
Abstract
The 2019-novel coronavirus has unfolded everywhere in the world and obliged a billion human beings in open confinement, whereas many treatments, and vaccines have been proposed towards this pandemic. The main protease (Mpro) is an attractive drug target due to the fact that it is the essential protein for virus invasion. This research tests in silico the effect of five vitamins towards Mpro, by employing molecular docking (MD), molecular dynamics simulation (MDS) with molecular mechanics–Poisson–Boltzmann surface area (MM–PBSA) studies. To achieve this work, we have applied some software’s as Autodock Vina, Discovery Studio Visualizer, APBS, and GROMACS. The inhibitors used were decided entirely on the basis of their importance in the production of red blood cells that prevent anemia, in lymphocyte immune system responses, in the regulation of reactive oxygen species production, such as tocopherol (vitamin E), thiamine (vitamin B1), pantothenic acid (vitamin B5), pyridoxine (vitamin B6), biotin (vitamin B7), and glutathione (GSH). The best inhibitor pose established at the highest repetition ratio (RR) and the minimal affinity energy value (MEV), then the best selected inhibitor was considered to MDS. The results indicate that GSH is the leading inhibitor model among the other tested vitamins in the active site of Mpro with a RR value of 94% and MEV of − 5.5 kcal/mol, its RMSD, RMSF, Rg, and hydrogen bonds show stability with Mpro. Furthermore, thiamine, biotin, and tocopherol are viewed as satisfying inhibitors to Mpro, but pyridoxine was observed as the weakest inhibitor. Based on our result, we could recommend the usage of glutathione and vitamin B family as a supportive strategy for feasible remedy of COVID-19 virus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Coronaviruses have long been one of the pathogens that affect human beings since it was discovered back in 1965 with its name, ‘corona,’ owing to the viruses' crown-like morphology, with its genus (Pyrc et al. 2007). There are now six species of coronaviruses identified, most of which attack the respiratory system, with the first identified being the cause of the common cold. The other types are likely to cause severe pneumonia, including severe acute respiratory syndrome-associated coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV), and the recent SARS-CoV-2 discovered first in Wuhan, China in December 2019, the latter of which has been the cause of the pandemic ravaging the world (Wu et al. 2020). The coronaviruses are single-stranded RNA viruses known for their high mutation rates measuring at 80–220 nm in diameter and are divided into four groups: alphaCoV (α), betaCoV (β), deltaCoV (γ), and gammaCoV (δ). The former two groups are infective to mammals while the latter two affect birds. The alphaCoVs are noted to have low pathogenicity, while the betaCoVs, like the SARS-CoV and the MERS-CoV reported pathogen, the SARS-CoV-2 was the focus of this study, it belongs to the betaCoV group, shares 79% and 50% of SARS-CoV and MERS-CoV, respectively (Guan et al. 2020; LeDuc and Barry 2004; Poutanen et al. 2003).
After infection with the SARS-CoV-2 patients will experience fever (38 °C or higher), dry hack, and tiredness. After an incubation under 14 days (Guan et al. 2020) it will in general be recovered without requiring any treatment for most conventional people (Chen et al. 2020), it cause respiratory difficulties; by infecting the lungs and cause the severe acute respiratory syndrome (SARS), It can inflame the respiratory tract. In severe cases, damage the alveoli. It spreads rapidly along the airways. The lining of the airways can be severely irritated. In some cases, lead to pneumonia, as the lungs fill with fluid causing inflammation and breathing difficulties. (Centers for Disease Control and Prevention n.d.; Ksiazek et al. 2003). Infection can be terribly deadly for older people or those with chronic conditions like diabetes and cardiovascular disease. Alternatively, age-related vulnerabilities—like malnutrition and frailty.
Many studies have provided evidence toward the importance of supplementation with vitamins and minerals against COVID-19 (Ahvanooei et al. 2021; Islam et al. 2021; Kumar et al. 2021; Schuetz et al. 2021; Shakoor et al. 2021; Srivastava et al. 2021) as indicated by their properties in boosting immunity, invulnerability and keeping up the neurological framework like vitamin B group, Vitamin B1 is an essential component for maintaining normal body homeostasis, it plays a fundamental role in reducing cellular oxidative stress, and metabolism via the thiamin diphosphate (ThDP) path (Bunik and Aleshin 2017), energy production, normal cellular functions, and development, (Reidling et al. 2010), its deficit was linked to neurological syndromes like Wernicke–Korsakoff syndrome caused by alcohol abuse; vitamin B1 based treatment was reported effective (Zubaran et al. 1997). Vitamin B5 is an essential nutrient used by the body to synthesis acyl carrier protein and coenzyme A (CoA); (Coates et al. 2010). CoA is essential for acetyl and acyl groups transfer, fatty acid synthesis and degradation (Thiamin 1998). It help generates energy from carbohydrates, fats, and proteins, vitamin B5 deficiency was reported in people with severe malnutrition, it can cause metabolic perturbation and neurodegeneration known as Huntington's disease (Patassini et al. 2019). Vitamin B6 and B7 are crucial coenzymes in the gluconeogenesis, fatty acid, amino acid, and sphingoid bases synthesis and more than 100 enzyme involved in the amino acids metabolism (Institute of Medicine Standing Committee on the Scientific Evaluation of Dietary Reference 1998). Vitamin B6 deficiency is often related to depression and confusion (Hawkins and Barsky 1948), seborrheic dermatitis (Mueller and Vilter 1950), microcytic anemia (decreased hemoglobin synthesis) (Snyderman et al. 1953), and epileptiform convulsions (Garty et al. 1962), low vitamin B6 levels is often related to depletion in neurotransmitters such as dopamine, serotonin, and γ-aminobutyrate due to its biochemical role as cofactor (PLP) for decarboxylases that are involved in neurotransmitter synthesis (Dakshinamurti and Stephens 1969). Vitamin E helps regulate the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS), and modulate signal transduction (Lee and Han 2018). Vitamin E deficiency cause neurologic abnormalities, especially ataxia characterized by sensory loss, and retinitis pigmentosa due to free radical mediated neuronal damage (Aslam et al. 2004). Glutathione is the main intracellular antioxidant. It has many functions, including protecting cells from oxidative stress, maintaining immune function and carcinogens. Low levels of glutathione are associated with an increased risk of cancer, cardiovascular disease and diabetes (Richie et al. 2015), and more seriously leads to idiopathic pulmonary fibrosis, human immunodeficiency virus disease (Herzenberg et al. 1997) and respiratory distress syndrome (White et al. 1994).
The present in silico study concentrated on the main protease (Mpro) known to be a core segment for the proteolytic development of the infection and confirmed as a potential objective viral protein (Anand et al. 2003; Jin et al. 2020). We utilized vitamins B1, B5, B6, B7, E, and GSH referred to cell reinforcements as expected inhibitors against Mpro, by performing docking via Autodock Vina and Discovery Studio Visualizer (DSV) packages followed by molecular dynamics simulation (MDS) and MM/PBSA by GROMACS 5.0.4 (http://www.gromacs.org/) (Abraham et al. 2015; Van Der Spoel et al. 2005). The root mean square deviation (RMSD), the root mean square fluctuations (RMSF), the radius of gyration (Rg), and hydrogen bond dissemination were computed to examine the stability of the complex Mpro–inhibitor.
Materials and Methods
Protein and Inhibitors Preparation
We worked with the crystal structure of the main protease (Mpro) (PDB ID 6lu7) as found in the Protein Data Bank (PDB; Jin Du 2020) and used as a receptor as shown in Fig. 1.

a The 3D structure of the main protease protein (PDB ID 6lu7) colored in blue, cyan and b zooming on the active site (colored in pink) and complexed with glutathione (presented with ball and stick) (Color figure online)
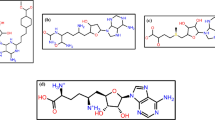
The five inhibitors considered in the present investigation are thiamine (vitamin B1), pantothenic acid (vitamin B5), pyridoxine (vitamin B6), Biotin (vitamin B7), α-tocopherol (vitamin E), and glutathione (GSH), their 2D structures are shown in Fig. 2.

The two-dimensional (2D) structures of the employed inhibitors
The three-dimensional structure (3D) of the six inhibitors was obtained from the PubChem database in SDF format (Kim et al. 2016).
Molecular Docking Settings
Molecular docking (MD) was performed using the Autodock Vina (Trott and Olson 2010) system via eight CPU work station that were used to make MD and set to provide full ligand (inhibitor) flexibility, MD was performed using blind type. MD consists of three main steps the first one; associated with the removal of ligands, heteroatoms, co-crystallized solvent, and unnecessary water molecules other than those involved in the active site described by Autodock Vina, in this case, three water molecules are stored inside a binding MPro packet. The second step consists of adding hydrogen and partial charges using Autodock Tools (ADT, version 1.5.4). The third step consists of setting the center of the grid box (GB) and the packet size of the grids connecting the x, y and z. The selected center setting and GB size were (x = − 26.28, y = 12.60, and z = 58.96), and (x = 52, y = 68, and z = 68), respectively, with grid points separated by one A° using ADT. The default settings are used unless the output conformation was set to one. The number of docking conformations is set to 50 poses. “Random Seed” is set to random. The optimum poses are selected based on small amounts of binding energy (MEV) in kcal/mol and the number of highly concentrated (RR) percentages (%) without polar hydrogen and hydrophobic interactions within the Mpro binding package. The results obtained for MD were analyzed using DSV, v 4.0 (Dassault Systèmes BIOVIA 2017; Linani et al. 2021; Serseg et al. 2021).
Molecular Dynamic Simulation
We have achieved the MDS exploiting GROMACS 5.0.4 (http://www.gromacs.org/) (Abraham et al. 2015) and the CHARMM36 force field (Huang et al. 2017; Malde et al. 2011) with the default water model in a working Ubuntu v20.04 station using 8 CPUs, with CUDA-based GPU acceleration, for 100 ns. All heteroatoms were deleted; the protein and ligand-network topologies were prepared separately (Zoete et al. 2011). A dodecahedron box was built as the most numerically efficient representation of the coordinates thereafter the sophisticated structures were placed in the center of the box and solved with water molecules, the ligands were checked for their bond orders, protonation, and tautomeric states, employing Avogadro v1.2.0 package (Hanwell et al. 2012) then uploaded into CGENFF server (Vanommeslaeghe et al. 2010) for validation and/or optimization of the high-penalty parameters and charges, the energy of each system was minimized (Pan and Roux 2008) to remove any steric clashes or unusual geometry by steepest descent algorithm using the input parameter EM file (Hess et al. 2008), to balance the solvent around the protein–ligand complex at the desired temperature and pressure, two phases of equilibration were carried out using two-step simulations (NVT and NPT) ensemble by LEAPFROG algorithm for 100 ps using a V-rescale modified Berendsen thermostat for both support parameter files, the protein was fixed in place while the solvent is allowed to move freely across it. After equilibrating the system, the MD production was run and defined with 50,000,000 steps. Finally, the results were analyzed for the RMSD, RMSF (root mean square fluctuation), hydrogen bond distribution, and Rg using VMD v1.9.3 program (Humphrey et al. 1996).
The Molecular Mechanics Poisson–Boltzmann Surface Area (MM–PBSA)
To achieve accurate binding free energies, we used for the calculation the known MM–PBSA method for GROMACS and APBS packages abbreviated “g_mmpbsa” tool (Kumari et al. 2014), the MM–PBSA (Eisenhaber et al. 1995; Pronk et al. 2013) is initiated under the Open Source Drug Discovery Consortium (OSDD; Bhardwaj et al. 2011), which is a collaborative platform to design and discover new drugs for neglected tropical diseases such as malaria, tuberculosis, leishmaniasis. The tool provides the calculation for three-energy components the first (a): the potential energy in vacuum as defined by the molecular mechanics (MM) model, which includes the Van der Waals, electrostatic interactions (Baker et al. 2001), and net non-bonded potential energy between the protein and inhibitor. The second (b): the polar solvation and non-polar solvation energies for solvent accessible surface area (SASA) model covers polar and non-polar solvation energies for unbound protein, unbound inhibitor, and protein–inhibitor complex. Third (3) the average binding energy calculation (ABEC). Include average and standard deviations of all energetic components including the binding energy (Kumari et al. 2014).
Results
Our results show that the SARS-CoV-2 Mpro is a medium-size proteases, it is a protein with two chains named A: composed of 306 residues and C: composed of 6 residues represents the peptide inhibitor named: n-[(5-methylisoxazol-3-yl)carbonyl]alanyl-l-valyl-n~1~-((1r,2z)-4(benzyloxy)-4-oxo-1([(3r)-2-oxopyrrolidin-3 yl]methyl)but-2-enyl)-l-leucinamide [12], this enzyme is with a total residues count of 312 amino acids and a molecular weight of 34 kDa (Fig. 1). Based on the repetition ratio (RR) and the minimum energy value (MEV) inside the limiting pocket of Mpro, the N3 interacts with the catalytic amino acids (CAA) and are His41, Leu141, Gly143, Ser144, Cys145, Glu166, Arg188, and Gln189, and all the internal interactions are conventional and carbon hydrogen bond type. These results show that glutathione is the best inhibitor among the other five vitamins with RR of 94% and with MEV of − 6.0 kcal/mol compared to the N3, in the other side, pyridoxine reported as the weakest inhibitor model with RR of 4% and MEV of − 4.8 kcal/mol. All results are summarized in Table 1.
Discussion
Molecular Docking
To comprehend the the mechanisms related with our inhibitors, we focus on the binding site investigation of the Mpro active site, from an underlying structural view. The active site forms an “I” shape and situated in a profound tunnel inside the enzyme in the upper side of chain A and shaped by the binding pocket (BP) amino acids. We characterized the BP relying upon the local inhibitor (N3) caught in an active catalysis moment. The BP amino acids recognized was His41, Cys44, Met49, Pro52, Tyr54, Tyr118, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166, Leu167, Pro168, His172, Phe181, Val186, ASP187, Arg188, Gln189, Thr190, Ala191, Gln192. We propose that all the inhibitors tested in this study bind covalently to the residue Cys145 or Leu141 in the Mpro active site through the determined interatomic distances, which may elucidate the high reactivity of the thiol group and the importance of these two amino acids in the active site.
To examine the interactions formed with our inhibitors, we characterized the (CAA) in each inhibitor model inside the dynamic site of Mpro, the CAA recorded against the vitamin B family (Fig. 2) including thiamine (vitamin B1), pantothenic acid (vitamin B5), Biotin (vitamin B7), and pyridoxine (vitamin B6) were (His41, Met49, Tyr54, Leu141, Asn142, Cys145, His163, His164, Met165, Glu166, His172, Asp187, Leu141, Gly143, Ser144), (Leu141, His163, His164, Asp187), (Leu141, Gly143, Ser144, Cys145), and (Cys145), respectively. We specified that the nucleophilic atoms reacting are the hydroxyl elements of the CAA addressed by the oxygen atom as the most negatively charged molecule toward the vitamin B family hydrogens, which introduced as the electrophilic atoms, the CAA answerable for the nucleophilic attack are Asn142 with 1.95 Å, Cys145 with 3.71 Å, for thiamine, pyridoxine, individually, and His164 for both biotin with 1.80 Å, and with pantothenic acid with 1.83 Å. These saved interactions were dominated hydrogen bond type. The thiamine (Fig. 3) was saved as the most preferred inhibitor in the active site of Mpro, among the other vitamin B family as indicated by the RR and MEV values (Table 1). We assume that by the structure flexibility, shape, and different chemical function properties, thus, assure its reactivity and stability which outline the high affinity to Mpro active site leveled by 14 interactions for example conventional hydrogen bond, carbon–hydrogen bond, and π–sulfur bond, these interactions were saved in total acquired conformations, and the favored bonds were saved in both sides of aminopyrimidine and thiazolium ring linked by a methylene bridge with Asn142, Cys145, and Tyr54. The second best inhibitor model to Mpro was biotin (Fig. 3), with a lower RR and MEV than thiamine (Table 1), resulting from the heterobicyclic structure that comprises 2-oxohexahydro-1H-thieno[3,4-d]imidazole that has a valeric acid substituent appended to the tetrahydrothiophene ring which limits the odds of flexibility inside the active site of Mpro and that was affirmed by the recorded interactions (Table 1) compared with the thiamine vitamin. All the interactions bind to the lactone group attached to the 2-oxohexahydro-1H-thieno[3,4-d]imidazole ring, which is explained by the electronegative field powered by the oxygen atom as the greatest localized negative charge in the biotin. Thus, the perfect hydrogen bond in all the inhibitors was saved with His164, at the atomic level, this amino acid could be very important in stabilizing the biotin in the active site. The third-best inhibitor model to Mpro was the pantothenic acid (Fig. 3) with lower RR compared with thiamine and biotin, in any case, pantothenic acid offers a similar MEV with biotin. Furthermore, a similar hydrogen bond was saved with His164 with a length of 1.83 Å. On the opposite side, the saved number of interactions was lower compared with the biotin, bringing about its lower affinity to Mpro while acting alone, which clarifies the pertinence of an expected relationship between the biotin and the pantothenic acid, and that could be significant in the inhibition mechanisms against Mpro.

The best inhibitor pose captured in the active site of Mpro presented in surface and colored in yellow, inhibitors are displayed with ball and stick, and the catalytic amino acids are shown in sticks and colored in green (Color figure online)
The last ranked inhibitor was the pyridoxine (Fig. 4) as the weakest inhibitor among the vitamin B family and all tested inhibitors in this study with RR of 4% and MEV of − 4.8 kcal/mol (Table 1), we recorded only one interaction with Cys145 of hydrophobic type, this interaction elucidates the environment between pyridoxine functional groups and BP amino acids which have a very little affinity, the interacting groups will be positioned in a manner to have the smallest contact surface with the active site.

The weakest inhibitors pose captured in the active site of Mpro presented in surface and colored in purple, inhibitors are presented in ball and stick, and the catalytic amino acids are presented in sticks and colored in green (Color figure online)
The CAA recorded in tocopherol (Fig. 4) were His41, Met49, Cys145, His163, Met165, Pro168, Arg188, Thr190, and Gln192, the nucleophilic atom reacting was in favor of the ketone function side of Arg188 and the hydroxyl function of the tocopherols represented by the hydrogen as the electrophilic atom, only one hydrogen bond was recorded in this context and that clarifies the native structure of the Arg since the other side of it ends with a guanidinium group which enables the formation of hydrogen bonds due to the delocalization of the positive charge of the nitrogen atom in the Arg bone structure. The RR of tocopherol recorded in Table 1 was very low compared with its MEV as the greatest value saved among all other inhibitors, and that is due to its high affinity with other sites since the most saved interactions were a hydrophobic type, which minimizes the chances of getting the exact conformation in the catalytic site of the Mpro.
The best inhibitor model to Mpro active site is the glutathione or γ-l-glutamyl-l-cysteinyl-glycine (Fig. 3), compared with the reference N3, with the best saved RR and MEV (Table 1). Where the CAA recorded was His41, Leu141, Gly143, Ser144, Cys145, Glu166, Arg188, and Gln189, as per our outcomes, glutathione presents the most elevated affinity to Mpro, this affinity is confirmed by different evidences such as interaction type, stereochemical properties, functional groups and similarity with the active site volume. The glutathione is profoundly located in the active site tunnel. All the internal connections are made of conventional hydrogen bond type that will guarantee its adaptability and strength since it shaped by triple amino acids (cysteine, glutamic acid, and glycine) appended through peptide linkage and as of now introduced in the active site which increases the reactivity and viability, this factor favorites covalently the fixation of glutathione. We propose a mechanism of action of glutathione inside the active site of Mpro based on the largest localized negative charge as indicated by the most intense red color (Fig. 5) and the proton being transferred has the most intense positive charge character indicated by the most intense blue color (Fig. 5) and the best hydrogen bond saved with Leu141.

Proposed mechanism of the glutathione with Leu141 in the active site of Mpro
The 2-step mechanism proposed in Fig. 5 clarifies the development of glutathione and leucine complex. The oxygen atom of Leu141 is the electron source and the hydroxide atom of the glutathione is the electron sink. To avoid overloading the valence of the hydrogen molecule the hydroxide bond should be broken to shape the glutathione ion. This latter is regularly balanced by resonance delocalization. Then, the π bond will move to fulfill the valence in the oxygen atom of Leu141. In step 1, we got an oxygen atom and a positive charge carbon atom with only three bonds, which lead us to step 2, ester linkage bond will be formed to stabilizing the new complex consequently, blocking the active site of Mpro.
Molecular Dynamics Simulation
To confirm the MDS results for the best-obtained inhibitor, MDS for 100 ns was carried out for glutathione and N3 as the reference ligand in the Mpro. MDS is a method used to investigate the folding (Berendsen et al. 1995) and motion of millions of atoms within larger or smaller molecule systems in a different period (Lindahl et al. 2001), the overall movements within a complex protein–ligand depend on the duration of the simulation. We focus on the RMSD, RMSF, and the atomic radial distribution function Rg along with the hydrogen bond distribution analysis since it is employed to study the protein–ligand complex atomic stability, deviations of the global structure, and atoms fluctuations (Sargsyan et al. 2017). The analysis of RMSD and RMSF usually provides an important information about stability and flexibility. High deviation and fluctuation of proteins during a simulation may show structure deformation and weak stability (Yu et al. 2021). Moreover, Rg analysis indicates the global shape and structure compactness of the viral protein during the simulation; it should be constant in all simulation periods. A short simulation period for 10 ns was run for both glutathione and N3, the latter was the native ligand in the Mpro.
RMSD
To test and compare the primary effects to Mpro, the N3 result shows two major deviation peaks visible on its RMSD plot (Fig. 6a), 0.3 nm (3 Å) was the maximum-recorded deviation at period 7.9 ns of the simulation at 0 ns (Fig. 6a) while 0.1 nm (1 Å) was the minimum-recorded deviation at period 0.1 ns just after the simulation beginning, the deviation persists during the 10 ns simulation explaining some instability in the whole system. However, the complex Mpro–N3 deviations plots stay superimposed during the simulation period, moreover, the glutathione maximum-recorded deviation was 0.34 nm (3.4 Å) at the period of 4.3 ns of the simulation (Fig. 6b) while 0.17 nm (1.7 Å) was the minimum-recorded deviation at period 1.2 ns just after the simulation beginning, the deviation stabilizes rapidly after 6 ns simulation. After confirming the glutathione effectiveness, a long simulation period for 100 ns was run, the results show consistent in the RMSD plot for both Mpro and glutathione with time, 0.35 nm (3.5 Å) was the maximum-recorded deviation at the period of 60 ns simulation (Fig. 6) while 0.1 nm (1 Å) was the minimum-recorded deviation at period 80 ns (Fig. 6b). Eventually, the system reaches the equilibrium state within 80 ns simulation and confirms the protein stability from its original conformation with lower deviations in the presence of glutathione. The deviations peaks that were observed during the simulation time between 40 and 80 ns in both plots (Fig. 6b) indicating high flexibility region due to the presence of two conformations, it refers to the glutathione as the second conformation because it is originally a polypeptide.

Plots of the RMSD backbone atoms of the viral protein in black and ligands in red, a the N3 heavy atoms vs. b the glutathione heavy atoms during the 10 ns simulation, and c the glutathione heavy atoms during the 100 ns simulation (Color figure online)
RMSF
The results show multiple fluctuations in N3, higher RMSF values likely are the loop regions with more conformational flexibility, however, the structure could destabilize due to this over flexibility (over 4 Å) with plus 14 fluctuations (Fig. 7b); In addition, it can be a factor of making the N3 reversible inhibitor in real time which is unfavorable case in the COVID-19 treatment, the fluctuations were less in the case of glutathione, the maximum recorded value was 2.5 Å with only 7 fluctuations (Fig. 7a) compared to N3 (Fig. 7b), we qualify the obtained glutathione data as moderate results compared with N3, to confirm our results, we superimposed the RMSF plots of the Mpro backbone atoms in the presence of both glutathione and N3, the results show similarity in all region for both ligands with fewer fluctuations in the case of glutathione (Fig. 7c), the Mpro seems more equilibrate in the presence of glutathione than the N3, in this case, the structure could be more stabilized with the glutathione due to its strong interaction, making of it possible irreversible inhibitor to the viral protein.

Plots of the RMSF viral protein backbone atoms and ligands in green, a the glutathione atoms, b the N3 atoms, and c the superposition of complex Mpro (glutathione in green and N3 in black) during 100 ns simulation (Color figure online)
Radius of Gyration
The radius of gyration indicates the global shape of our protein during the simulation in the presence of our inhibitors. The Rg stays very constant in the whole simulation for both inhibitors with slight changes in Rg of N3 (Fig. 8a), the glutathione Rg starts at 4 Å and stays consistent in all simulation, moreover, the N3 Rg starts at 6 Å and raised to 7 Å, the complex Mpro–glutathione Rg was stable comparing the complex Mpro–N3 (Fig. 8b) indicating that the Mpro structure was conserved during the simulation period in the presence of glutathione compared to the reference ligand N3, in this case, the viral protein conserves its structure.

Plots of the radius of gyration (Rg) for both ligands and protein during the 10 ns simulation, a the Rg of glutathione in blue and N3 in red and b the Rg of the complex Mpro–glutathione in green and the Mpro–N3 in black (Color figure online)
Hydrogen Bond
To achieve and confirm the Mpro–glutathione complex stability, and the irreversible attachment to it, intermolecular hydrogen bonds analysis are mandatory to check, thus, hydrogen bonds were evaluated for their number and distribution, and were shown in Fig. 9, the number of the hydrogen bond was decreasing in both cases (Fig. 9), with a faster rate in the N3. However, at a slower rate in the case of glutathione. Therefore, the glutathione hydrogen bonds distribution was high (> 20) and close between a donors–acceptor atom (~ 2.7 Å) (Fig. 9a). Furthermore, the N3 hydrogen bonds distribution was less (< 16) with higher distances between a donors–acceptor atom (~ 3.0 Å, Fig. 9b). We confirm from this the irreversible linkage of glutathione towards the Mpro.

Plots of the hydrogen bond distribution (donor–acceptor distances and number) of the glutathione (a) vs. N3 (b) in purple during the simulation (Color figure online)
MM–PBSA Energy Calculation
The polar solvation energy was significant in both inhibitors, the N3 plot (Fig. 10) shows consistent variation from − 10,000 to − 9000 kJ/mol compared to the glutathione with more changes from − 9800 to − 8800, this reflects its high stability as a protein part more than ligand, this describes the binding polar energy found (Table 2). The apolar solvation (SASA) is an important tool to evaluate the protein stability to the hydrophobic residues during the simulation period. The SASA energy was relatively similar and ranged from 350 to 380 kJ/mol for both inhibitors (Fig. 10), the inhibitors binding contributes to the values of the SASA; as a result, the binding of inhibitors does not affect the folding of the protein (Kumar et al. 2020). The vacuum MM energy shows that glutathione Van der Waals energy was less compared to the N3 with a higher obtained value (Table 2), it may reflect its strong binding affinity. However, the glutathione electrostatic energy was slightly increased, as revenge to N3. The overall energy shows a regular variation in glutathione (~ 0.2 kJ/mol) than N3 (> 0.4 kJ/mol) (Fig. 10), clearly, the hydrophobic interactions (Djafri et al. 2020) play an important role in both cases.


Plots of the different interactions energy of both Mpro complexed with a glutathione in red and b N3 in blue (Color figure online)
Conclusion
Throughout this in silico study, we have presented glutathione, the B vitamin family and vitamin E as possible inhibitors of the main protease of SARS-CoV-2, which can theoretically decrease the spread of infection. The proposed treatment strategy could help improve the immune system and inhibit main proteases. Today, glutathione is used as a treatment in many diseases such as respiratory infections, asthma and severe respiratory disorders, in immune deficiencies such as HIV and in liver diseases. The original peptide structure of glutathione gives it an advantage over other inhibitors. We provided a deep MDS to study the major deviations and fluctuations of the protease–glutathione complex over time. Based on our results, we recommend the exceptional use of glutathione as a valuable dual impact for preventive activity and a viable treatment beyond antisymptomatic viral drugs. The daily use of glutathione and the B vitamin family as a supplement may be an answer to restrict and decrease the viral load.
References
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25. https://doi.org/10.1016/j.softx.2015.06.001
Ahvanooei MR, Norouzian MA, Vahmani P (2021) Beneficial effects of vitamins, minerals, and bioactive peptides on strengthening the immune system against COVID-19 and the role of cow’s milk in the supply of these nutrients. Biol Trace Elem Res 27:1–14
Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R (2003) Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300(5626):1763–1767
Aslam A, Misbah SA, Talbot K, Chapel H (2004) Vitamin E deficiency induced neurological disease in common variable immunodeficiency: two cases and a review of the literature of vitamin E deficiency. Clin Immunol 112(1):24–29
Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98(18):10037–10041
Berendsen HJC, van der Spoel D, van Drunen R (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun 91(1–3):43–56
Bhardwaj A, Scaria V, Raghava GPS, Lynn AM, Chandra N, Banerjee S et al (2011) Open source drug discovery—a new paradigm of collaborative research in tuberculosis drug development. Tuberculosis 91(5):479–486
Bunik VI, Aleshin VA (2017) Chapter 11—analysis of the protein binding sites for thiamin and its derivatives to elucidate the molecular mechanisms of the noncoenzyme action of thiamin (vitamin B1). In: Atta ur R (ed) Studies in natural products chemistry, vol 53. Elsevier, Amsterdam, pp 375–429
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y et al (2020) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395(10223):507–513. https://doi.org/10.1016/s0140-6736(20)30211-7
Coates PM, Betz JM, Blackman MR, Cragg GM, Levine M, Moss J, White JD (2010) Encyclopedia of dietary supplements. CRC Press, Boca Raton
Centers for Disease Control and Prevention (n.d.) Information about the Pfizer–BioNTech COVID-19 Vaccine
Dakshinamurti K, Stephens MC (1969) Pyridoxine deficiency in the neonatal rat. J Neurochem 16(11):1515–1522
Dassault Systèmes BIOVIA (2017) BIOVIA workbook, release 2017; BIOVIA pipeline pilot. Dassault Systèmes, San Diego
Djafri A, Perveen F, Benhalima N, Khelloul N, Rahmani R, Djafri A et al (2020) Experimental spectral characterization, Hirshfeld surface analysis, DFT/TD-DFT calculations and docking studies of (2Z, 5Z)-5-(4-nitrobenzylidene)-3-N (2-methoxyphenyl)-2-N′(2-methoxyphenylimino) thiazolidin-4-one. Heliyon 6(12):e05754
Eisenhaber F, Lijnzaad P, Argos P, Sander C, Scharf M (1995) The double cubic lattice method: efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J Comput Chem 16(3):273–284
Garty R, Yonis Z, Braham J, Steinitz K (1962) Pyridoxine-dependent convulsions in an infant. Arch Dis Child 37(191):21
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX et al (2020) Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. https://doi.org/10.1056/NEJMoa2002032
Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR (2012) Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform 4(1):1–17
Hawkins WW, Barsky J (1948) An experiment on human vitamin B6 deprivation. Science 108(2802):284–286
Herzenberg LA, De Rosa SC, Dubs JG, Roederer M, Anderson MT, Ela SW et al (1997) Glutathione deficiency is associated with impaired survival in HIV disease. Proc Natl Acad Sci USA 94(5):1967–1972
Hess B, Kutzner C, Van Der Spoel D, Lindahl E (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4(3):435–447
Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, De Groot BL et al (2017) CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 14(1):71–73
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14(1):33–38
Institute of Medicine Standing Committee on the Scientific Evaluation of Dietary Reference, I (1998) Vitamin B6. In: Dietary reference intakes for thiamin, riboflavin, niacin, vitamin B6, folate, vitamin B12, pantothenic acid, biotin, and choline. National Academies Press, Washington, DC
Islam MT, Quispe C, Martorell M, Docea AO, Salehi B, Calina D et al (2021) Dietary supplements, vitamins and minerals as potential interventions against viruses: perspectives for COVID-19. Int J Vitam Nutr Res. https://doi.org/10.1024/0300-9831/a000694
Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng CJN (2020) Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 582(7811):289–293. https://doi.org/10.1038/s41586-020-2223-y
Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A et al (2016) PubChem substance and compound databases. Nucleic Acids Res 44(D1):D1202-1213. https://doi.org/10.1093/nar/gkv951
Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S et al (2003) A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 348(20):1953–1966. https://doi.org/10.1056/NEJMoa030781
Kumar A, Choudhir G, Shukla SK, Sharma M, Tyagi P, Bhushan A, Rathore M (2020) Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. J Biomol Struct Dyn 39(10):1–11
Kumar P, Kumar M, Bedi O, Gupta M, Kumar S, Jaiswal G et al (2021) Role of vitamins and minerals as immunity boosters in COVID-19. Inflammopharmacology 29:1–16
Kumari R, Kumar R, Open Source Drug Discovery Consortium, Lynn A (2014) g_mmpbsa—a GROMACS tool for high-throughput MM–PBSA calculations. J Chem Inf Model 54(7):1951–1962
LeDuc JW, Barry MA (2004) SARS, the first pandemic of the 21st century. Emerg Infect Dis 10(11):e26. https://doi.org/10.3201/eid1011.040797_02
Lee GY, Han SN (2018) The role of vitamin E in immunity. Nutrients. https://doi.org/10.3390/nu10111614
Linani A, Benarous K, Bou-salah L, Yousfi M (2021) Hispidin, Harmaline, and Harmine as potent inhibitors of bovine xanthine oxidase: gout treatment, in vitro, ADMET prediction, and SAR studies. Bioorg Chem. https://doi.org/10.1016/j.bioorg.2021.104937
Lindahl E, Hess B, Van Der Spoel D (2001) GROMACS 3.0: a package for molecular simulation and trajectory analysis. Mol Model Annu 7(8):306–317
Malde AK, Zuo L, Breeze M, Stroet M, Poger D, Nair PC et al (2011) An automated force field topology builder (ATB) and repository: version 1.0. J Chem Theory Comput 7(12):4026–4037
Mueller JF, Vilter RW (1950) Pyridoxine deficiency in human beings induced with desoxypyridoxine. J Clin Investig 29(2):193–201
Pan AC, Roux B (2008) Building Markov state models along pathways to determine free energies and rates of transitions. J Chem Phys 129(6):064107
Patassini S, Begley P, Xu J, Church SJ, Kureishy N, Reid SJ et al (2019) Cerebral vitamin B5 (d-pantothenic acid) deficiency as a potential cause of metabolic perturbation and neurodegeneration in Huntington’s disease. Metabolites 9(6):113
Poutanen SM, Low DE, Henry B, Finkelstein S, Rose D, Green K et al (2003) Identification of severe acute respiratory syndrome in Canada. N Engl J Med 348(20):1995–2005
Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R et al (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29(7):845–854
Pyrc K, Berkhout B, Van Der Hoek L (2007) The novel human coronaviruses NL63 and HKU1. J Virol 81(7):3051–3057
Reidling JC, Lambrecht N, Kassir M, Said HM (2010) Impaired intestinal vitamin B1 (thiamin) uptake in thiamin transporter-2-deficient mice. Gastroenterology 138(5):1802–1809. https://doi.org/10.1053/j.gastro.2009.10.042
Richie JP, Nichenametla S, Neidig W, Calcagnotto A, Haley JS, Schell TD, Muscat JE (2015) Randomized controlled trial of oral glutathione supplementation on body stores of glutathione. Eur J Nutr 54(2):251–263. https://doi.org/10.1007/s00394-014-0706-z
Sargsyan K, Grauffel C, Lim C (2017) How molecular size impacts RMSD applications in molecular dynamics simulations. J Chem Theory Comput 13(4):1518–1524
Schuetz P, Gregoriano C, Keller U (2021) Supplementation of the population during the COVID-19 pandemic with vitamins and micronutrients—how much evidence is needed? Swiss Med Wkly 151(1920):w20522
Serseg T, Benarous K, Yousfi M (2021) Hispidin and Lepidine E: two natural compounds and folic acid as potential inhibitors of 2019-novel coronavirus main protease (2019-nCoVMpro), molecular docking and SAR study. Curr Comput Aided Drug Des 17(3):469–479
Shakoor H, Feehan J, Al Dhaheri AS, Ali HI, Platat C, Ismail LC et al (2021) Immune-boosting role of vitamins D, C, E, zinc, selenium and omega-3 fatty acids: could they help against COVID-19? Maturitas 143:1–9
Snyderman SE, Holt LE Jr, Carretero R, Jacobs K (1953) Pyridoxine deficiency in the human infant. Am J Clin Nutr 1(3):200–207
Srivastava A, Gupta RC, Doss RB, Lall R (2021) Trace minerals, vitamins and nutraceuticals in prevention and treatment of COVID-19. J Diet Suppl. https://doi.org/10.1080/19390211.2021.1890662
Thiamin R (1998) Dietary reference intakes for thiamin, riboflavin, niacin, vitamin B6, folate, vitamin B12, pantothenic acid, biotin, and choline1. National Academies Press, Washington, DC
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461. https://doi.org/10.1002/jcc.21334
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC (2005) GROMACS: fast, flexible, and free. J Comput Chem 26(16):1701–1718
Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J et al (2010) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31(4):671–690
White AC, Thannickal VJ, Fanburg BL (1994) Glutathione deficiency in human disease. J Nutr Biochem 5(5):218–226. https://doi.org/10.1016/0955-2863(94)90039-6
Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG et al (2020) A new coronavirus associated with human respiratory disease in China. Nature 579(7798):265–269. https://doi.org/10.1038/s41586-020-2008-3
Yu Z, Kan R, Ji H, Wu S, Zhao W, Shuian D et al (2021) Identification of tuna protein-derived peptides as potent SARS-CoV-2 inhibitors via molecular docking and molecular dynamic simulation. Food Chem 342:128366
Zoete V, Cuendet MA, Grosdidier A, Michielin O (2011) SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 32(11):2359–2368
Zubaran C, Fernandes JG, Rodnight R (1997) Wernicke-Korsakoff syndrome. Postgrad Med J 73(855):27. https://doi.org/10.1136/pgmj.73.855.27
Acknowledgements
The authors wish to thank Dr. Oleg Trott from the Scripps Research Institute for providing the Autodock Vina Program. S. Goumri-Said thank the Office of Research at Alfaisal University in Saudi Arabia for funding this research work through Internal Project Number C20431.
Funding
This research received no external funding.
Author information
Authors and Affiliations
Contributions
AL, KB, LB-S, MY and SG-S: Conceptualization; AL, KB, LB-S, MY: methodology; AL, KB, LB-S, MY: software; AL, KB, LB-S, MY and SG-S: validation; AL, KB, LB-S, MY and SG-S: formal analysis; KB, TS, MY and SG-S: investigation; KB, TS, MY and SG-S: resources; KB, TS, MY and SG-S: data curation; KB, TS: writing—original draft preparation; KB, TS, MY and SG-S: writing—review and editing; KB, TS: visualization; KB: supervision; SG-S: project administration. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest, financial or otherwise.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Linani, A., Benarous, K., Bou-Salah, L. et al. Exploring Structural Mechanism of COVID-19 Treatment with Glutathione as a Potential Peptide Inhibitor to the Main Protease: Molecular Dynamics Simulation and MM/PBSA Free Energy Calculations Study. Int J Pept Res Ther 28, 55 (2022). https://doi.org/10.1007/s10989-022-10365-6
Accepted:
Published:
DOI: https://doi.org/10.1007/s10989-022-10365-6