of the substituent than 1 and in consequence the pyridine ring in 4 is a worse transmitter. This may result from the fact that cSAR(X) describes rather local electron properties of substituents, whereas SESE describes its global effect within the molecule.

Relationships between cSAR(NO2) and SESE for the rotamers of methylaminonitropyridine derivatives
Conclusion
The molecular geometries and the internal rotational barriers of the methylamino group for nitropyridines are calculated with the application of a DFT method at the B3LYP/6-311++G(3df,3pd) level. The analysis of the geometries indicates that 3(180) and 6(0) are planar molecules. Intramolecular hydrogen bonds are observed in 1(180) and 4(180). The nitro groups in these compounds are twisted due to the steric effect. The calculated internal rotational barriers of the methylamino group of all of the analyzed molecules depend on the rotation and position of the substituent, except for 4 (due to steric hindrance between two neighboring groups). The clear effect of substituent rotation on the aromatic ring is observed in all of the compounds. Despite a different physical background, quantum chemistry–based HOMA, NICS1(zz), SESE, and cSAR(X) exhibit mutual tendencies in most cases. The best tendencies are always found for the meta- and para-substituted systems, whereas for the ortho-substituted derivatives the tendencies are worse. Whether measured by HOMA or NICS(1zz), an increase in ED properties induces a decrease in the aromatic character of the ring. Furthermore, the dependencies of cSAR(NO2) on cSAR(NHCH3) demonstrate the intensity of communications between NO2 and NHCH3. The slopes for all of the series are negative, indicating that an increase in the donor power of the substituent methylamino group is associated with an increase in the acceptor power of the nitro group. Moreover, the analyzed dependency of SESE values on the torsional angle of the methylamino group indicates that an increase in the electron-attracting power of the substituent leads to a decrease in energy, i.e., destabilization of ortho-compounds. In conclusion, the results of this study indicate that SESE and cSAR characteristics can be used for designing changes in π-electron delocalization patterns with the pyridine ring.
References
Hayes C, Katz MA, Davane JG, Hsieh JTC, Wolfe FDL, Potter PJ, Blight AR (2003) J Clin Pharmacol 43:379–385
Camp H, Derc J (2001) Handbook of Am Chem Soc
Yoon SJ, Lee SE, Kim ND, Park YK, Lee GH, Kim JW, Park SJ, Park HJ, Shin DH (2004) US Pat 6:795
Fukui K, Imamura A, Nagata C (1960) Bul Chem So Jpn 33:122–123
Okabayashi IT (1953) J Ferment Technol 31:373–375
Bertier G, Defranceschi M, Lazaretti P, Tsoucaris G, Zanasi R (1992) J Mol Struct 254:205–218
Zyss J, Chemla DS, Nicoud JF (1981) J Chem Phys 74:4800–4811
Munn RW, Smith SPB (1992) Adv Mater Opt Electron 1:65–71
Kondo T, Akase F, Kumagai M, Ito R (1995) Opt Rev 2:128–131
Bosshard C, Sutter K, Gunter P (1989) J Opt Soc Am B6:721–725
Campanelli AR, Domenicano A, Ramondo F, Hargittai I (2004) J Phys Chem A 108:4940–4948
Exner O (1978) In: Chapman NB, Shorter F (eds) Correlation analysis in organic chemistry-recent advances. London, Plenum Press
Hansch C, Leo A, Taft RW (1991) Chem Rev 91:165–195
Stasyuk OA, Szatylowicz H, Fonseca Guerra C, Krygowski TM (2015) Struct Chem 26:905–913
Sołtysiak P, Zarychta B, Spaleniak G, Ejsmont K (2019) J Mol Struct 1186:317–324
Gurzyński Ł, Puszko A, Chmurzyński L (2007) J Chem Thermodyn 39:1667–1674
Abdel Hamid A, Kanan S, That Z (2015) Res Chem Intermed 41:6859–6875
Abdel Hamid A (2018) IntechOpen 43-55
Hęclik K, Dobrowolski JC (2017) J Phys Org Chem 30:3656–3667
Daszkiewicz Z, Domański A, Kyzioł JB (1997) Chem Pap 51:22–28
Oxford Diffraction (2008) CrysAlis CCD and CrysAlis RED Versions 1.171.32.29 Oxford Diffraction Ltd. Abingdon. England
Sheldrick GM (2015) Acta Cryst A71:3–8
George MS (2015) Crystal structure refinement with . Acta Crystallographica Section C Structural Chemistry 71 (1):3–8
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2010) Gaussian 09, revision D.01, Gaussian, Inc., Wallingford CT
Becke AD (1988) Phys Rev A38:3098–3100
Becke AD (1993) J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Phys Rev B Condens Matter 37:785–789
Krishnan R, Binkley JS, Seeger R, People JA (1980) J Chem Phys 72:650–654
Frisch MJ, People JA, Binkley JS (1984) J Chem Phys 80:3265–3269
Kruszewski J, Krygowski TM (1972) Tetrahedron Lett 13(36):3839–3842
Krygowski TM (1993) J Chem Inf Comput Sci 33:70–78
Krygowski TM, Cyrański MK (1996) Tetrahedron 52:10255–10264
Schleyer PR, Maerker C, Dransfield A, Jiao van Eikema Hommes H (1996) J Am Chem Soc 118:6317–6318
Schleyer PR, Monohar M, Wang Z, Kiran B, Puchta R, Jiao van Eikema Hommes H (2001) Org Lett 3:2465–2468
Corminboeuf C, Heine T, Seifert G, Schleyer PR, Weber J (2004) Phys Chem Chem Phys 6:273–276
Charton M (1981) Prog Phys Org Chem 13:119–251
Sadlej-Sosnowska N (2007) Chem Phys Lett 447:192–196
Sadlej-Sosnowska N (2007) Pol J Chem 81:1123–1134
Hirshfeld (1977) Theor Chim Acta 44:129–138
Lorenz E, Mączka M, Hermanowicz K, Waśkowska A, Puszko A, Hanuza J (2005) Vib Spectrosc 37:195–207
Lorenc J, Hanuza J, Janczak J (2012) Vib Spectrosc 59:59–70
Yufit DS, Turner MJ, Howard JAK (2006) Acta Crystallogr E62:1237–1239
Palusiak M, Simon S, Solà M (2006) J Organomet Chem 71:5241–5248
Krygowski TM, Zachara-Horeglad JE, Palusiak M, Pelloni S, Lazzeretti P (2008) J Organomet Chem 73:2138–2145
Szatyłowicz H, Siodła T, Stasyuk OA, Krygowski TM (2016) Phys Chem Chem Phys 18:11711–11721
Olga AS, Halina S, Celia FG, Tadeusz MK (2015) Theoretical study of electron-attracting ability of the nitro group: classical and reverse substituent effects. Struct Chem 26 (4):905–913
Pareras G, Palusiak M, Duran M, Solà S, Simon S (2018) J Phys Chem A 122:2279–2287
Acknowledgments
The calculations referred to herein were carried out with the use of resources provided by Wrocław Centre for Networking and Supercomputing (http://wcss.pl) grant no. 311.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 32 kb).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sołtysiak, P., Dziuk, B., Zarychta, B. et al. The substituent effect of π-electron delocalization in N-methylamino-nitropyridine derivatives: crystal structure and DFT calculations. Struct Chem 31, 1185–1196 (2020). https://doi.org/10.1007/s11224-020-01514-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-020-01514-y