
Abstract
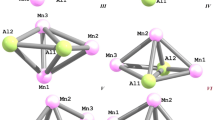
By using DFT method in the OPBE/TZVP level, key parameters of molecular structures of five-atomic heteronuclear clusters having Al2M3 composition where M = Fe or Cu (bond lengths, bond angles, and torsion (dihedral) angles) have been calculated. Each of these clusters was found to exist in eight modifications different substantially in their total energy. It has been noted that, in general, the molecular structures of Al2Fe3 and Al2Cu3 differ very significantly between each other both in terms of geometric parameters and in external form; nevertheless, the most stable modifications of these metal clusters are externally similar and differ only in the number of M–M bonds.


Similar content being viewed by others

References
Maroun F, Ozanam F, Magnussen OM, Behm RJ (2001) The role of atomic ensembles in the reactivity of bimetallic electrocatalysts. Science 293:1811–1814
Eberhardt W (2002) Clusters as new materials. Surf Sci 500(1):242–270
Dong D, Xiao-Yu K, Jian-Jun G, Ben-Xia Z (2009) First-principle study of AunFe (n=1–7) clusters. J Mol Struct 902(1):54–58
Kilimis DA, Papageorgiou DG (2010) Density functional study of small bimetallic Ag–Pd clusters. J Mol Struct 939(1):112–117
Yang JX, Guo JJ, Die D (2011) Ab initio study of AunIr (n=1–8) clusters. Comput Theor Chem 963(3):435–438
Zhao S, Ren Y, Wang J, Yin W (2011) Density functional study of NO binding on small AgnPdm (n+m≤5) clusters. Comput Theor Chem 964(2):298–303
Hong L, Wang H, Cheng J, Huang X, Sai L, Zhao J (2012) Atomic structures and electronic properties of small Au–Ag binary clusters: effects of size and composition. Comput Theor Chem 993(1):36–44
Liu X, Tian D, Meng C (2013) DFT study on stability and H2 adsorption activity of bimetallic Au79−nPdn (n=1–55) clusters. Chem Phys 415(1):179–185
Ma L, Wang J, Hao Y, Wang G (2013) Density functional theory study of FePdn (n=2–14) clusters and interactions with small molecules. Comput Mater Sci 68(1):166–173
Singh NB, Sarkar U (2014) A density functional study of chemical, magnetic and thermodynamic properties of small palladium clusters. Mol Simul 40(15):1255–1264
Wen JQ, Xia T, Zhou H, Wang JF (2014) A density functional theory study of small bimetallic PdnAl (n=1–8) clusters. J Phys Chem Solids 75(4):528–534
Bouderbala W, Boudjahem AG, Soltani A (2014) Geometries, stabilities, electronic and magnetic properties of small PdnIr (n = 1–8) clusters from first-principles calculations. Mol Phys 112(13):1789–1798
Chaves AS, Rondina GG, Piotrowski MJ, Da Silva JLF (2015) Structural formation of binary PtCu clusters: a density functional theory investigation. Comput Mater Sci 98(2):278–286
Ling W, Dong D, Shi-Jian W, Zheng-Quan Z (2015) Geometrical, electronic, and magnetic properties of CunFe (n=1–12) clusters: a density functional study. J Phys Chem Solids 76(1):10–16
Al-Odail F, Mazher J, Abuelela AM (2017) A density functional theory study of structural, electronic and magnetic properties of small PdnAg (n = 1–8) clusters. Comput Theor Chem 1125(1):103–111
Schaefer A, Horn H, Ahlrichs R (1992) Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J Chem Phys 97(4):2571–2577
Schaefer A, Huber C, Ahlrichs R (1994) Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J Chem Phys 100(8):5829–5835
Hoe WM, Cohen A, Handy NC (2001) Assessment of a new local exchange functional OPTX. Chem Phys Lett 341(3–4):319–328
Perdew JP, Burke K, Ernzerhof M (1997) Generalized gradient approximation made simple. Phys Rev Lett 78(7):1396–1396
Paulsen H, Duelund L, Winkler H, Toftlund H, Trautwein AX (2001) Free energy of spin-crossover complexes calculated with density functional methods. Inorg Chem 40(9):2201–2203
Swart M, Groenhof AR, Ehlers AW, Lammertsma K (2004) Validation of exchange−correlation functionals for spin states of iron complexes. J Phys Chem A 108(25):5479–5483
Swart M, Ehlers AW, Lammertsma K (2004) Performance of the OPBE exchange-correlation functional. Mol Phys 102(23):2467–2474
Swart M (2007) Metal–ligand bonding in metallocenes: differentiation between spin state, electrostatic and covalent bonding. Inorg Chim Acta 360(1):179–189
Gaussian 09, Revision A.01, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li H, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, and Fox DJ, Gaussian, Inc., Wallingford CT, 2009
Mikhailov OV (2013) Synthesis of 3d-element metalmacrocyclic chelates into polypeptide biopolymer medium and their molecular structures. Inorg Chim Acta 394(1):664–684
Mikhailov OV, Chachkov DV (2013) Molecular structures of (5456) metalmacrocyclic chelates with 7-imino-1-oxa-3,6,8,11-tetraazacyclo-dodecanetetrathione-4,5,9,10 formed at template synthesis according to DFT OPBE/TZVP method data. Inorg Chim Acta 408(1):246–250
Mikhailov OV, Chachkov DV, Grigorieva ON (2013) Structures of metal-macrocyclic compounds arising from "self-assembly" in ion 3d-element- dithiooxamide- 2-hydroxysubstituted acetaldehyde ternary systems. Inorg Chim Acta 408(1):199–203
Funding
The present study was carried out with financial support in the framework of draft No. 4.5784.2017/8.9 to the competitive part of the state task of the Russian Federation on the 2017–2019 years. All quantum-chemical calculations were performed at the Joint Supercomputer Center, Kazan Branch, Russian Academy of Sciences–Branch of Federal Scientific Center “Research Institute of System Investigations of the RAS.”
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Mikhailov, O.V., Chachkov, D.V. DFT calculation of molecular structures of Al2Fe3 and Al2Cu3 heterobinuclear clusters. Struct Chem 29, 1543–1549 (2018). https://doi.org/10.1007/s11224-018-1146-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-018-1146-9