
Abstract
The apocritan Hymenoptera show extraordinary features in mitochondrial genomes, but no complete sequence has been reported for the basal lineage, Evanioidea. Here, we sequenced the complete mitochondrial genome of Evania appendigaster. This genome is 17,817 bp long; with low A+T content, 77.8%, compared with other hymenopteran species. Four tRNA genes were rearranged, among which remote inversion is the dominant gene rearrangement event. Gene shuffling is caused by tandem duplication-random loss while remote inversion is best explained by recombination. The start codon of nad1 was found as TTG, which might be common across Hymenoptera. trnS2 and trnK use abnormal anticodons TCT and TTT, respectively, and the D-stem pairings in trnS2 are absent. The secondary structure of two rRNA genes are predicted and compared with those in other insects. Five long intergenic spacers were present, including a long intergenic spacer between atp8 and atp6, where these two genes overlap in the previously reported animal genomes. A conserved motif was found between trnS1 and nad1, which is proposed to be associated with mtTERM. The A+T-rich region is 2,325 bp long, among the longest in insects, and contains a tandem repeat region.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Animal mitochondrial genomes are about 16 Kb in size and contain 37 genes: 13 protein-coding genes, 22 transfer RNA genes (tRNA) and two ribosomal RNA genes (rRNA) [1, 2]. The genome is highly economized with few sections of noncoding DNA, intergenic regions, or repetitive sequences [3, 4], except for an A+T rich region, which contains essential regulatory elements for transcription and replication [5].
Gene arrangements are usually conserved within major lineages [2], but may be highly rearranged in certain groups [6–12]. Gene rearrangement events may serve as useful phylogenetic markers and models for evolutionary studies [13–16]. In apocritan Hymenoptera, frequent gene rearrangements have been observed from broad examinations of gene segments [10, 17] and whole genome sequences [18–22]. However, no informative arrangement pattern has been identified to date, for which there are two possible explanations: the one is that diversified gene arrangements have arisen independently among different hymenopteran lineages, and the other is that limited sampling is concealing potentially synapomorphic rearrangements. The apocritan lineage shows other extraordinary features in the mitochondrial genome, such as high A+T content [23, 24], diversified gene rearrangement events, and the involvement of recombination in gene rearrangement [17].
Evaniidae is proposed to be one of the most basal lineages in Hymenoptera [25, 26]. Presently, no complete mitochondrial genome has been sequenced from members of this family or its presumed sister groups, the Aulacidae and Gasteruptiidae. Here, we present the complete mitochondrial genome of Evania appendigaster (Hymenoptera: Evaniidae) and give a thorough description of its genome features in comparison to other hymenopteran species.
Materials and methods
DNA extraction, PCR amplification and sequencing
Total genomic DNA was extracted using the DNeasy tissue kit (Qiagen, Hilden, Germany) from a leg of an E. appendigaster adult.
A range of universal insect mitochondrial primers [27, 28] and hymenopteran mitochondrial primers were used to amplify the regions of cox1-cox2, cob-rrnL, rrnL-rrnS. Species-specific primers were designed based on sequenced fragments and combined in various ways to bridge the gap of cox2-cob and rrnS-cox1. Six fragments of 575–8626 bp were amplified, covering the whole mitochondrial genome (Table 1). The PCR and sequencing procedures followed the methods in Wei et al. [23].
Genome annotation and secondary structure prediction
tRNA genes were initially identified using the tRNAscan-SE search server [29] with default parameters. Sequences longer than 100 bp between the identified tRNA genes were used as queries in BLAST searches in GenBank for identification of protein-coding and rRNA genes. The exact initiation and termination codons were identified in ClustalX version 2.0 [30] using reference sequences from other insects, following the criteria in Wei et al. [23]. Finally, the tRNA search was carried out again for the large intergenic regions using a reduced cutoff score. Twenty-one of the 22 typical animal mitochondrial tRNA genes were found using the previous steps, except for trnS2, which was identified by alignment. A+T content and codon usage were calculated using MEGA version 4.0 [31].
All tRNA secondary structures were predicted using the tRNAscan-SE search server [29] except for trnS2, which was predicted manually. rRNA structures were predicted by comparison and algorithm-based methods as in Wei et al. [23].
Results and discussion
Genome structure and base composition
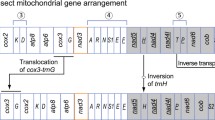
The complete mitochondrial genome of E. appendigaster is 17,817 bp (GenBank accession No. FJ593187), which is among the largest animal mitochondrial genomes yet sequenced [1]. All of the 37 typical animal mitochondrial genes were identified (Fig. 1; Table 2).

Organization of Evania appendigaster mitochondrial genome. Gene abbreviations are as follows: cox1, cox2, and cox3 refer to the cytochrome oxidase subunits, cob refers to cytochrome b, nad1-nad6 refer to NADH dehydrogenase components, and rrnL and rrnS refer to ribosomal RNAs. Transfer RNA genes are denoted by one letter symbol according to the IPUC-IUB single-letter amino acid codes. L1, L2, S1 and S2 denote tRNA Leu(CUN), tRNA Leu(UUR), tRNA Ser(AGY) and tRNA Ser(UCN), respectively. AT indicates A+T-rich region. Gene names with lines indicate that the genes are coded on the minority strand while those without lines are on the majority strand
There are in total 31 overlapping nucleotides between neighboring genes in nine locations and the length of overlapping sequence is 1–7 bp, while there are in total 943 bp intergenic nucleotides in 13 locations and the length of intergenic spacers is 1–534 bp, excluding the A+T-rich region (Table 2).
The A+T content of E. appendigaster mitochondrial genome are lower than all other sequenced hymenopteran species, and there are more A and C than T and G in the majority strand (Table 3). A higher A+T content was found in parasitic wasps (Apocrita) compared with nonparasitic wasps (Symphyta) in partial mitochondrial genes [24] and whole genome sequences [18–20, 22, 32, 33].
Gene rearrangement
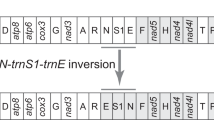
Gene arrangement of the E. appendigaster mitochondrial genome is similar to other apocritan species. Gene rearrangement events have been classified as translocation, local inversion (inverted in the local position), gene shuffling (local translocation) and remote inversion (translocated and inverted) [17]. Four tRNA genes are rearranged, which are remote inversions of trnW, trnC and trnS1 and gene shuffling of trnM (Fig. 1). Rearrangement of tRNA genes is common in the hymenopteran mitochondrial genome, especially those in tRNA clusters, such as in the junctions of A+T-rich region-nad2, nad2-cox1, cox2-atp8 and nad3-nad5 [10, 17, 23]. However, the rearrangements in the E. appendigaster mitochondrial genome are novel. In vertebrates, gene shuffling is the dominant gene rearrangement event [34], while in Hymenoptera, equal numbers of gene shuffling, inversion and translocation events have been observed at the cox2-atp8 junction [10]. In the E. appendigaster mitochondrial genome, remote inversion was found to be the dominant gene rearrangement event.
Gene shuffling is usually explained by the tandem duplication-random loss (TDRL) model [17, 35]. Evidence of the TDRL model includes a derived pattern of gene order, pseudogene and the position of intergenic spacer, the last two of which are the expected intermediate steps in changing mitochondrial gene order under this model. In the derived tRNA cluster between the A+T-rich region and nad2, all neighboring genes are overlapped or directly adjacent except for trnQ and nad2, where there is a 22 bp intergenic spacer (Table 2). Under the TDRL model, it is unlikely to randomly delete the duplicated or original genes to produce a pattern in which remnant adjacent genes overlap. Thus, it is unlikely that trnC and trnS1 were rearranged by TDRL, while it is possible that trnM was rearranged by tandem duplication of the trnI-trnQ-trnM cluster followed by deletion of trnI-trnQ and trnM in the two boundaries in an intermediate state before the insertion of trnC and trnS1. This region is located to one side of the A+T rich region that is thought to contain two replication origins [36], so an illicit-primer may be responsible for the duplication of the original tRNA cluster. The 22 bp intergenic spacer between trnQ and nad2 may be a remnant region after deletion of the second copy of trnM. Recombination may be involved in remote inversions and is the most plausible explanation for local inversions in apocritan mitochondrial genomes.
Protein-coding genes
The size of the protein-coding genes in the E. appendigaster mitochondrial genome is similar to their corresponding orthologs in other insects. The genes with the highest A+T content in the hymenopteran mitochondrial genome are usually nad6 or atp8. In E. appendigaster, the A+T content of atp8 is 69.1%, amongst the lowest ones, and this is the result of lower A+T content in the 3′ sequence of atp8.
All protein-coding genes start with ATN codons (two with ATA, four with ATT, one with ATC, and five with ATG) except for nad1, which uses TTG as start codon (Table 1). cox1 is usually found to use nonstandard start codons in insects, such as TCG, ACC, CGA, CTA, CCG and AAA [37, 38]. In E. appendigaster, cox1 uses the usual start codon ATG, 3 bp after the end of trnY, and the translated amino acid sequence aligned well with orthologs in other Hymenoptera. All examined species in Lepidoptera have been found to use R as the initial amino acid for cox1 [39], whereas in Hymenoptera all species uses the ATN start codon [18, 19, 21–23, 32] except for Vanhornia eucnemidarum [20]. In E. appendigaster, three ATA lying in or 6 bp downstream from trnL1 are possible start codons for nad1. However, we proposed TTG directly after trnL1 as the start codon for nad1. This would minimize intergenic spacer and avoid overlapping between trnL1 and nad1 [37, 40]. We examined nad1 start codons in the 11 previously reported hymenopteran species, and the results revealed that either the intergenic spacers or the overlapping regions would be reduced in Perga condei [32], Vanhornia eucnemidarum [20] and three Nasonia species [21] if TTG is assigned as the start codon (Fig. 2). In Diadegma semiclausum [23], Polistes humilis [22] and three bee species [18, 33], no TTG codon is found near the initial region of nad1. In two vespid species, Abispa ephippium and Polistes humilis, trnL1 is rearranged and rrnL is left upstream nad1. In A. ephippium, a TTG codon is present 3 bp downstream the identified start codon ATA. Since there is no standard way to define the exact boundaries of rRNAs, the criteria of reducing intergenic spacer and overlapping region could not be applied to assign the start codon. In conclusion, our results suggest that TTG is a possible start codon for nad1 in Hymenoptera [37, 40, 41].

Determination of nad1 start codons in Evania appendigaster and other reported hymenopteran mitochondrial genomes. The box indicates the newly assigned start codons, and the shaded regions the previously assigned start codons. Sequences of tRNA are marked by solid lines, intergenic spacers by dotted lines and rrnL by dashed lines
Nine protein-coding genes use the termination codon TAA. Four protein-coding genes use incomplete stop codons: nad1 and nad2 use the truncated termination codon TA, and cox3 and nad4 use T, which is commonly reported in other invertebrates [18, 42]. The relative synonymous codon usage values show a biased use of A and T nucleotides in E. appendigaster (Table 4).
tRNA genes
The length of tRNAs ranges from 61 to 70 bp. All tRNA genes have a typical cloverleaf structure except for trnS2 (Fig. 3). trnS2 could not be identified and folded using conventional tRNA search methods such as tRNAscan-SE. We manually found the location of trnS2 by comparisons with those identified in other insects and then determined the exact boundaries according to the secondary structure folded by eye. The D-stem pairings in the DHU arm are absent in E. appendigaster trnS2, which has also been reported in other insects [6, 18, 37, 43] and the rest of Metazoa [44, 45]. Since this atypical trnS2 is common in Coleoptera, Sheffield et al. [37] built an updated covariance model for automated annotation, which also performs well in other insects.

Predicted secondary structures for the 22 typical tRNA genes of Evania appendigaster mitochondrial genome. Base-pairing is indicated as follows: Watson–Crick pairs by lines, wobble GU pairs by dots and other noncanonical pairs by circles
A total of 28 unmatched base pairs exist in the E. appendigaster mitochondrial tRNA secondary structures, 19 of which are G–U pairs, eight U–U and one A–A. The number of mismatches is relatively high in the E. appendigaster mitochondrial tRNAs compared with other insects, and even within Metazoa [46]. Mismatches in regions where the tRNA genes overlap with adjacent downstream genes could be corrected by 3′-RNA editing [47–50]. The 5′-parts of tRNA accepter stems are also found in Acanthamoeba [51] and some fungi [52]. Of the 28 mismatches, only four in trnQ, trnR and trnS1 are located in the overlapping regions in the accepter stem, indicating that other mechanisms might be involved to escape the effects of Muller’s ratchet in the E. appendigaster mitochondrial genome [53].
trnS2 and trnK use abnormal anticodons TCT and TTT, respectively, which have been found to be correlated with gene rearrangement [23].
rRNA genes
rrnL has a length of 1274 bp, with an A+T content of 79.7%. rrnS has a length of 747 bp, with an A+T content of 76.0%. The gene sizes are normal, but the A+T contents are lower than their counterparts in other hymenopteran species.
Both rrnL and rrnS conform to the secondary structure models proposed for these genes from other insects [23, 39, 54–56]. Forty-nine helices are present in E. appendigaster rrnL as in D. melanogaster [55] and A. mellifera [54], belonging to six domains (Fig. 4). H837 usually forms a long stem structure with a small loop in the terminal [23, 37, 54], but it forms a shorter stem and a larger loop in E. appendigaster as that in D. melanogaster [55]. The deduced structures of H2347 and H2520 are variable [54, 57, 58], but in E. appendigaster they are more similar to those from A. mellifera by Gillespie et al. (2006) than those from other insects [57, 58].

Predicted rrnL secondary structure in Evania appendigaster mitochondrial genome. Tertiary interactions and base triples are shown connected by continuous lines. A 5′ half of rrnL; B 3′ half of rrnL. Symbols for base-pairings are as in Fig. 2
The secondary structure of rrnS contains 29 helices present in D. virilis [56] and A. mellifera [54], belonging to three domains (Fig. 5). Helix H39 could not be predicted, where a circle was formed by H27, H47, H367 and H500, and the sequences in between. Helix 47 is variable among different lepidopteran species, but the terminal portion of this stem is conserved [37], and in E. appendigaster, two loops were formed similar to D. virilis but different from two other hymenopteran species, D. semiclausum and A. mellifera, where a larger loop is present. H673 is well conserved in moths, where one stem with a bulge in the terminal is present [39, 59], and in E. appendigaster, two stem-loop structures are present as in D. virilis [56] and D. semiclausum [23], but different from that in A. mellifera [54], in which this structure is similar to moths. The structure of H1074 has been discussed in honey bee [54, 60, 61], and our predicted structure in E. appendigaster is consistent with that of Page (2000) and Gillespie et al. (2006).

Predicted rrnS secondary structure in Evania appendigaster mitochondrial genome. Symbols are as in Fig. 3
Non-coding regions
One of the most interesting features in the E. appendigaster mitochondrial genome is the presence of five major non-coding regions of more than 20 bp: spacer 1 is 22 bp between trnQ and nad2, spacer 2 is 534 bp between trnK and trnD, spacer 3 is 244 bp between atp8 and atp6, spacer 4 is 94 bp between cob and nad1, and spacer 5 is 2325 bp between rrnS-trnW and trnC-trnM-trnI. Long intergenic spacers have been identified in several insect mitochondrial genomes [18, 20, 23, 40, 62, 63]. Although intergenic spacers appeared to be unique to individual species [37], conserved motifs have been found across all insects, and are proposed to be associated with mtTERM [37, 39, 64].
Spacer 1 shows limited conservation among hymenopteran species which possess it. In Hymenoptera, the tRNAs directly upstream nad2 are variable because of frequent gene rearrangements of the tRNAs between A+T-rich region and nad2 [23], therefore this spacer is unlikely to have any function in translation or transcription. However, we suggested that it is the product of gene rearrangement as in that in D. semiclausum [23]. Spacer 2 has an A+T content of 96.8%, composed of seven tandem repeat units “GTAATTTTAT”, twelve “AATAATAATATT”, eight “AATAATAATATTAAT”, an initial sequence “TTATTAATAAACCTTAAATTAAAAATTAATTA”, and a terminal sequence “AATAATAATAT(TAA)8(TA)33AT”. Spacer 3 has an A+T content of 76.2% and contains no repeat sequence although it is 224 bp long. As far as we know, no intergenic nucleotides between atp6 and atp8 have been found in the previously reported insect mitochondrial genomes, and furthermore, it is a common feature of metazoan mitochondrial genomes that atp8 and atp6 overlap [65]. It has been proposed that the secondary structure of the transcribed mRNA may facilitate cleavage between the abutting proteins [38, 66, 67]. We could map secondary structure as those in other insects [38, 46] (Fig. 6), which indicated that the presence of spacer 3 in E. appendigaster would not affect the cleavage of atp6 and atp8. Spacer 4 was found in another six hymenopteran species (Fig. 7). This intergenic spacer region may correspond to the binding site of mtTERM, a transcription attenuation factor [64], as evidenced by a 7 bp motif (ATACTAA) conserved across Lepidoptera [39] and a 5 bp (TACTA) motif conserved across Coleoptera [37]. In Hymenoptera, we found a 6 bp conserved motif (THACWW), which shows high similarity to those in Lepidoptera and Coleoptera. In P. condei and D. semiclausum, although there is only a 2 bp intergenic spacer and a 7 bp overlapping region between trnS1 and nad1, respectively, we could still find conserved motifs in both species nearby regions between trnS1 and nad1. This may indicate wrong annotations of this region in both genomes, or the existence of the motif within genes. Spacer 5 is proposed as the A+T-rich region because of its location between rrnS-trnW and trnC-trnM-trnI and high A+T content (85.6%). It is one of the longest A+T-rich regions in the sequenced insect mitochondrial genomes [23, 68]. Twenty-three tandemly arranged units of “GTCATTATTTAATATAAAATA” are present in the middle of the A+T-rich region. This region, characterized by five elements [2, 5], is believed to function in the initiation of replication and control of transcription. However, these elements in the E. appendigaster mitochondrial genome are not arranged in the conserved pattern.

mRNA loops for genes atp8-atp6 in Evania appendigaster mitochondrial genome. The box indicates start codon of atp6

Alignment of the intergenic spacers between trnS1 and nad1 (Spacer 4) across Evania appendigaster and other reported hymenopteran mitochondrial genomes. Shaded region indicates the conserved motif and some of these unconserved intergenic nucleotides are replaced by dots
References
Wolstenholme DR (1992) Animal mitochondrial DNA: structure and evolution. Int Rev Cytol 141:173–216
Boore JL (1999) Animal mitochondrial genomes. Nucleic Acids Res 27:1767–1780
Harrison RG (1989) Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol Evol 4:6–11
Lunt DH, Whipple LE, Hyman BC (1998) Mitochondrial DNA variable number tandem repeats (VNTRs): utility and problems in molecular ecology. Mol Ecol 7:1441–1455
Zhang DX, Hewitt GM (1997) Insect mitochondrial control region: a review of its structure, evolution and usefulness in evolutionary studies. Biochem Syst Ecol 25:99–120
Shao RF, Barker SC (2003) The highly rearranged mitochondrial genome of the plague thrips, Thrips imaginis (Insecta: Thysanoptera): convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol Biol Evol 20:362–370
Shao R, Campbell NJH, Barker SC (2001) Numerous gene rearrangements in the mitochondrial genome of the wallaby louse, Heterodoxus macropus (Phthiraptera). Mol Biol Evol 18:858–865
Shao RF, Campbell NJH, Schmidt ER, Barker SC (2001) Increased rate of gene rearrangement in the mitochondrial genomes of three orders of hemipteroid insects. Mol Biol Evol 18:1828–1832
Negrisolo E, Minelli A, Valle G (2004) Extensive gene order rearrangement in the mitochondrial genome of the centipede Scutigera coleoptrata. J Mol Evol 58:413–423
Dowton M, Austin AD (1999) Evolutionary dynamics of a mitochondrial rearrangement “hot spot” in the Hymenoptera. Mol Biol Evol 16:298–309
Covacin C, Shao R, Cameron SL, Barker SC (2006) Extraordinary amounts of gene rearrangement in the mitochondrial genomes of lice (Insecta: Phthiraptera). Insect Mol Biol 15:63–68
Cameron SL, Johnson KP, Whiting MF (2007) The mitochondrial genome of the screamer louse Bothriometopus (Phthiraptera: Ischnocera): effects of extensive gene rearrangements on the evolution of the genome. J Mol Evol 65:589–604
Boore JL, Brown WM (1998) Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Opin Genet Dev 8:668–674
Boore JL, Lavrov DV, Brown WM (1998) Gene translocation links insects and crustaceans. Nature 392:667–668
Dowton M, Castro LR, Austin AD (2002) Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: the examination of genome ‘morphology’. Invertebr Syst 16:345–356
Kurabayashi A, Sumida M, Yonekawa H, Glaw F, Vences M, Hasegawa M (2008) Phylogeny, recombination, and mechanisms of stepwise mitochondrial genome reorganization in mantellid frogs from Madagascar. Mol Biol Evol 25:874–891
Dowton M, Castro LR, Campbell SL, Bargon SD, Austin AD (2003) Frequent mitochondrial gene rearrangements at the hymenopteran nad3-nad5 junction. J Mol Evol 56:517–526
Crozier RH, Crozier YC (1993) The mitochondrial genome of the honeybee Apis mellifera: complete sequence and genome organization. Genetics 133:97–117
Cha SY, Yoon HJ, Lee EM, Yoon MH, Hwang JS, Jin BR, Han YS, Kim I (2007) The complete nucleotide sequence and gene organization of the mitochondrial genome of the bumblebee, Bombus ignitus (Hymenoptera: Apidae). Gene 392:206–220
Castro LR, Ruberu K, Dowton M (2006) Mitochondrial genomes of Vanhornia eucnemidarum (Apocrita: Vanhorniidae) and Primeuchroeus spp. (Aculeata: Chrysididae): evidence of rearranged mitochondrial genomes within the Apocrita (Insecta: Hymenoptera). Genome 49:752–766
Oliveira D, Raychoudhury R, Lavrov DV, Werren JH (2008) Rapidly evolving mitochondrial genome and directional selection in mitochondrial genes in the parasitic wasp Nasonia (Hymenoptera: Pteromalidae). Mol Biol Evol 25:2167–2180
Cameron SL, Dowton M, Castro LR, Ruberu K, Whiting MF, Austin AD, Diement K, Stevens J (2008) Mitochondrial genome organization and phylogeny of two vespid wasps. Genome 51:800–808
Wei SJ, Shi M, He JH, Sharkey MJ, Chen XX (2009) The complete mitochondrial genome of Diadegma semiclausum (Hymenoptera: Ichneumonidae) indicates extensive independent evolutionary events. Genome 52:308–319
Dowton M, Austin AD (1995) Increased genetic diversity in mitochondrial genes is correlated with the evolution of parasitism in the Hymenoptera. J Mol Evol 41:958–965
Dowton M, Austin A, Dillon N, Bartowsky E (1997) Molecular phylogeny of the apocritan wasps: the Proctotrupomorpha and Evaniomorpha. Syst Entomol 22:245–255
Sharkey MJ (2007) Phylogeny and classification of Hymenoptera. Zootaxa 1668:521–548
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P (1994) Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am 87:651–701
Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT (2006) Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu Rev Ecol Syst 37:545–579
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Castro LR, Dowton M (2005) The position of the Hymenoptera within the Holometabola as inferred from the mitochondrial genome of Perga condei (Hymenoptera: Symphyta: Pergidae). Mol Phylogen Evol 34:469–479
Silvestre D, Arias MC (2006) Mitochondrial tRNA gene translocations in highly eusocial bees. Genet Mol Biol 29:572–575
Macey JR, Larson A, Ananjeva NB, Fang ZL, Papenfuss TJ (1997) Two novel gene orders and the role of light-strand replication in rearrangement of the vertebrate mitochondrial genome. Mol Biol Evol 14:91–104
San Mauro D, Gower DJ, Zardoya R, Wilkinson M (2006) A hotspot of gene order rearrangement by tandem duplication and random loss in the vertebrate mitochondrial genome. Mol Biol Evol 23:227–234
Goddard JM, Wolstenholme DR (1980) Origin and direction of replication in mitochondrial DNA molecules from the genus Drosophila. Nucleic Acids Res 8:741–757
Sheffield NC, Song H, Cameron SL, Whiting MF (2008) A comparative analysis of mitochondrial genomes in Coleoptera (Arthropoda: Insecta) and genome descriptions of six new beetles. Mol Biol Evol 25:2499–2509
Fenn JD, Cameron SL, Whiting MF (2007) The complete mitochondrial genome of the Mormon cricket (Anabrus simplex: Tettigoniidae: Orthoptera) and an analysis of control region variability. Insect Mol Biol 16:239–252
Cameron SL, Whiting MF (2008) The complete mitochondrial genome of the tobacco hornworm, Manduca sexta,(Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 408:112–123
Bae JS, Kim I, Sohn HD, Jin BR (2004) The mitochondrial genome of the firefly, Pyrocoelia rufa: complete DNA sequence, genome organization, and phylogenetic analysis with other insects. Mol Phylogen Evol 32:978–985
Mitchell SE, Cockburn AF, Seawright JA (1993) The mitochondrial genome of Anopheles quadrimaculatus species A: complete nucleotide sequence and gene organization. Genome 36:1058–1073
Masta SE, Boore JL (2004) The complete mitochondrial genome sequence of the spider Habronattus oregonensis reveals rearranged and extremely truncated tRNAs. Mol Biol Evol 21:893–902
Beard CB, Hamm DM, Collins FH (1993) The mitochondrial genome of the mosquito Anopheles gambiae: DNA sequence, genome organization, and comparisons with mitochondrial sequences of other insects. Insect Mol Biol 2:103–124
Chimnaronk S, Jeppesen MG, Suzuki T, Nyborg J, Watanabe K (2005) Dual-mode recognition of noncanonical tRNAs Ser by seryl-tRNA synthetase in mammalian mitochondria. EMBO J 24:3369–3379
Steinberg S, Cedergren R (1994) Structural compensation in atypical mitochondrial tRNAs. Nat Struct Biol 1:507–510
Kim I, Cha SY, Yoon MH, Hwang JS, Lee SM, Sohn HD, Jin BR (2005) The complete nucleotide sequence and gene organization of the mitochondrial genome of the oriental mole cricket, Gryllotalpa orientalis (Orthoptera: Gryllotalpidae). Gene 353:155–168
Yokobori S, Paeaebo S (1995) tRNA editing in metazoans. Nature 377:490
Yokobori S, Paabo S (1995) Transfer RNA editing in land snail mitochondria. Proc Natl Acad Sci USA 92:10432–10435
Yokobori S, Paabo S (1997) Polyadenylation creates the discriminator nucleotide of chicken mitochondrial tRNA(Tyr). J Mol Biol 265:95–99
Tomita K, Ueda T, Watanabe K (1996) RNA editing in the acceptor stem of squid mitochondrial tRNA(Tyr). Nucleic Acids Res 24:4987–4991
Lonergan KM, Gray MW (1993) Editing of transfer RNAs in Acanthamoeba castellanii mitochondria. Science 259:812–816
Laforest MJ, Roewer I, Lang BF (1997) Mitochondrial tRNAs in the lower fungus Spizellomyces punctatus: tRNA editing and UAG ‘stop’ codons recognized as leucine. Nucleic Acids Res 25:626–632
Borner GV, Yokobori S, Morl M, Dorner M, Paabo S (1997) RNA editing in metazoan mitochondria: staying fit without sex. FEBS Lett 409:320–324
Gillespie JJ, Johnston JS, Cannone JJ, Gutell RR (2006) Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): structure, organization, and retrotransposable elements. Insect Mol Biol 15:657–686
Cannone JJ, Subramanian S, Schnare MN, Collett JR, D’Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Müller KM (2002) The comparative RNA web(CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics 3. doi: 10.1186/1471-2105-3-2
Schnare MN, Damberger SH, Gray MW, Gutell RR (1996) Comprehensive comparison of structural characteristics in eukaryotic cytoplasmic large subunit (23S-like) ribosomal RNA. J Mol Biol 256:701–719
Buckley TR, Simon C, Flook PK, Misof B (2000) Secondary structure and conserved motifs of the frequently sequenced domains IV and V of the insect mitochondrial large subunit rRNA gene. Insect Mol Biol 9:565–580
Misof B, Fleck G (2003) Comparative analysis of mt LSU rRNA secondary structures of Odonates: structural variability and phylogenetic signal. Insect Mol Biol 12:535–548
Niehuis O, Naumann CM, Misof B (2006) Identification of evolutionary conserved structural elements in the mt SSU rRNA of Zygaenoidea (Lepidoptera): a comparative sequence analysis. Org Divers Evol 6:17–32
Hickson RE (1996) Conserved sequence motifs, alignment, and secondary structure for the third domain of animal 12S rRNA. Mol Biol Evol 13:150–169
Page RDM (2000) Comparative analysis of secondary structure of insect mitochondrial small subunit ribosomal RNA using maximum weighted matching. Nucleic Acids Res 28:3839–3845
Dotson EM, Beard CB (2001) Sequence and organization of the mitochondrial genome of the Chagas disease vector, Triatoma dimidiata. Insect Mol Biol 10:205–215
Cornuet JM, Garnery L, Solignac M (1991) Putative origin and function of the intergenic region between COI and COII of Apis mellifera L. mitochondrial DNA. Genetics 128:393–403
Taanman JW (1999) The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta 1410:103–123
Campbell NJH, Barker SC (1999) The novel mitochondrial gene arrangement of the cattle tick, Boophilus microplus: fivefold tandem repetition of a coding region. Mol Biol Evol 16:732–740
Bibb MJ, Van Etten RA, Wright CT, Walberg MW, Clayton DA (1981) Sequence and gene organization of mouse mitochondrial DNA. Cell 26:167–180
Clary DO, Wolstenholme DR (1985) The mitochondrial DNA molecule of Drosophila yakuba: nucleotide sequence, gene organization, and genetic code. J Mol Evol 22:252–271
Lewis DL, Farr CL, Kaguni LS (1995) Drosophila melanogaster mitochondrial DNA: completion of the nucleotide sequence and evolutionary comparisons. Insect Mol Biol 4:263–278
Acknowledgments
We thank Jing-xian Liu for his collecting the specimens, Dr. Andrew R. Deans (North Carolina State University, USA) for his identification of the species, and Prof. Michael J. Sharkey (University of Kentucky, USA) for his improving the manuscript. Funding for this study was provided jointly by the National Science Fund for Distinguished Young Scholars (No. 30625006), 973 Program (No. 2006CB102005), the National Science Foundation of China (30499341, 30570193, 30700063, 30871675), the National S&T Infrastructure Project (2005DKA21402, 2005DKA21105), and the National Special Basic Research Funds (2006FY110500-3, 2006FY120100).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wei, Sj., Tang, P., Zheng, Lh. et al. The complete mitochondrial genome of Evania appendigaster (Hymenoptera: Evaniidae) has low A+T content and a long intergenic spacer between atp8 and atp6 . Mol Biol Rep 37, 1931–1942 (2010). https://doi.org/10.1007/s11033-009-9640-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-009-9640-1