
Abstract
The Trigonidiidae, a family of crickets, comprises 981 valid species with only one mitochondrial genome (mitogenome) sequenced to date. To explore mitogenome features of Trigonidiidae, six mitogenomes from its two subfamilies (Nemobiinae and Trigonidiinae) were determined. Two types of gene rearrangements involving a trnN-trnS1-trnE inversion and a trnV shuffling were shared by Trigonidiidae. A long intergenic spacer was observed between trnQ and trnM in Trigonidiinae (210−369 bp) and Nemobiinae (80–216 bp), which was capable of forming extensive stem-loop secondary structures in Trigonidiinae but not in Nemobiinae. The anticodon of trnS1 was TCT in Trigonidiinae, rather than GCT in Nemobiinae and other related subfamilies. There was no overlap between nad4 and nad4l in Dianemobius, as opposed to a conserved 7-bp overlap commonly found in insects. Furthermore, combined comparative analysis and transcript verification revealed that nad1 transcripts ended with a U, corresponding to the T immediately preceding a conserved motif GAGAC in the superfamily Grylloidea, plus poly-A tails. The resultant UAA served as a stop codon for species lacking full stop codons upstream of the motif. Our findings gain novel understanding of mitogenome structural diversity and provide insight into accurate mitogenome annotation.
Similar content being viewed by others


Introduction
The typical mitochondrial genome (mitogenome) of insects is a circular molecule ranging in size from 15 kb to 18 kb1. It harbors 37 genes including two ribosomal RNA (rRNA) genes, 22 transfer RNA (tRNA) genes, and 13 protein-coding genes (PCGs). These genes are mainly arranged in a conserved order that is regarded as the ancestral gene arrangement of insects. The mitogenome also contains non-coding regulatory regions, including a control region with initiation sites for replication and transcription1 and, in most insect orders, an intergenic spacer (IGS) between nad1 and trnS2 with a conserved binding site for the transcription termination factor2,3. During transcription, insect mitogenome is first transcribed into polycistronic primary transcripts, which are processed into mature transcripts including 33 single gene transcripts and two bicistronic transcripts encoding the atp8/atp6 and nad4l/nad4 gene pairs4. Typically, a 7-bp overlap exists between the two genes forming the bicistronic transcripts3. Insect mitogenome provides an abundant source of sequence data for population and phylogenetic studies1,3,5,6,7,8. In addition, structural features such as gene rearrangements and stem-loop secondary structures are used as supplementary markers to trace evolutionary history8,9.
The Trigonidiidae is a worldwide distributed family of crickets. It encompasses two subfamilies, Nemobiinae (ground crickets) and Trigonidiinae (sword-tailed crickets), and 981 extant species10. Many members are well-known for calling songs and are kept as pets for entertainment in Asian and European countries, especially in China and Japan11,12. Despite the high species diversity of the family Trigonidiidae, its mitogenome information is severely underrepresented. To date, only the complete mitogenome of Trigonidium sjostedti (Chopard, 1925)6 from Trigonidiinae has been sequenced within the whole family. It shows unique structural features, including particularly the occurrence of two types of gene rearrangements: an inversion of trnN-trnS1-trnE encoded on the majority strand to trnE-trnS1-trnN on the minority strand and a shuffling of trnV to the position between rrnS and the control region. The inversion is also present in two closely related families, Gryllidae and Phalangopsidae, and it is thus proposed that the inversion is a synapomorphy of the three families13. To consolidate this proposal, an expanded mitogenome sampling is critical especially for Trigonidiidae with the single representative mitogenome available so far. As for the trnV shuffling, it is not reported in closely related families except for Ornebius bimaculatus (Shiraki, 1930)13. Still, it remains yet to be explored whether the trnV shuffling in Trigonidiidae is specific to T. sjostedti or conserved at a higher taxonomic level. Worse still, there has not been even an attempt to investigate mitogenome features of the other subfamily Nemobiinae. There is thus an urgent need of mitogenome sequencing of Trigonidiidae, which can shed light on the phylogenetic distribution and evolutionary origin of these rearrangements and other features.
To fill this gap, three representative mitogenomes (Dianemobius fascipes (Walker, 1869), D. furumagiensis (Ohmachi & Furukawa, 1929), and Polionemobius taprobanensis (Walker, 1869)) from Nemobiinae and three (Homoeoxipha nigripes Xia & Liu, 1992, Natula pravdini (Gorochov, 1985), and Svistella anhuiensis He, Li & Liu, 2009) from Trigonidiinae were sequenced and annotated in this study. A detailed comparison of structural features among these mitogenomes was performed and, to place the analysis in a broader context, additional mitogenomes currently available for Grylloidea (a superfamily of crickets) was included. Comparative analysis reveals gene rearrangement patterns and a series of other shared and unique structural features, contributing to our understanding of structural diversity of insect mitogenomes. More importantly, we provide a means to ensure accurate annotation of nad1 stop codons for Grylloidea.
Methods
High-throughput sequencing
D. fascipes and P. taprobanensis samples were collected in field from Shandong, China, while D. furumagiensis, S. anhuiensis, H. nigripes, and N. pravdini were purchased from a flower and bird market in Shanghai, China. To determine the 3′ end of nad1 transcript, an additional species Truljalia hibinonis (Matsumura, 1917) from the family Gryllidae was collected from Shanghai, China. These samples were preserved in 100% ethanol at 4 °C prior to mitogenome sequencing and were preserved directly at −80 °C before RNA analysis. Genomic DNA was extracted from a single specimen for each species using a DNeasy Blood & Tissue kit (Qiagen, Hilden, Germany). The DNA was utilized for 150-bp paired-end sequencing on the Illumina HiSeq. 2500 platform (Novogene, Beijing, China) following the manufacturer′s protocol. After removal of poor-quality reads with Trimmomatic14, the remaining reads were used to assemble the full mitogenome by SOAPdenovo-Trans15 and MITObim v1.916 with parameter setting as per Ma and Li13.
PCR amplification and Sanger sequencing
To avoid short-read misassembly of non-coding regions mainly due to potential presence of long tandem repeats13, PCR amplification (see Table S1 for PCR primers) followed by Sanger sequencing was performed spanning the control region and the IGS between trnQ and trnM. The amplification was performed with LA Taq™ (TaKaRa, Dalian, China) under the following conditions: 95 °C for 1 min; 30 cycles of 98 °C for 10 s, 50 °C for 10 s, and 65 °C for 3 min; 65 °C for 5 min. Purified PCR products were sequenced using 3730xl DNA Analyser (Applied Biosystems, CA, USA).
Mitogenome annotation
Mitogenome annotation was performed on the MITOS webserver (http://mitos.bioinf.uni-leipzig.de/index.py)17 followed by manual correction. To validate nad1 stop codons, the 3′ end of nad1 transcripts in D. furumagiensis, N. pravdini, P. taprobanensis, S. anhuiensis, and T. hibinonis was amplified with a SMARTer RACE 5′/3′ Kit (Clontech, CA, USA) and sequenced with Sanger sequencing. Specifically, total RNA was extracted from −80 °C stored samples with TRIzol (Invitrogen, CA, USA) and used for first-strand cDNA synthesis. Rapid amplification of cDNA ends (RACE) was performed with gene-specific primers (Table S1) and the following cycling conditions: 94 °C for 30 s; 5 cycles of 94 °C for 30 s, 68 °C for 30 s, and 72 °C for 60 s; 27 cycles of 94 °C for 30 s, 65 °C for 30 s, and 72 °C for 60 s. Purified PCR products were sequenced directly using 3730xl DNA Analyser (Applied Biosystems, CA, USA). To test whether nad4 and nad4l were expressed in a bicistronic mRNA, 5′ RACE of nad4 transcript and 3′ RACE of nad4l transcript of D. furumagiensis were performed employing the above methods (primers are listed in Table S1). The mitogenome sequences were deposited in GenBank (Table 1).
Sequence analysis
Tandem repeats were identified using the online Tandem Repeats Finder with default setting18. Secondary structures of IGSs and trnS1 were predicted with the Mfold19 and the MITOS webserver17, respectively, and drawn with VARNA20. Nucleotide composition was calculated with DAMBE21. AT skew [(A − T)/(A + T)] and GC skew [(G − C)/(G + C)]22 were used to measure strand asymmetry in nucleotide compositions. To conduct a comparative analysis at a higher taxonomic level, 16 more mitogenomes in the superfamily Grylloidea were downloaded from GenBank and combined with the six newly sequenced mitogenomes (Table 1). DNA sequences were aligned using MUSCLE23 with manual refinement for nad1 and neighboring regions to arrange the incomplete stop codon T and the immediately adjacent motif GAGAC (see Results and Discussion) in a successive manner.
Phylogenetic analysis
Four additional mitogenomes (Gryllotalpa orientalis Burmeister, 1838, NC_006678; Myrmecophilus manni Schimmer, 1911, NC_011301; Troglophilus neglectus Krauss, 1879, NC_011306; Comicus campestris Irish, 1986, NC_028062) from closely related superfamilies (Gryllotalpoidea, Rhaphidophoroidea, and Schizodactyloidea) were selected to root the tree of the superfamily Grylloidea. The 37 genes were each aligned in MUSCLE23 and trimmed in Gblocks24 before concatenation into a single dataset. Mutational saturation test in DAMBE21 by plotting observed substitutions against GTR-corrected genetic distances indicated severe saturation of third codon positions of the PCGs, which was also observed in a previous study13. The third codons were thus removed from phylogenetic analysis. Partition schemes and substitution models for each gene and codon position were selected in PartitionFinder v2.1.125. Four partitions each with GTR + I + G substitution model was set for the Bayesian tree reconstruction using MrBayes v3.226. The analysis was run for 10 million generations with a sampling frequency of 1000 generations and a burnin of 25%. A maximum likelihood tree was inferred using RAxML v8.2.1027 with the GTRGAMMA substitution model and four partitions identified in PartitionFinder v2.1.125. Branch support was assessed by 1000 bootstrap replicates.
Results and Discussion
Mitogenome organization
Mitogenomes of six representative species belonging to the family Trigonidiidae were determined in this study. Of them, five full-length mitogenome sequences were obtained, ranging in size from 15,350 bp in D. furumagiensis to 16,494 bp in S. anhuiensis (Table 1). These sizes fell within those of typical insect mitogenomes1,28. P. taprobanensis had a larger size of more than 16,641 bp, mainly due to the occurrence of multiple 84-bp tandem repeats in the IGS between trnK and trnD; however, the whole IGS was not sequenced through (see below). Like other insects, the six species showed a strong bias toward A + T, accounting for 70.34–78.77% of nucleotide composition. Moreover, highly negative GC skew values (an excess of C relative to G) and different AT skew patterns between the two subfamilies (more T for Nemobiinae and more A for Trigonidiinae) were observed (Table 1). All six mitogenomes encoded the typical set of 37 genes for insects, i.e. 13 PCGs, 22 tRNA genes, and two rRNA genes. Nine PCGs and 11 tRNA genes were encoded on the majority strand while the other genes were on the minority strand.
Gene rearrangements and phylogenetic distribution
Relative to the ancestral gene arrangement of insects1, which occurs in the majority of insect mitogenomes, the six mitogenomes possessed two types of gene rearrangements including an inversion of trnN-trnS1-trnE encoded on the majority strand to trnE-trnS1-trnN on the minority strand and a shuffling of trnV to the position between rrnS and the control region (Fig. 1). The same gene rearrangements have also been observed in T. sjostedti6, the sole mitogenome of the family Trigonidiidae available in GenBank before our work. These species formed a monophyletic clade in the phylogenetic tree (Fig. 2), supporting the monophyly of the family Trigonidiidae29. The gene rearrangement pattern seems therefore to be a shared character for the family Trigonidiidae. The trnN-trnS1-trnE inversion has also been reported in all currently sequenced mitogenomes of two closely related families, Gryllidae and Phalangopsidae, which were supported to be sister to Trigonidiidae here (Fig. 2) and in previous studies13,29. Such a phylogenetic distribution pattern suggests that the trnN-trnS1-trnE inversion event happened in the most recent common ancestor of Gryllidae, Phalangopsidae, and Trigonidiidae, corroborating the proposal by Ma and Li13. Among all other sequenced mitogenomes of the suborder Ensifera, the trnV shuffling occurs exclusively in O. bimaculatus of the family Mogoplistidae13. The non-monophyly of the species with the trnV shuffling (Fig. 2) suggests that this rearrangement occurred independently twice in history, one in the common ancestor of Trigonidiidae and the other in O. bimaculatus.

Linearized representation of mitochondrial gene order. The two types of gene rearrangements (the trnN-trnS1-trnE inversion and the trnV shuffling) in the six mitogenomes are indicated. Genes encoded on the minority strand are shaded. tRNA genes are labeled with the one-letter code for the corresponding amino acid.

The Bayesian tree of the superfamily Grylloidea. Bayesian posterior probabilities and the maximum likelihood support values are indicated at nodes. Species with the trnV shuffling rearrangement are underlined, while the clade with the trnN-trnS1-trnE inversion is labeled with an arrow. The clade with an anticodon TCT for trnS1 is shaded.
Protein-coding genes
Most PCGs started with ATG, ATT, and ATA, which were typical start codons for insect mitochondrial PCGs. The exceptions were cox1 starting with CCG in all but P. taprobanensis, nad3 with TTG in P. taprobanensis, and nad1 with CTG in D. furumagiensis and TTG in D. fascipes, H. nigripes, P. taprobanensis, and S. anhuiensis. These anomalous start codons were proposed previously13,30 and among them, CCG for cox113 and TTG for nad14,31 have been validated by transcript information.
Both complete (TAA and TAG) and partial stop codons (T and TA) were employed by PCGs in the six mitogenomes. As in other mitogenomes, these partial stop codons always immediately preceded a tRNA gene and could be converted into a complete stop codon (TAA) by the addition of poly-A tails during polyadenylation after tRNA cleavage4,32. Nevertheless, it seemed problematic to annotate nad1 stop codons based on sequence alignments alone due to high variability in their 3′-end sequences (Fig. 3) and a utilization of a partial stop codon (T) not immediately followed by a tRNA gene13. Since RNA data are useful to characterize the 3′-ends of mitochondrial transcripts4,33, 3′ RACE and Sanger sequencing of nad1 transcripts were performed for D. furumagiensis, N. pravdini, P. taprobanensis, and S. anhuiensis in our present study. An additional species T. hibinonis was also sampled to provide further evidence. We found that they all terminated with a U plus a poly-A tail (Fig. 3), rendering the formation of a complete stop codon for P. taprobanensis and S. anhuiensis. The same observation was also noticed in the cases of two Ornebius species in our former study13. Ending at this position resulted in untranslated nucleotides in nad1 transcripts in D. furumagiensis, N. pravdini, and T. hibinonis (Fig. 3). The presence of untranslated nucleotides is also reported in mitochondrial transcripts including nad1 in Drosophila4. The U abutting poly-A tails corresponded to the T flanked by GAGAC, a highly conserved motif in the mitogenomes across the superfamily Grylloidea (Fig. 3). These findings suggest that the conserved motif could provide recognition signals for the precise cleavage between the U and GAGAC during mRNA maturation in Grylloidea. Although we did not find the same motif in other insects, a conserved binding site for transcription termination factor is located between nad1 and trnS2 in most insect orders2,3. It is probable that the conserved motif in Grylloidea also functions through binding the transcription termination factor to stop transcription, enabling the direct formation of the 3′-end of nad1 mRNA. Notably, there were indeed in-frame codons TAG and TAA located between the conserved motif and downstream trnS2 in P. taprobanensis and S. anhuiensis, respectively, but they were cleaved from mature transcripts as revealed by the 3′ RACE (Fig. 3), excluding the possibility that they acted as stop codons. Accordingly, the previously designated complete stop codons in Grylloidea, including TAA in T. sjostedti6, which were located downstream of the conserved motif, were expected to be removed during posttranscriptional processing. In these cases, nad1 should be revised to terminate with the T immediately preceding the conserved motif. Collectively, we pinpoint the precise 3′-ends of nad1 transcripts in Grylloidea and provide experimental evidence for the GAGAC-flanked T as a partial stop codon in cases of the absence of a full stop codon upstream of this motif.

Alignments of nad1 ends, partial trnS2, and the IGS. Sequences are from the minority strand. Complete (TAA and TAG) and partial stop codons (T) are highlighted in grey. The 3′ ends of nad1 transcripts were verified by 3′ RACE and Sanger sequencing for the five underlined species in our study and two Ornebius species in Ma and Li13. The position of poly-A tails in mature nad1 transcripts is indicated by an arrow. Dots represent nucleotides identical to those in X. marmoratus, and dashes refer to gaps in the alignment.
An identical 7-bp overlap was found between nad4 and nad4l in four of the six mitogenomes sequenced in our study, and the overlap was highly homologous in Grylloidea (Fig. 4). The presence of a 7-bp overlap at this location is almost a common feature in insects3 and the two genes can form a mature bicistronic transcript4. Few exceptions to the 7-bp overlap have been reported, e.g. a 40-bp IGS in a lepidopteran species34. Here, we found two more exceptions, i.e. no overlap or IGS between nad4 and nad4l in D. fascipes and D. furumagiensis. Further, 5′ RACE of nad4 transcript and 3′ RACE of nad4l transcript in D. furumagiensis supported a single mRNA comprised of the two tightly adjoining genes (Fig. 4). Our finding demonstrates that nad4 and nad4l in D. fascipes and D. furumagiensis are still transcribed and processed into a mature bicistronic mRNA as in other insects4.

Sequence alignments of the beginning of nad4 and the end of nad4l. Sequences are from the minority strand. Proposed start codons of nad4 and stop codons of nad4l are boxed and shaded, respectively. In D. furumagiensis, nad4 and nad4l genes exist in a bicistronic mRNA as evidenced by transcript-end sequencing.
Transfer and ribosomal RNA genes
The tRNA genes ranged in size from 60 bp (trnC in P. taprobanensis) to 72 bp (trnI in N. pravdini). Compared with the regular cloverleaf secondary structure of tRNAs35, trnS1 exhibited different structural features (Fig. 5). The acceptor stem was comprised of 6 bp in the three species of the subfamily Nemobiinae. The anticodon stem lacked 1 bp in N. pravdini and had an extra unpaired nucleotide in S. anhuiensis and T. sjostedti. A stable dihydrouridine arm was missing in the subfamily Trigonidiinae. Instead, it was replaced by an unpaired stretch of 15 nucleotides in N. pravdini and 12 nucleotides in H. nigripes, S. anhuiensis, and T. sjostedti. The lack of a stable dihydrouridine arm is a common feature for trnS1 in nearly all Metazoa36. By contrast, the same dihydrouridine arm consisting of one G-T and two A-T pairs was observed in the three species of Nemobiinae but they all lacked a typical dihydrouridine loop.

Atypical cloverleaf structures of trnS1 in the family Trigonidiidae.
With the exception of trnS1, the tRNA genes each shared the same anticodon among the six species. For trnS1, the anticodon was TCT in Trigonidiinae including T. sjostedti, whereas it was GCT in the three species of Nemobiinae (Fig. 5). Moreover, we found a conserved GCT anticodon for trnS1 in all other species of the suborder Ensifera hitherto sequenced. These findings indicate that GCT is an ancestral anticodon while TCT is a derived form. Furthermore, the monophyly of Trigonidiinae in our (Fig. 2) and a previous study29 suggests a single G-to-T substitution event that occurred in the common ancestor of the subfamily Trigonidiinae.
The two rRNA genes (rrnL and rrnS) ranged in size from 1,273 bp to 1,312 bp and 753 bp to 797 bp, respectively. Their nucleotide composition was highly A + T-biased (73.76–79.07% for rrnL and 70.51–79.71% for rrnS). Both genes adjoined each other and were encoded by the minority strand.
Non-coding regions
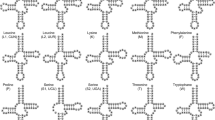
Non-coding regions including IGSs and the control region were identified in the six mitogenomes. Strikingly, a long IGS was detected between trnQ and trnM with a length of 210–369 bp for Trigonidiinae and 80–216 bp for Nemobiinae. Although the primary sequences varied substantially, the IGSs in Trigonidiinae were all capable of forming stable stem-loop secondary structures with a delta G value of −74.00~−51.45 kcal/mol (Fig. 6). A consecutive and long stem was recognized in S. anhuiensis (72 bp) and T. sjostedti (52 bp), and the stem was even longer with multiple mismatches or bulges in H. nigripes and N. pravdini. On the contrary, no obvious stem-loop structures were detected in the IGSs of Nemobiinae (delta G = −6.48~−1.87 kcal/mol). The IGS is 94 bp in Xenogryllus marmoratus (Haan, 1844)37 with a delta G of −1.67 kcal/mol and is no more than 23 bp in all other sequenced mitogenomes of Grylloidea. There is even 1-bp overlap between trnQ and trnM in T. hibinonis38. At a higher taxonomic level of the suborder Ensifera, the longest IGS (465 bp) between trnQ and trnM is reported in a Deflorita species7, but it could not form stable stem-loop structures (delta G = −15.61 kcal/mol).

Predicted secondary structures of the IGS between trnQ and trnM in Trigonidiidae. Delta G values are shown in parentheses.
The assembled control region sequences were validated by PCR amplification followed by Sanger sequencing. The control region ranged in length from 516 bp to 1,416 bp for the six species and showed a strong preference for A and T (81.12–88.45%). A 410-bp motif was found to be tandemly repeated 3.0 times in S. anhuiensis, and similar repeats larger than 100 bp were not detected in the five other species. The occurrence of tandem repeats is widely favored to be a consequence of strand slippage and mispairing during mtDNA replication39. The repeat motif in S. anhuiensis was capable of forming multiple stem-loop secondary structures (delta G = −42.61 kcal/mol), which could enhance the probability of replication slippage by stabilizing slipped strands or blocking the polymerase40.
A novel non-coding region larger than 893 bp was exclusively identified between trnK and trnD in P. taprobanensis. More than five 84-bp tandem repeats were sequenced at each end of this region but the entire repeat region was not sequenced through with short-read data from Illumina sequencing. It is suggested to validate non-coding regions possibly containing repeats via PCR amplification followed by Sanger sequencing13. After many attempts, however, we failed to amplify this region. Instead, we obtained a massive smear of electrophoretic bands, a prevalent observation for PCR amplification of repetitive DNAs41. The exact copy number of these repeats remained therefore unknown.
Conclusion
Comparative analysis of the six representative mitogenomes of the family Trigonidiidae and those from related species reveals common and unique structural features including gene rearrangements, large-sized IGSs with secondary structures, altered tRNA anticodons, and no overlap between adjacent nad4 and nad4l. These findings add to our knowledge of structural diversity of insect mitogenomes. Transcript end sequencing demonstrates that mature nad1 mRNA terminates with a U, corresponding to the T immediately followed by a highly conserved motif GAGAC in Grylloidea, plus a poly-A tail generated through polyadenylation. The GAGAC-flanked T acts as a partial stop codon in cases where a full stop codon upstream of this motif lacks. Our study thus provides a guide toward reliable annotation of nad1 stop codons in Grylloidea and has implications for understanding the process of mitochondrial transcript maturation.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Cameron, S. L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117 (2014).
Cameron, S. L. & Whiting, M. F. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 408, 112–123 (2008).
Cameron, S. L. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research. Syst. Entomol. 39, 400–411 (2014).
Stewart, J. B. & Beckenbach, A. T. Characterization of mature mitochondrial transcripts in Drosophila, and the implications for the tRNA punctuation model in arthropods. Gene 445, 49–57 (2009).
Ma, C. et al. Mitochondrial genomes reveal the global phylogeography and dispersal routes of the migratory locust. Mol. Ecol. 21, 4344–4358 (2012).
Song, N., Li, H., Song, F. & Cai, W. Z. Molecular phylogeny of Polyneoptera (Insecta) inferred from expanded mitogenomic data. Sci. Rep. 6, https://doi.org/10.1038/Srep36175 (2016).
Zhou, Z. et al. Towards a higher-level Ensifera phylogeny inferred from mitogenome sequences. Mol. Phylogenet. Evol. 108, 22–33 (2017).
Cameron, S. L., Lo, N., Bourguignon, T., Svenson, G. J. & Evans, T. A. A mitochondrial genome phylogeny of termites (Blattodea: Termitoidae): robust support for interfamilial relationships and molecular synapomorphies define major clades. Mol. Phylogenet. Evol. 65, 163–173 (2012).
Dowton, M., Castro, L. R. & Austin, A. D. Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: the examination of genome ‘morphology’. Invertebr. Syst. 16, 345–356 (2002).
Cigliano, M. M., Braun, H., Eades, D. C. & Otte, D. Orthoptera Species File. http://Orthoptera.SpeciesFile.org (2019).
Ryan, L. G. Insect Musicians and Cricket Champions: a Cultural History of Singing Insects in China and Japan. (China Books & Periodicals, Inc., 1996).
Costa-Neto, E. M. Entertainment with insects: singing and fighting insects around the world. A brief review. Etnobiología 3, 20–28 (2003).
Ma, C. & Li, J. Comparative analysis of mitochondrial genomes of the superfamily Grylloidea (Insecta, Orthoptera) reveals phylogenetic distribution of gene rearrangements. Int. J. Biol. Macromol. 120, 1048–1054 (2018).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Xie, Y. L. et al. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30, 1660–1666 (2014).
Hahn, C., Bachmann, L. & Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 41, https://doi.org/10.1093/nar/gkt371 (2013).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003).
Darty, K., Denise, A. & Ponty, Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 25, (1974–1975 (2009).
Xia, X. H. DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 35, 1550–1552 (2018).
Xia, X. H. DNA replication and strand asymmetry in prokaryotic and mitochondrial genomes. Curr. Genomics 13, 16–27 (2012).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T. & Calcott, B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773 (2017).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Simon, S. & Hadrys, H. A comparative analysis of complete mitochondrial genomes among Hexapoda. Mol. Phylogenet. Evol. 69, 393–403 (2013).
Chintauan-Marquier, I. C. et al. Laying the foundations of evolutionary and systematic studies in crickets (Insecta, Orthoptera): a multilocus phylogenetic analysis. Cladistics 32, 54–81 (2016).
Sheffield, N. C., Hiatt, K. D., Valentine, M. C., Song, H. J. & Whiting, M. F. Mitochondrial genomics in Orthoptera using MOSAS. Mitochondr. DNA 21, 87–104 (2010).
Besansky, N. J., Krzywinski, J. & Grushko, O. G. Analysis of the complete mitochondrial DNA from Anopheles funestus: An improved dipteran mitochondrial genome annotation and a temporal dimension of mosquito evolution. Mol. Phylogenet. Evol. 39, 417–423 (2006).
Ojala, D., Montoya, J. & Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474 (1981).
Markova, S., Filipi, K., Searle, J. B. & Kotlik, P. Mapping 3 ‘ transcript ends in the bank vole (Clethrionomys glareolus) mitochondrial genome with RNA-Seq. BMC Genomics 16, https://doi.org/10.1186/s12864-015-2103-2 (2015).
Liu, Q. N. et al. The complete mitochondrial genome of fall armyworm Spodoptera frugiperda (Lepidoptera: Noctuidae). Genes & Genomics 38, 205–216 (2016).
Kinouchi, M., Kanaya, S., Ikemura, T. & Kudo, Y. Detection of tRNA based on the cloverleaf secondary structure. Genome Inform. 11, 301–302 (2000).
Jühling, F. et al. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 40, 2833–2845 (2012).
Ma, C., Zhang, L. & Li, J. Characterization of the complete mitochondrial genome of a bush cricket Xenogryllus marmoratus (Insecta: Orthoptera). Mitochondrial DNA Part B-Resour. 4, 172–173 (2019).
Li, J. J. et al. Phylogeny and acoustic signal evolution of a pure tone song katydid Pseudophyllus titan (Orthoptera: Tettigoniidae) based on the complete mitogenome. Mitochondrial DNA Part A 30, 385–396 (2019).
Levinson, G. & Gutman, G. A. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol. Biol. Evol. 4, 203–221 (1987).
Savolainen, P., Arvestad, L. & Lundeberg, J. mtDNA tandem repeats in domestic dogs and wolves: mutation mechanism studied by analysis of the sequence of imperfect repeats. Mol. Biol. Evol. 17, 474–488 (2000).
Hommelsheim, C. M., Frantzeskakis, L., Huang, M. M. & Ulker, B. PCR amplification of repetitive DNA: a limitation to genome editing technologies and many other applications. Sci. Rep. 4, https://doi.org/10.1038/srep05052 (2014).
Acknowledgements
The study was supported by the Natural Science Foundation of Beijing (5182031), the Joint Fund of the Natural Science Foundation of China and the Karst Science Research Center of Guizhou Province (U1812401), the National Natural Science Foundation of China (31400325), the Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2015-IAR), and the earmarked fund for Modern Agro-Industry Technology Research System (CARS-44) in China.
Author information
Authors and Affiliations
Contributions
C.M. designed the project, analyzed the data, and wrote the manuscript. Y.W. performed the experiments and analyzed the data. L.Z. performed the experiments. J.L. supervised the study and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, C., Wang, Y., Zhang, L. et al. Mitochondrial genome characterization of the family Trigonidiidae (Orthoptera) reveals novel structural features and nad1 transcript ends. Sci Rep 9, 19092 (2019). https://doi.org/10.1038/s41598-019-55740-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-55740-4
- Springer Nature Limited