
Abstract
The applicability of popular and efficient B3LYP hybrid density functional and medium-size Pople-type basis set in combination with computationally expensive anharmonic model to obtain more accurate theoretical structure, vibrational frequencies and GIAO NMR parameters of cytosine was tested. We report on prediction of cytosine equilibrium (R e ) and rovibrationally averaged (R v ) structures and vibrational frequencies in the gas phase and DMSO solution using density functional theory combined with 6-311++G** basis set. The harmonic and anharmonic vibrational frequencies (using second-order vibrational perturbation theory, VPT2) were critically discussed. In comparison with initial harmonic data, a significantly better agreement between scaled and anharmonic frequencies and experiment was observed. Proton and carbon nuclear magnetic shieldings were calculated at R e and R v structures of cytosine in the gas phase and DMSO solution using BHandH and B3LYP density functionals combined with 6-311++G**, aug-cc-pVTZ-J and STO-3Gmag basis sets. The obtained NMR results were compared with available experimental data and discussed at length.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
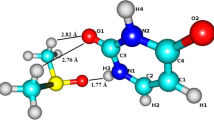
DNA molecule is one of the most important biologically active compounds. It encodes the genetic instructions used in the development and functioning of all known living organisms. The information in DNA is stored as a code made up of four nitrogen bases. The formation of DNA base pairs plays a crucial role in the realization of the main role of DNA, which is the storage and replication of genetic information [1]. Therefore, a detailed knowledge about structure and properties of single building blocks of DNA is of great importance. One of the DNA bases is cytosine. Its atom numbering is shown in Fig. 1.

Atom numbering in cytosine
Despite the fact that cytosine may exist in various tautomeric forms, we will focus on keto-amino structure (see Fig. 2), which is presumably the most stable one in the gas phase [2].

Selected resonance forms of cytosine for the most stable keto-amino structure
The question of cytosine tautomeric stability in the gas phase, low-temperature matrices and polar solution is not clear [2]. Several theoretical calculations at density functional theory (DFT) and second-order perturbation Moller–Plesset theory (MP2) level, using a relatively incomplete basis sets (like 6-311G(2d,2p) or 6-311++G** and with simplified inclusion of solvent in Ref. [2], suggest the keto-amino form being the most stable form in the gas phase and solution. On the other hand, the recent MP2/6-311++G** work by Alonso and coworkers [3] proposes the trans enol-amino form as the most stable form in the gas phase (lower by only about 1.19 kcal/mol from the corresponding keto-amino form). However, the detailed earlier theoretical work by Zeegers-Huyskens et al. [4] clearly indicates the amino-oxo tautomer as the most stable form in the gas phase. In addition, they stressed its predominance (by a factor of ten) in water.
Vibrational (IR/Raman) and NMR spectroscopic techniques, additionally supported by computational methods, have been used as very efficient tools for characterization of biological molecules [5]. On the other hand, current theoretical methods support interpretation of complex NMR, Raman and IR spectra, and combined experimental and computational studies are in routine use [6–8]. Unfortunately, the use of harmonic approximation for prediction of vibrational frequencies suffers from neglecting the effect of anharmonicity [6, 9]. The simplest remedy to bring theoretical harmonic frequencies (often overestimating experiment by 5–10 %) close to measured values is by using a uniform scaling factor [9]. However, the value of a proper scaling factor depends on the method of calculation and basis set quality. Several optimal scaling factors have been reported [9–12].
A more theoretically sound approach involves the inclusion of an anharmonic potential. Several methods including anharmonicity, for example, the second-order vibrational perturbational theory (VPT2) [13, 14], vibrational self-consistent field (VSCF) [15–17] and vibrational configuration interaction (VCI) [18–20] are available. Unfortunately, these approaches are significantly more computationally demanding and therefore are practically limited to small- and medium-size molecules.
Similarly, accurate modeling of 13C and 1H NMR spectra relays primarily on the selected theory level and completeness and flexibility of the basis set used [21–23]. Besides, the inclusion of zero-point vibration corrections and solvent effects should further improve the agreement between theory and experiment [24–28].
Cytosine has been the subject of numerous experimental and theoretical studies. Its structural parameters of single molecule were investigated by ab initio methods both in the gas phase [29–32] and in solution [31, 33–35]. Several studies focused on cytosine and other nucleobase tautomers [36–39]. In addition, hydrated complexes of cytosine were studied theoretically [29, 40]. IR studies of cytosine have been carried out in the gas phase [41, 42], argon [43] and N2 [44] matrixes, aqueous solutions [45, 46] and in the solid state [47]. Calculated harmonic [31, 48–50] and anharmonic [30] vibrations were also reported. Most vibrational studies were conducted using a simple harmonic model combined with DFT and MP2 calculations. Rasheed et al. [30] reported on HF, B3LYP and MP2 calculated anharmonic vibrational spectra of cytosine using the VSCF and CC-VSCF methods. The authors observed a good agreement between DFT and MP2 anharmonic wavenumbers and experiment. 1H [51], 13C [51, 52], 15N [52] and 17O [53] NMR chemical shifts of cytosine measured in DMSO solution have been reported. However, we are not aware of high-level theoretical prediction of the corresponding NMR parameters. The only available report published the B3LYP/6-311G(2d,2p) calculated proton and carbon chemical shifts in the gas phase and solution using TMS as theoretical Ref. [2]. Besides, the authors did not verify the accuracy of their predictions with experiment.
The aim of this work was to discuss the impact of time-consuming anharmonic model on the accuracy of the predicted structural and spectroscopic properties of isolated cytosine molecule (shown in Fig. 2b as keto-enol tautomer) in vacuum and DMSO solution using DFT calculations. Cytosine is selected as an example of well-characterized experimentally and theoretically real-size biomolecule containing 10–15 atoms. Obviously, a molecule of this size could be a subject of benchmark calculations, for example, at the coupled cluster level and very large basis sets. However, such calculations are extremely expensive, and the calculation cost could scale very steep (N7 or N8 of number of basis sets) with the size of atomic system. An example of such anharmonic studies on structure and anharmonic vibrations of uracil was reported [54, 55].
Thus, we want to see whether there is an improvement in prediction of cytosine structure, vibrational parameters and also NMR chemical shifts using a popular and efficient B3LYP density functional [56, 57] and a medium-size Pople-type basis set 6-311++G** in the gas phase by replacing a standard harmonic model with anharmonic one. Finally, we want to test the advantage of including solvent effects within harmonic and anharmonic models on the accuracy of the selected cytosine structural and spectroscopic parameters. A simple polarized continuum model (PCM [58, 59]) will be used to account for DMSO solvent.
Theoretical approach
The unconstrained cytosine geometry optimization, vibrational analysis and NMR calculations were performed using Gaussian 09 [60] software. B3LYP hybrid density functional [56, 57] combined with 6-311++G** basis set was used to fully optimize free cytosine equilibrium geometry (R e ) in the gas phase and in DMSO solution. In order to include an impact of solvent on selected properties of dissolved cytosine, the self-consistent reaction field (SCRF) calculations within the polarized continuum model (PCM) [58, 59] were selected.
The harmonic and anharmonic vibration calculations (yielding rovibrationally averaged R v structure) were carried out in vacuum and in DMSO at B3LYP/6-311++G** level of theory using the VPT2 method [13, 14]. All vibrational calculations yielded only positive vibrations ensuring minimum energy structures.
In our studies, we preferred to use PCM as a simple model of solvent impact on the structure and spectroscopic properties of cytosine. This very rough approach works well for solvents of low polarity and nonpolar solute molecules. We are aware about the limitations of PCM model, but the use of a super molecule model with explicit DMSO molecules, in particular for the VPT2 calculations, is very expensive computationally.
Finally, the B3LYP/6-31++G** calculated cytosine R e and R v geometries in the gas phase and in DMSO were used for all subsequent prediction of nuclear shieldings using the gauge-independent atomic orbital (GIAO) [61, 62] approach. For calculation of nuclear magnetic shieldings, we selected two density functionals—B3LYP and BHandH. The latter functional was selected because our earlier studies indicated its good performance in predicting proton, carbon and fluorine NMR parameters [63]. Since the GIAO NMR parameters are very sensitive to the completeness and quality of the used basis set [6, 24, 64], we selected three basis sets. Initially, we used the same basis set as for geometry optimization (Pople-type 6-311++G**). Next, we selected aug-cc-pVTZ-J basis set, tailored by Sauer et al. [65, 66] for accurate calculations of indirect spin–spin coupling constants. However, this basis set also enabled prediction of carbon nuclear shieldings in a set of small molecules close to complete basis set limit [24, 67–70]. This basis set was downloaded from Environmental Molecular Sciences Laboratory (EMSL) exchange basis set library [71, 72]. Finally, we selected somehow smaller and more compact STO-3Gmag basis set, designed by Leszczyński and coworkers [73] for efficient prediction of carbon shieldings in larger molecular systems. The latter basis sets was taken directly from their article [73].
Theoretical carbon and proton chemical shifts were referenced to benzene, calculated at the same level of theory, and the corresponding parameters were calculated as follows:
Besides, we used magnetic shielding of water [74], calculated at the same level of theory (B3LYP/6-311++G**) in the gas phase and DMSO, as reference for 17O chemical shifts. Thus, the corresponding 17O shieldings were 296.366 and 328.791 ppm. Taking into account magnetic shielding of liquid water (−36.1 ppm [75]) used as reference in experimental studies, the theoretical reference values were 260.266 and 292.691 ppm, respectively. Similarly, liquid nitromethane (shielding of −112.56 ppm or chemical shift of 380.2 ppm relative to neat ammonia [76]) is used as reference in 15N NMR spectroscopy. Our calculated 15N shieldings for nitromethane in the gas phase and DMSO were −152.622 and −166.051 ppm, respectively.
Besides, as suggested by the reviewer, we applied empirically derived linear correlations [77] between theoretical nuclear shieldings (B3LYP/6-311++G** results) and experimental carbon and nitrogen chemical shifts to derive theoretical chemical shifts. This approach does not involve a theoretical reference molecule and takes advantage of “averaging” about 395 and 56 chemical shifts for 13C and 15N, respectively.
The accuracy of theoretical predictions is often expressed by the root-mean-square (RMS) deviation from experimental values. In this work, we applied the following formula for RMS calculation:
In case of cytosine frequency modes and selected structural parameters, xi corresponds to 33 vibrations and eight bond lengths between non-hydrogen atoms. However, only four carbon and two proton chemical shifts are available for statistics. So, from statistical point of view, the NMR data should be discussed in terms of averaged deviations of calculated values from experiment. However, for consistency, we decided to use RMS as a rough measure of prediction quality in the current study.
Results and discussion
Structure in the gas phase and DMSO solution
As mentioned in Introduction, we decided to study the trans keto-amino cytosine tautomer (Fig. 2b). The MP2/6-311++G** results in Ref. [3] indicate the possible existence of three forms, differing by only 1–2.5 kcal/mol, and the remaining two forms are significantly less stable (by 3.67 and 5.36 kcal/mol). Thus, the relatively low level of theory does not warrant conclusive information about the most stable form of cytosine in vacuum.
In Table 1 are compared the selected equilibrium (R e ) and rovibrationally averaged (R v ) cytosine interatomic distances calculated at the B3LYP/6-311++G** level of theory in the gas phase and DMSO solution with the available experimental X-ray values [78]. The total performance of theory is given by RMS deviations from experiment (see Table 1). We are aware that the comparison between theoretical numbers in the gas phase or solution with experimental data measured in the solid state is somehow artificial (H-bonding and crystal packing forces are not considered in single molecule calculations), but unfortunately, there are no other available experimental studies in the literature. The agreement between theory and experiment for the selected CC, CN and CO is better visualized using a graphical presentation of bond length deviations from experiment (see Fig. 3).

Deviations of selected equilibrium (R e ) and rovibrationally averaged (R v ) cytosine bonds, calculated at the B3LYP/6-311++G** level of theory, in the gas phase and DMSO solution from the experimental X-ray values
One could expect that DMSO solvent, due to its strong tendency to H-bonding, should produce geometry more resembling than that for crystalline cytosine. In particular, in comparison with the gas phase data, the C=O and N–H bonds should be more elongated in both the DMSO solution and in the crystalline state. This is in agreement with our results showing that the RMS values in the gas phase increase from 0.020 to 0.030 Å and are about 50 % larger than in DMSO (RMS raises from 0.010 to 0.015 Å, see Table 1; Fig. 3).
It is apparent from Fig. 3 that the C5–C6 and C6–N1 bond lengths are predicted very accurately, and the worse results are produced for C4–N8 and C2–N1 bonds. It is known that the R v structures should generally show more elongated bonds. Interestingly, both in the gas phase and DMSO solution, the overall R v structures are in worse agreement with experimental data (RMS is higher by about 50 %) than the initial R e structures.
The observed accuracy of predicted bond lengths in DMSO is somehow related to the relative higher “content” of resonance structures A and C in comparison with the neutral form B (see Fig. 2). The C5–C6 and C6–N1 bonds do not take part in the resonance structures and therefore are predicted very accurately both in the gas phase and DMSO, using harmonic and anharmonic modeling. In contrast, the C2=O7 bond is very sensitive to the solvent presence and anharmonicity corrections. Similarly, the shortening of C2–N1, C2–N3 and C4–N8 is consistent with the effect of resonance.
Harmonic and anharmonic frequencies
In the next step, we will discuss the quality of theoretically predicted cytosine frequencies (Table S1) in comparison with experimental values, measured in low-temperature argon matrix [43]. Besides, we will compare our results with recent theoretical data [30], obtained at significantly lower level of theory (B3LYP/6-31G**). Thus, we will compare our B3LYP/6-311++G** calculated cytosine harmonic, scaled (with a single scaling factor of 0.9688) and anharmonic frequencies in vacuum and DMSO solution with recent harmonic, anharmonic VSCF, VCI [30] and experimental results [30, 43]. Instead of discussing all individual modes, we will concentrate here on the overall picture only. Thus, we will concentrate on RMS deviations between theoretical wavenumbers predicted in the gas phase and fifteen highest frequency experiment performed in low-temperature noble gas matrix (e.g., for an experimental setup resembling gas phase). Thus, going from raw frequencies in the gas phase to scaled and VPT2 anharmonic frequencies, a consistent improvement of results is visible from Table 2 (RMS drops from about 86 to 29 and 20 cm−1). It is also worth mentioning larger RMS values obtained for Rasheed and Ahmad [30] results: The corresponding RMS values for harmonic and anharmonic VSCF and CC-VSCF frequencies are 96, 53 and 39 cm−1. Besides, when we look at the diagnostic and typically most intense C=O stretch band in the IR spectrum (see Table 2), we observe a large improvement and the corresponding deviations from experiment are 49, −6 and 16 cm−1. Interestingly, the 33 calculated raw harmonic frequencies for cytosine in DMSO (see Table S1 in supplementary material) are of identical accuracy to those in the gas phase (RMS of 76 and 75 cm−1) and are improved by a similar amount using a single scaling factor (RMS of 60 and 58 cm−1). However, inclusion of anharmonicity and solvent significantly worsens the results (RMS increases from 50 to 90 cm−1).
It is apparent from Fig. 4 that the selected high-frequency anharmonic modes of cytosine in the gas phase are closer to experimental data measured in the low-temperature argon matrix than the theoretical results obtained from the DMSO solution. This tendency is particularly pronounced for C=O stretch.

Deviation of selected harmonic, scaled and anharmonic B3LYP/6-311++G** frequencies of cytosine in the gas phase and DMSO solution from experimental data in argon matrix [43]
1H and 13C NMR results
Finally, we will concentrate on the predicted proton and carbon chemical shifts using B3LYP and BHandH density functionals and their comparison with the reported experimental values [51, 52] in DMSO-d6. In this case, we will look at the impact of basis set quality, inclusion of rovibration effects and solvent effect. Here we will only consider the accuracy of the calculated chemical shift of four different carbon atoms (C2, C4, C5 and C6) and two protons (C5H and C6H). Obviously, our simplified PCM model cannot account for specific H-bonding, shifting the –NH and –NH2 signals by 3–5 ppm, and observed in experimental spectra, recorded in DMSO at room temperature. Thus, we will initially exclude from the discussion all nitrogen and oxygen data, as well as exchangeable protons, involved in strong hydrogen bonds.
In Fig. 5 are shown deviations of B3LYP and BHandH predicted carbon and proton chemical shifts, calculated for the gas phase structures, from experiment performed in solution. The use of R v geometry improves the agreement for B3LYP calculated C2, C4, C5, C5H and C6H chemical shifts and does not influence the accuracy for C6 (however, the reverse sign of deviation is observed). However, there is no clear dependence for BHandH calculated chemical shifts obtained for R v structure.

Deviations of B3LYP and BHandH predicted carbon and proton chemical shifts from experiment (DMSO-d6 solution at room temperature). Theoretical values are calculated for R e and R v structures in the gas phase and referenced to benzene
The importance of solvent inclusion for prediction of cytosine chemical shifts is apparent from Fig. 6. First, the B3LYP calculated C2, C4, C5, C5H and C6H chemical shifts at R e geometry in DMSO are closer to experiment than the corresponding gas phase values (see also Fig. 5). Besides, a comparable agreement for C6 is observed. However, the respective results predicted with BHandH do not show a uniform improvement upon including solvent effect. In addition, the use of R v geometry and PCM model does not improve consistently the theoretical results in comparison with gas phase calculations. In particular, a combination of BHandH density functional, solvent impact and rovibrationally averaged geometry lowers the predictive power of theory.

In order to get a general picture of the performance of different models on the accuracy of NMR chemical shifts in Table 3, we gathered the corresponding RMS values. These results also show the performance of three selected basis sets used in GIAO NMR calculations.
First, we notice an improvement of gas phase BHandH and B3LYP results calculated at R v structure of cytosine in comparison with the corresponding proton and carbon chemical shifts, predicted at R e geometry. An opposite situation is observed in DMSO solution. The best agreement for proton chemical shifts is observed for aug-cc-pVTZ-J (in DMSO) calculations at R e geometry (RMS of 0.262 and 0.253 ppm for BHandH and B3LYP). The best results for carbon chemical shifts are predicted when using B3LYP/6-311++G** calculations in DMSO (RMS of 1.530 ppm). Somehow worse result is obtained from B3LYP/STO-3Gmag calculations of 13C chemical shifts at R v structure in DMSO (RMS of 1.840 ppm). Thus, the improvement of basis set quality does not produce better agreement with experiment in case of proton and carbon chemical shifts of cytosine. Unfortunately, this trend for BHandH and B3LYP density functionals is opposite to coupled cluster calculated GIAO NMR results (in case of CCSD(T), a continuous improvement of predicting power is observed toward the complete basis set limit [24, 27]). However, when we consider typical chemical shift ranges observed for proton and carbon spectra (10 and 200 ppm), the best RMS values from Table 3 will correspond to 0.21 and 0.55 %, respectively. Thus, the result for cytosine chemical shifts points out very accurate predictions using DFT calculations.
Another way of looking at performance of theoretical models is to analyze the quality of correlation between calculated and experimental chemical shifts and the corresponding parameters of least square fit. For brevity, such correlations including both proton and carbon results are included in Figs. S1–S4 in the supplementary material. Here we only shortly mention the general conclusions from all these graphs: Very nice linear correlations, indicating good reproduction of experimental NMR parameters by theory, were obtained (y = ax + b, with slope (a) close to 0.5 and r 2 values close to 1). Besides, the parameter b in all these graphs was close to zero.
In order to asses the total performance of GIAO B3LYP/6-311++G** calculations in the gas phase and DMSO in case of more theoretically difficult nuclei, e.g., 15N and 17O, we also gathered in Table 4 the corresponding nuclear shieldings and chemical shifts for these isotopes (all nuclear shieldings were referenced to benzene, nitromethane and water). Besides, we also included carbon and nitrogen chemical shifts derived from empirical linear formulas reported by Blanco and coworkers [77]. This approach does not need to use a separate calculation for a reference molecule.
It is apparent from Table 4 that the direct (and popular) referencing of carbon data both in the gas phase and DMSO solution leads to somehow better reproduction of experiment (RMS of 4.46 vs. 5.20 ppm in the gas phase and 1.53 vs. 1.90 ppm in DMSO) in comparison with empirical relation [77]. On the other hand, the empirical formula for nitrogen works significantly better (RMS of 19.77 vs. 15.12 in the gas phase and 25.20 vs. 7.81 ppm in DMSO). Thus, the inclusion of DMSO solvent improves prediction of carbon chemical shieldings but worsens nitrogen chemical shifts with respect to values measured in DMSO. The advantage of including solvent is particularly important in case of 17O NMR chemical shift (deviation of 42.58 in the gas phase vs. −13.69 ppm in DMSO).
However, we are aware that the experimental nitrogen and oxygen chemical shifts are also recorded in different conditions (solvent, temperature or solid state), but the presence of hydrogen bonding is not taken into account in our calculations. Thus, the absolute deviation between theory and experiment in case of nitrogen and oxygen chemical shifts could be significantly larger than in case of carbon and proton data (15N and 17O appear at significantly larger range of chemical shifts than 13C or 1H).
Conclusions
The use of affordable B3LYP/6-311++G** level of theory enabled very fast and reliable prediction of equilibrium structure of cytosine. The RMS deviations of R e bond lengths between non-hydrogen atoms from experimental values, measured using X-ray technique, were fairly small (RMS of 0.010–0.020 Å). The VPT2 predicted rovibrational structure in the gas phase, and DMSO solution (within PCM solvent model) was significantly more expensive computationally. Besides, the agreement between the R v structure and X-ray experiment was slightly worse (RMS of 0.015–0.030 Å). However, anharmonic frequencies reproduced significantly better the fifteen highest frequency experimental values, measured in low-temperature argon matrices than the raw, harmonic data (RMS of 20 vs. 86 cm−1). Obviously, a simple uniform scaling also improved the results significantly (RMS of about 30 cm−1).
Typical BHandH and B3LYP calculations with popular 6-311++G** basis set for cytosine R e structure in the gas phase resulted in very inaccurate cytosine proton chemical shifts (RMS of 0.854 and 0.708 ppm). The use of R v instead of R e cytosine structure in the gas phase generally improved proton chemical shifts, and the only exception was for BHandH/aug-cc-pVTZ-J. DFT-predicted proton chemical shifts in DMSO were consistently more accurate when using R v instead of R e cytosine structure for the tested 6-311++G**, aug-cc-pVTZ-J and STO-3Gmag basis sets. Besides, addition of solvent using PCM model improves the predicted cytosine proton shifts calculated for both R e and R v structures. Only in case of B3LYP/STO-3Gmag calculations at R e geometry, it leads to worse agreement with experiment. The overall best results for cytosine protons are observed for both BHandH and B3LYP density functionals combined with aug-cc-pVTZ-J basis set when using R v structure in DMSO (RMS of 0.225 and 0.206 ppm).
The use of R v structure in the gas phase improves the accuracy of carbon chemical shifts. This tendency was particularly pronounced for B3LYP density functional (RMS of 4.172 decreased to 1.655 ppm for B3LYP/6-311++G**). Furthermore, the inclusion of solvent improves carbon chemical shifts calculated for cytosine R e structure. The best agreement with experiment (RMS of 1.530 ppm) was observed for B3LYP/6-311++G** predicted 13C chemical shifts using R e structure of cytosine in DMSO. Moreover, in most cases, the combination of PCM calculations and anharmonic correction yielded worse agreement between predicted carbon chemical shifts and the corresponding experimental values. In case of cytosine carbon chemical shifts, the improvement of basis set quality did not produce better agreement with experiment. Thus, probably due to cancelation of different errors, the use of inexpensive R e structure with PCM solvent model predicted by B3LYP/6-311++G** calculations produced the best carbon shieldings.
The obtained NMR results strongly suggest caution when mixing different correction techniques in order to improve the predictive power of DFT. Thus, the semi-empirical nature of the used density functionals seems to be the source of limitations when using DFT as predicting tool in calculation of GIAO NMR parameters.
References
Sivakova S, Rowan SJ (2005) Nucleobases as supramolecular motifs. Chem Soc Rev 34:9–21
Sathyabama V, Anandan K, Kanagaraju R (2009) Quantum chemical studies of solvent effects on cytosine tautomers. J Mol Struct (Theochem) 897:106–110
Alonso JL, Vaquero V, Peña I, López JC, Mata S, Caminati W (2013) All five forms of cytosine revealed in the gas phase. Angew Chem Int Ed 52:2331–2334
Chandra AK, Michalska D, Wysokińsky R, Zeegers-Huyskens T (2004) Theoretical study of the acidity and basicity of the cytosine tautomers and their 1:1 complexes with water. J Phys Chem A 108:9593–9600
Jalkanen KJ, Elstnerb M, Suhaic S (2004) Amino acids and small peptides as building blocks for proteins: comparative theoretical and spectroscopic studies. J Mol Struct (Theochem) 675:61–7715
Foresman JB, Frisch A (1996) Exploring chemistry with electronic structure methods. Gaussian Inc, Pittsburg
Kjær H, Sauer SPA, Kongsted J (2010) Benchmarking NMR indirect nuclear spin-spin coupling constants: SOPPA, SOPPA(CC2), and SOPPA(CCSD) versus CCSD. J Chem Phys 133:144106
Kupka T, Lin H-M, Stobiński L, Chen C-H, Liou W-J, Wrzalik R, Flisak Z (2010) Experimental and theoretical studies on corals. I. Toward understanding the origin of color in precious red corals from Raman and IR spectroscopies and DFT calculations. J Raman Spectrosc 41:651–658
Scott AP, Radom L (1996) Harmonic vibrational frequencies: an evaluation of Hartree–Fock, Møller–Plesset, quadratic configuration interaction, density functional theory, and Semiempirical scale factors. J Phys Chem 100:16502–16513
Merrick JP, Moran D, Radom L (2007) An evaluation of harmonic vibrational frequency scale factors. J Phys Chem A 111:11683–11700
Alecu IM, Zheng J, Zhao Y, Truhlar DG (2010) Computational thermochemistry: scale factor databases and scale factors for vibrational frequencies obtained from electronic model chemistries. J Chem Theory Comput 6(9):2872–2887
Tantirungrotechai Y, Phanasant K, Roddecha S, Surawatanawong P, Sutthikhum V, Limtrakul J (2006) Scaling factors for vibrational frequencies and zero-point vibrational energies of some recently developed exchange-correlation functionals. J Mol Struct Theochem 760:189–192
Barone V (2004) Vibrational zero-point energies and thermodynamic functions beyond the harmonic approximation. J Chem Phys 120:3059–3065
Barone V (2005) Anharmonic vibrational properties by a fully automated second-order perturbative approach. J Chem Phys 122:014108
Bowman JM (1978) Self-consistent field energies and wave functions for coupled oscillators. J Chem Phys 68:608–610
Gerber RB, Ratner MA (1979) A semiclassical self-consistent field (SC SCF) approximation for eigenvalues of coupled-vibration systems. Chem Phys Lett 68:195–198
Carney GD, Sprandel LI, Kern CW (1978) Variational approaches to vibration–rotation spectroscopy for polyatomic molecules. Adv Chem Phys 37:305–379
Bowman JM, Christoffel K, Tobin F (1979) Application of SCF CI theory to vibrational motion in polyatomic molecules. J Phys Chem 83:905–920
Christoffel KM, Bowman JM (1982) Investigations of self consistent field, SCF CI and virtual state configuration interaction vibrational energies for a model three-mode system. Chem Phys Lett 85:220–224
Carter S, Bowman JM, Handy NC (1998) Extensions and tests of “multimode”: a code to obtain accurate vibration/rotation energies of many-mode molecules. Theor Chem Acc 100:191–198
Gauss J, Stanton JF (1995) Gauge-invariant calculation of nuclear magnetic shielding constants at the coupled-cluster singles and doubles level. J Chem Phys 102:251–253
Gauss J (2002) Analytic second derivatives for the full coupled-cluster singles, doubles, and triples model: nuclear magnetic shielding constants for BH, HF, CO, N2, N2O, and O3. J Chem Phys 116:4773–4776
Auer A, Gauss J, Stanton JF (2003) Quantitative prediction of gas-phase 13C nuclear magnetic shielding constants. J Chem Phys 118:10407–10417
Kupka T, Stachów M, Nieradka M, Kaminský J, Pluta T (2010) Convergence of nuclear magnetic shieldings in the Kohn–Sham limit for several small molecules. J Chem Theor Comput 6:1580–1589
Ruud K, Astrand P-O, Taylor PR (2000) An efficient approach for calculating vibrational wave functions and zero-point vibrational corrections to molecular properties of polyatomic molecules. J Chem Phys 112:2668–2683
Ruud K, Astrand P-O, Taylor PR (2001) Zero-point vibrational effects on proton shieldings: functional-group contributions from ab initio calculations. J Am Chem Soc 123:4826–4833
Kupka T, Stachów M, Nieradka M, Kaminský J, Pluta T, Sauer SPA (2011) From CCSD(T)/aug-cc-pVTZ-J to CCSD(T) complete basis set limit isotropic nuclear magnetic shieldings via affordable DFT/CBS calculations. Magn Reson Chem 49:231–236
Książek A, Borowski P, Woliński K (2009) Theoretical analysis of solvent effects on nitrogen NMR chemical shifts in oxazoles and oxadiazoles. J Magn Reson 197:153–160
Chandra AK, Nguyen MT, Zeegers-Huyskens Th (2000) Theoretical study of the protonation and deprotonation of cytosine. Implications for the interaction of cytosine with water. J Mol Struct 519:1–11
Rasheed T, Ahmad S (2010) Approximate solution of the mode–mode coupling integral: application to cytosine and its deuterated derivative. Spectrochim Acta Part A 77:446–456
Carbonniere P, Thicoipe S, Very T, Assfeld X (2012) Vibrational analysis beyond the harmonicity from ab initio molecular dynamics: case of cytosine in its anhydrous and aqueous forms. Int J Quantum Chem 112:2221–2230
Tomić K, Tatchen J, Marian CM (2005) Quantum chemical investigation of the electronic spectra of the keto, enol, and keto-imine tautomers of cytosine. J Phys Chem A 109:8410–8418
Alemán C (1999) Hydration of cytosine using combined discrete/SCRF models: influence of the number of discrete solvent molecules. Chem Phys 244:151–162
Alemán C (1999) Solvation of cytosine and thymine using a combined discrete/SCRF model. Chem Phys Lett 302:461–470
Shishkin OV, Gorb L, Leszczyński J (2000) Does the hydrated cytosine molecule retain the canonical structure? A DFT study. J Phys Chem B 104:5357–5361
Fogarasi G (1997) High-level electron correlation calculations on some tautomers of cytosine. J Mol Struct 413:271–278
Kobayashi R (1998) A CCSD(T) study of the relative stabilities of cytosine tautomers. J Phys Chem A 102:10813–10817
Fogarasi G (2002) Relative stabilities of three low-energy tautomers of cytosine: a coupled cluster electron correlation study. J Phys Chem A 106:1381–1390
Trygubenko SA, Bogdan TV, Rueda M, Orozco M, Luque FJ, Sponer J, Slavicek P, Hobza P (2002) Correlated ab initio study of nucleic acid bases and their tautomers in the gas phase, in a microhydrated environment and in aqueous solution: part 1. Cytosine Phys Chem Chem Phys 4:4192–4203
Van Mourik T, Benoit DM, Price SL, David CC (2000) Ab initio and diffusion Monte Carlo study of uracil–water, thymine–water, cytosine–water, and cytosine–(water)2. Phys Chem Chem Phys 2:1281–1290
Ferro D, Bencivenni L, Teghil R, Mastromarino R (1980) Vapour pressures and sublimation enthalpies of thymine and cytosine. Thermochim Acta 42:75–83
Nir E, Müller M, Grace LI, de Vries MS (2002) REMPI spectroscopy of cytosine. Chem Phys Lett 355:59–64
Szczesniak M, Szczepaniak K, Kwiatkowski JS, KuBulat K, Person WB (1988) Matrix isolation infrared studies of nucleic acid constituents. 5. Experimental matrix-isolation and theoretical ab initio scf molecular orbital studies of the infrared spectra of cytosine monomers. J Am Chem Soc 110(25):8319–8333
Radchenko ED, Sheina GG, Smorygo NA, Blagoi YP (1984) Experimental and theoretical studies of molecular structure features of cytosine. J Mol Struct 116:387–396
Aamouche A, Baron MH, Berthier G, Coulombeau C, Ghomi M, Grajcar L, Henriet C, Jobic H, Turpin PY (1997) Neutron inelastic scattering, optical spectroscopies and scaled quantum mechanical force fields for analyzing the vibrational dynamics of pyrimidine nucleic acid bases: 3. cytosine. J Phys Chem A 101:10063–10074
Florián J, Baumruk V, Leszczyński J (1996) IR and raman spectra, tautomeric stabilities, and scaled quantum mechanical force fields of protonated cytosine. J Phys Chem 100:5578–5589
Susi H, Ard JS (1974) Planar valence force constants and assignments for pyrimidine derivatives. Spectrochim Acta A 30:1843–1853
Kwiatkowski JS, Leszczyński J (1996) Molecular structure and vibrational ir spectra of cytosine and its thio and seleno analogues by density functional theory and conventional ab initio calculations. J Phys Chem 100:941–953
Santamaria R, Charro E, Zacarias A, Castro M (1999) Vibrational spectra of nucleic acid bases and their Watson-Crick pair complexes. J Comp Chem 20(5):511–530
Subramanian V, Chitra K, Venkatesh K, Sanker S, Ramasami T (1997) Comparative study on the vibrational IR spectra of cytosine and thiocytosine by various semi-empirical quantum mechanical methods. Chem Phys Lett 264:92–100
http://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi; http://www.sigmaaldrich.com/spectra/fnmr/FNMR001198.PDF
López C, Claramunt RM, Alkorta I, Elguero J (2000) Solution and solid state (CPMAS) NMR studies of the tautomerism of six-membered heterocyclic compounds related to 2-pyridones. Spectroscopy 14:121–126
Wu G, Dong D, Ida R, Reen N (2002) A solid-state 17O nuclear magnetic resonance study of nucleic acid bases. J Am Chem Soc 124(8):1768–1777
Puzzarini C, Biczysko M, Barone V (2011) Accurate anharmonic vibrational frequencies for uracil: the performance of composite schemes and hybrid CC/DFT model. J Chem Theory Comput 7:3702–3710
Puzzarini C, Barone V (2011) Extending the molecular size in accurate quantum-chemical calculations: the equilibrium structure and spectroscopic properties of uracil. Phys Chem Chem Phys 13:7189–7197
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Miertus S, Tomasi J (1982) Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem Phys 65:239–245
Tomasi J, Mennucci B, Cammi R (2005) Quantum mechanical continuum solvation models. Chem Rev 105:2999–3093
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ (2009) Gaussian 09, Revision A.02. Gaussian, Wallingford
Ditchfield R (1974) Self-consistent perturbation theory of diamagnetism. Mol Phys 27:789–807
Woliński K, Hinton JF, Pulay P (1990) Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J Am Chem Soc 112:8251–8260
Kupka T (2009) Magn Reson Chem 47(11):959–970
Kupka T, Ruscic B, Botto RE (2002) Toward Hartree–Fock and density functional complete basis-set predicted NMR parameters. J Phys Chem A 106:10396–10407
Provasi PF, Aucar GA, Sauer SPA (2001) The effect of lone-pairs and electronegativity on the indirect nuclear spin-spin coupling constants in CH2X (X=CH2, NH, O, S). Ab initio calculations using optimized contracted basis sets. J Chem Phys 115:1324–1334
Enevoldsen T, Oddershede J, Sauer SPA (1998) Correlated calculations of indirect nuclear spin-spin coupling constants using second order polarization propagator approximations: SOPPA and SOPPA (CCSD). Theor Chem Acc 100:275–284
Kendall RA, Dunning TH Jr, Harrison RJ (1992) Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J Chem Phys 96:6796
Woon DE, Dunning TH Jr (1993) Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J Chem Phys 98:1358
Dunning TH Jr (1989) Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J Chem Phys 90:1007
Feller D (1992) Application of systematic sequences of wave functions to the water dimer. J Chem Phys 96:6104–6114
Feller D (1996) The role of databases in support of computational chemistry calculations. J Comp Chem 17(13):1571–1586
Schuchardt KL, Didier BT, Elsethagen T, Sun L, Gurumoorthi V, Chase J, Li J, Windus TL (2007) Basis set exchange: a community database for computational sciences. J Chem Inf Model 47(3):1045–1052
Voronkov E, Rossikhin V, Okovytyy S, Shatckih A, Bolshakov V, Leszczyński J (2012) Novel physically adapted STO##-3G basis sets. Efficiency for prediction of second-order electric and magnetic properties of aromatic hydrocarbons. Int J Quantum Chem 112:2444–2449
Puzzarini C, Cazzoli G, Harding ME, Vázquez J, Gauss J (2009) A new experimental absolute nuclear magnetic shielding scale for oxygen based on the rotational hyperfine structure of H 172 O. J Chem Phys 131(23):1–11
Wasylishen RE, Mooibroek S, Macdonald JB (1984) A more reliable oxygen-17 absolute chemical shielding scale. J Chem Phys 81:1057–1059
Witanowski M, Biedrzycka Z, Grela K, Wejroch K (1998) Nitrogen NMR shieldings of nitroalkanes as a structural and conformational probe. Magn Reson Chem 36(998):S85–S92
Blanco F, Alkorta I, Elguero J (2007) Statistical analysis of 13C and 15N NMR chemical shifts from GIAO/B3LYP/6-311++G** calculated absolute shieldings. Magn Reson Chem 45(9):797–800
Voet D, Rich A (1970) The crystal structures of purines, pyrimidines and their intermolecular complexes. Prog Nucleic Acid Res Mol Biol 10:183–265
Acknowledgments
Roksana Wałęsa is recipient of a PhD fellowship from a project funded by the European Social Fund. Calculations were carried out in the Wrocław Centre for Networking and Supercomputing (http://www.wcss.wroc.pl), and in the Academic Computer Centre, CYFRONET, AGH, Kraków. T. K. and M. B. were supported by the Faculty of Chemistry, UO project (8/WCH/2014-S).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Wałęsa, R., Kupka, T. & Broda, M.A. Density functional theory (DFT) prediction of structural and spectroscopic parameters of cytosine using harmonic and anharmonic approximations. Struct Chem 26, 1083–1093 (2015). https://doi.org/10.1007/s11224-015-0573-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-015-0573-0