
Abstract
Context
NLRP9 is a member of nucleotide-binding domain leucine-rich repeat-containing receptors and is found to be associated with many inflammatory diseases. In the current scenario, the identification of promising anti-inflammatory compounds from natural sources by repurposing approach is still relevant for the early prevention and effective management of the disease.
Methods
In the present study, we docked bioactives of Ashwagandha (Withanoside IV, Withanoside V, Withanolide A, Withanolide B, and Sitoindoside IX) and two control drugs against bovine NLRP9 protein. ADME/T analysis was used to determine the physiochemical properties of compounds and standard drugs. Molecular modeling was used to evaluate the correctness and quality of protein structures. In silico docking analysis revealed Withanolide B had the highest binding affinity score of −10.5 kcal/mol, whereas, among control drugs, doxycycline hydrochloride was most effective (−10.3 kcal/mol). The results of this study revealed that bioactives of Withania somnifera could be promising inhibitors against bovine NLRP9. In the present study, molecular simulation was used to measure protein conformational changes over time. The Rg value was found to be 34.77A°. RMSD and B-factor were also estimated to provide insights into the flexibility and mobile regions of protein structure. A functional protein network interaction was constructed from information collected from non-curative sources as protein-protein interactions (PPI) that play an important role in determining the function of the target protein and the ability of the drug molecule. Thus, in the present situation, it is important to identify bioactives with the potential to combat inflammatory diseases and provide strength and immunity to the host. However, there is still a need to study in vitro and in vivo to further support these findings.
Similar content being viewed by others


Introduction
The innate immune system is a ubiquitous and ancestral defense system that protects the host against microbial infections and other potential threats. In mammals, the first molecules involved in the innate immune defense system that protects microbial agents are the Toll-like receptors (TLRs) [1,2,3]. However, recent studies have reported that in bacterial-induced inflammation, Nod-like receptors (NODs) act as intracellular regulators [4, 5]. The nucleotide-binding oligomerization domain-like receptors (NOD-like receptors), also known as nucleotide-binding domain leucine-rich repeat-containing receptors (NLRs), are a group of ancestral germline-encoded intracellular proteins that serve as important pattern recognition receptors (PRRs) to identify a wide array of pathogen-associated molecular patterns (PAMPs), environmental stress, and damage-associated molecular patterns (DAMPs) [6]. This process of recognition activates a protein supramolecular complex in the cytosol known as the inflammasome [7]. This protein complex acts as a signal and triggers an inflammatory response that mediates the synthesis of proinflammatory cytokine molecules (IL-1β AND IL-18) [8, 9]. NLRs have been found in a diverse range of species including plants, all of which have their origin from an ancestral protein containing nucleotide-binding domain (NBD) [10]. In the intracellular immune surveillance of invading pathogens, NLRs of animals and plants are heavily involved via domains that share conserved activation principles. The genome of humans and mice encodes at least 22 and 34 NLR genes [11]. NLRs are exclusively found in the cytosol and nucleus wherein they detect microbial pathogens [12, 13]. Notably, there are several members of the NLR family like NLRP1, NLRP3, NLRP5, NLRP6, and NLRP9. Among these protein molecules, inflammasomes are formed by NLRP1, NLRP3, and NLRP4 respectively that can arbitrate immune responses against stress, pathogens, flagellins, M2 proteins (viruses), and damage [14]. However, in recent years, a few less-characterized NLR members have emerged as potentially effective signaling molecules involved in defense and inflammation. Among them, NLRP9 is an emerging NLR signaling molecule and a member of the NOD-like cytoplasmic receptor family. Originally, NLRP9 was supposed to function and express solely by reproductive organs [15]. Earlier research studies have reported that bovine, human, and murine NLRP9 is selectively expressed by ovaries, oocytes, and testes [16, 17] and is likely to be involved in the development of the preimplantation embryo [18, 19]. Recent studies have reported that NLRP9 in the intestine can initiate the formation of inflammasome to curb damage and replication of rotavirus infection [20]. NLRP9 is linked with various inflammatory diseases such as Alzheimer’s disease [21 ], lung injury [22], multiple sclerosis [23], and Helicobacter pylori infection [24]. Currently, anti-inflammatory drugs are commonly used in clinical practice to treat inflammation; however, their long-term use causes adverse reactions and drug resistance [25, 26]. In such a scenario, drugs with higher safety should be used as much as possible. Therefore, finding natural plant compounds that possess more pharmacological properties without toxic effects can serve as good curative to replace traditional drugs and lead to the development of new natural anti-inflammatory drugs. In the present study, natural bioactives of Withania somnifera (Ashwagandha) like Withanoside IV, Withanoside V, Withanolide A, Withanolide B, and Sitoindoside IX were selected. Withania somnifera has been found to possess anti-inflammatory, immunomodulatory, antioxidant, antidepressant, anticancer, and neuroprotective properties [27,28,29,30,31,32,33].
The recent advances in computational approaches have significantly developed the rationale for identifying and designing pharmacologically active natural molecules that can target proteins of interest. The natural compounds obtained from plants can be repurposed by computational approaches to prove their potential. Here, in the present study, we have used, Withanoside IV, Withanoside V, Withanolide A, Withanolide B, and Sitoindoside IX as potential anti-inflammatory natural compounds to target bovine NLRP9 protein by using computational approaches. In the present study, ADMET (absorption, distribution, metabolism, excretion, and toxicity) analysis of three natural compounds and two control drugs gentamicin and doxycycline hydrochloride was done. Further, the present study also aimed to determine sequence alignment, molecular modeling, calculation of free binding energy, inhibition constant, and residue network interactions along with molecular docking and simulation against bovine NLRP9 protein.
Materials and methods
Ligand selection
In the present study, ligands such as Withanoside IV, Withanoside V, Withanolide A, Withanolide B, and Sitoindoside IX along with two control drugs (Gentamicin and doxycycline hydrochloride) were selected. The three-dimensional structures of ligands in simple data format (SDF) were obtained from the PubChem server (https://pubchem.ncbi.nlm.nih.gov/compound; last accessed on 21 May 2022).
ADMET studies
ADMET (Adsorption, Distribution, Metabolism, Excretion, and Toxicity) analysis is important for determining the pharmacodynamic properties of the molecule. SWISSADME (www.swissadme.ch/; last accessed on 21 May 2022) a web-based online server was used for determining ADMET properties of natural compounds and drugs with the probable best matches [34, 35]. In SWISSADME, ligand smiles were uploaded that were obtained from PubChem.
Toxicity analysis
An online web tool ProTox-II did the prediction of toxicity analysis (https://ox-new.charite.de/protox_II/; last accessed on 21 May 2022).
Homology modeling
SWISSMODEL an online tool (https://swissmodel.expasy.org/; last accessed on 21 May 2022) was used for modeling building using protein’s amino acid sequence (fasta format). The accuracy of the protein model was determined by the PROCHECK tool (https://saves.mbi.ucla.edu/; last accessed on 21 May 2022) [36, 37].
Receptor/protein preparation
The preparation of the target protein was done as per the procedure of Tallei et al. [38]. In the present study, NLRP9 served as the target receptor and whose 3D crystal structure was retrieved from RCSB Protein Databank (http://www.rcsb.org/pdb/; last accessed on 21 May 2022) with PDB ID of 7WBT. AutoDock Vina (UCSF-Chimera©, version 1.13) software was used to perform docking as per the procedure of Trott and Olson, [39]. A Python prescription 0.8 suite consisting of AutoDock Vina determined the analysis of target protein with ligands. In the present study, compounds that acted as ligands were prepared in AutoDock Vina by first uploading the ligands separately in the input server and then selecting them as molecules to be docked. After these steps, the addition of charges/hydrogen and torsion angles were computed for the ligand molecules. The docked ligand molecules were then saved in pdbqt format for further analysis. The protein molecule was initially prepared in AutoDock Vina by removing molecules of water, followed by the addition of polar hydrogen atoms and computing Gasteiger charges from the target protein. The docked protein was consequently saved as a pdbqt file and then subjected to the docking process. In AutoDock Vina, the grid size was set to 40 × 40 × 40 (x, y, z) points with a spacing of 0.375 A° and the center dimensions (x = 8.333, y = −11.151, z = −31.135 A°) respectively. In the docking process, AutoDock Vina used information on both ligand and protein along with details of grid box characteristics in the configuration file. In the docking procedure, both the ligands and protein are considered rigid. After the completion of the docking process, 10 configuration files with the 10 best docking poses for each protein-ligand complex were generated. The compounds with the lowest binding energy (kcal/mol) and minimum root mean square deviation (RMSD) were taken as the most suitable docking pose. Discovery Studio (version 2.4.1, 2016) was used for the visualization of protein-ligand complexes.
Inhibition constant
In this study, the inhibition constant (Ki) was calculated from the binding energy (ΔG) by using the below formula
Ki (μM) = exp (ΔG/RT), where R is the gas constant (1.985 × 10−3 kcal/mol−1 K−1) and T (temperature = 298.15 K) respectively.
Molecular simulation
Molecular dynamics on Web (MDWeb) a web-based server was used to perform simulation in the high throughput range (http://mmb.irbbarcelona.org/MDWeb/; accessed on 21 May 2022). It provides simulation analysis for the three most popular MD packages (NAMD, Gromacs, and Amber). In the input type, a single structure protein file with pdb format was selected and uploaded on the web server. In the simulation process, coarse-grained MD: normal mode analysis (NMA) C-alpha was chosen. In NMA, a linear algorithm with a force constant (Kcal/mol A°2) of 40 A° and a cutoff value of 8.0 A° for a time of 100 ps was taken to calculate the radius of gyration (Rg), trajectory root mean square deviation (RMSD), RMSD values per residue, atomic fluctuation (BF values per residue), hydrophobicity index, and hydrogen bond interactions.
Protein Contacts Atlas
It is an interactive web-based database (http://pca.mggroup.bio/; accessed on 02 September 2022) that enables visualization of the interaction between ligands, small molecules, proteins (single or complex), and nucleic acids. This tool uses a network of residue-residue interactions and helps to visualize molecule structure at atomic resolutions. The protein contact atlas is a precomputed database of crystal structures that is determined either by experimental or computational approaches so far.
Delphi Web server
Delphi is an online tool (http://compbio.clemson.edu/sapp/delphiwebserver/; accessed on 02-September 2022) that uses a finite difference approach to solve the Poisson-Boltzmann solver equation for determining electrostatic and potential energies for biomolecules. In analysis, a force field amber approach was selected for protonation and a pH of 7.0 respectively. The other field parameters were a linear Poisson-Boltzmann approach was used with a grid size of 135, resolution of 1A°, grid box is centered (A) −9.64, 11.02, −28.36, percent of the box occupied [40], internal dielectric constant (4.0), external dielectric constant (80.0), salt concentration (0.15 M, absolute temperature (K) 297.33 and a probe radius of 1.40. The type of energies selected in the output results was columbic, solvation, and grid energy along with visualization of the potential map.
STRING
The string is a precomputed database (http://string-db.org; accessed on 02 September 2022) that was used to evaluate protein-protein interactions that include both physical as well as functional relations. A scoring system provides scores for every protein-protein interaction wherein these scores (edge weights in every network) describe confidence scores with values ranging from 0 to 1. In the present study, STRING was used to study network summary (showing current interactions), gene co-expression (during expression correlation of genes coding proteins), co-occurrence (gene families whose patterns show similar occurrence across genomes), and gene fusion (sometimes genes may fuse up into single open reading frames).
MoDEL
The molecular dynamics extended library (MoDEL) is a precomputed database of atomistic molecular dynamics trajectories (http://mmb.irbbarcelona.org/MoDEL; accessed on 02 September 2022). In the present study, the MoDEL tool used the NAMD approach with standard amber force field (parm99 tip3P), simulation time (10,000 ps), integration time (0.0020 ps), temperature (300.0 K), box dimensions (90.079 Å, 90.079 Å, 90.079 Å), and salt concentration (94.83mM) respectively.
Results
ADMET analysis
In the present study, the pharmacokinetic (PK) and physiochemical properties of natural compounds of Ashwagandha (Withanoside IV, Withanoside V, Withanolide A, Withanolide B, and Sitoindoside IX) along with control drugs (Gentamicin and Doxycycline hydrochloride) were done. In the present study, Withanolide A and Withanolide B followed Lipinski’s rule of five; however, Sitoindoside IX showed 2 violations (MW>500, number of hydrogen bond acceptors >10). Similarly, Withanoside IV, and Withanoside V, did not follow Lipinski’s rule as the properties were not within the acceptable range. The pivotal prerequisite to determining the apparent efficacy of an oral drug is determined by its intestinal absorption. In the present study, Withanolide A, Withanolide B, and Sitoindoside IX showed high GI absorption, whereas the rest of the compounds had low GI absorption. About two-thirds of known drugs are metabolized by the 57 isozymes that make up the human cytochrome P450 family (phase I enzymes), with five isozymes accounting for 80% of this process: isozymes 1A2, 3A4, 2C9, 2C19, and 2D6. The liver is where the majority of these CYPs that are in control of phase I reactions are concentrated. In the present study, the output value is the probability of being a substrate or an inhibitor, ranging from 0 (No) to 1 (Yes). The evaluation of synthetic accessibility assesses the ease of synthesis of a large number of chemical compounds that are expected to lead to a breakthrough in drug discovery. This method assigns scores ranging from 1 (easy to make) to 10 (difficult to make). In the present study, synthetic accessibility scores of Withanoside IV, Withanoside V, Withanolide A, Withanolide B, and Sitoindoside IX were 8.88, 8.84, 6.39, 6.34, and 8.12 respectively. The ADMET analysis results are presented in Table 1.
Toxicity analysis
ProTox-II predicts the toxicities of small molecules which is an important part of the development process of drug designing. In the present study, different factors were considered for toxicity analysis such as LD50, predicted toxicity class, immunotoxicity, hepatotoxicity, mutagenicity, carcinogenicity, and cytotoxicity. Levels of toxicity were grouped into different classes according to:
-
Class I: fatal if swallowed (LD50 ≤ 5 mg/kg);
-
Class II: fatal if swallowed (5 mg/kg < LD50 ≤ 50 mg/kg);
-
Class III: toxic if swallowed (50 mg/kg < LD50 ≤ 300 mg/kg);
-
Class IV: harmful if swallowed (300 mg/kg < LD50 ≤ 2000 mg/kg);
-
Class V: may be harmful if swallowed (2000 mg/kg < LD50 ≤ 5000 mg/kg).
-
Class VI: Non-toxic
ProTox-II was used to evaluate some toxicity endpoints, including hepatotoxicity, carcinogenicity, immunotoxicity, cytotoxicity, and mutagenicity. The predictive models are constructed using information from both in vitro (such as from Tox21 tests, Ames bacterial mutation experiments, hepG2 cytotoxicity assays, and immunotoxicity assays) and in vivo assays (carcinogenicity, hepatotoxicity). In the present study, selected bioactives (Withanoside IV, Withanoside V, and Withanolide A) were inactive for hepatotoxicity, carcinogenicity, and mutagenicity; however, they were predicted to have immunotoxicity and cytotoxicity. Similarly, active activity was shown by Withanolide B for carcinogenicity, immunotoxicity, and cytotoxicity. Moreover, Sitoindoside IX reported active activity for immunotoxicity and cytotoxicity. Among the drugs, gentamicin showed inactive prediction for all but active for immunotoxicity, whereas doxycycline hydrochloride showed active prediction for hepatotoxicity and immunotoxicity and inactive for rest. The predicted probabilities of toxicity analysis are shown in Table 2.
Molecular modeling
In the present study, bovine NLRP9 showed a MolProbity score of 1.23 which determines the correctness and quality of protein structure. The ϕ and ψ scores of the residues are plotted on the background (Fig. 1). In bovine NLRP9, 96.23% of the residues were lying in the Ramachandran favored region. The clash score was 0.90 for residues (B788 LYS-B817 GLU), (B197 LYS-B496 GLU), 0.11% for Ramachandran outliers, and 1.82% for rotamer outliers. Similarly, 11 C-Beta Deviations, 1 bad bond, and 81 bad angles were found in the protein structure. The Ramachandran plots for Gly, Pro, and pre-Pro residues are shown in Fig. 1A–D. The three-dimensional geometry of the protein model was determined by the PROCHECK web tool, which calculated the Ramachandran plot and generated results for residues showing regions with different colors, i.e., red (favored), yellow (additionally allowed), pale yellow (generously allowed), and white colored areas (disallowed) (Fig. 1E). In bovine NLRP9 protein, a total of 1798 residues were found, out of which 1480 (88.8%) of the residues lied in the most favored region (A, B, L), 181 (10.9%) residues in additional allowed regions (a,b,l,p). Moreover, 5 (0.3%) residues were found in generously allowed regions (~a, ~b, ~l, ~p), and none of the residues were found in disallowed regions (Fig. 1E). In bovine NLRP9, 1666 (100%), non-glycine and non-proline residues were found, and 8 residues were the end residues (excluding glycine and proline) (Fig. 1E). The number of glycine residues (shown as triangles) was 66, and proline residues were 58 respectively (Fig. 1E). QMEAN Z-Score of NLRP9 protein was 0.90 (Fig. 1F), and the values of approximately zero specify superior quality between experimental and modeled structures.

Ramachandran plots of NLRP9: (A) All chains. (B) Glycine. (C) Proline. (D) Pre-proline. (E) PROCHECK residue analysis with plot statistics. (F) QMEAN Z-scores
The ProSA tool was used to determine the overall quality and structure validity of bovine NLRP9 protein and gave a Z-score of −13.38. The Z-score determines the total energy deviation of protein structures as compared to acquired energy distribution from random configurations. The complete ProSA results obtained are shown in Fig. 2. The group structures retrieved from numerous sources (NMR, X-ray) are differentiated by different colors (Fig. 2A). Only plots that have a size of fewer than 1000 residues represent those chains. The average energies of residue lying over a sliding window are plotted in the window as a measure of central residue. Due to the larger size of the protein chain (default: 40), a window size of 10 is used (Fig. 2B). In the increasing order of energy, residues are colored from blue to red (Fig. 2C).

Bovine NLRP9 receptor-binding domain Z-score and plot of residue scores. (A) ProSA-web z-scores of all protein chains. (B) Energy plot. (C) Highest and lowest energies of protein
The six graphs on the main-chain parameters plot show how the structure (represented by the solid square) compares with well-refined structures at a similar resolution (Fig. 3). The dark band in each graph represents the results from the well-refined structures; the central line is a least-squares fit to the mean trend as a function of resolution, while the width of the band on either side of it corresponds to a variation of one standard deviation about the mean. In some cases, the trend is dependent on the resolution, and in other cases, it is not. For structures solved by NMR a nominal resolution of 2.0Å is used.

Plot statistics of main chain parameters of bovine NLRP9 protein
The quality of the Ramachandran plot is shown in Fig. 3A, and this feature measures the protein residues percentage that lies in the favored or core region. Our protein model showed that 88.8% of residues are present in the favored region. Fig. 3B represents the planarity of the peptide bond that is measured by determining the standard deviation of the protein structure’s omega torsion angles. A perfectly planar peptide bond is represented by smaller values that cluster tightly around the ideal 180° structure. In our study, an omega standard deviation value of 4.9 was found which is considered better for a protein structure. The bad non-bonded interactions measure the number of bad contacts (wherein the distance of the closest approach is less than or equal to 2.6A °) per 100 residues. In our study, the parameter value of 0.2A° for non-bonded interactions was found (Fig. 3C). In this study, alpha carbon tetrahedral distortion that gives a measure of the standard deviation of zeta torsion angle was found to be 0.9A° (Fig. 3D). The main chain hydrogen bond energy feature determines the hydrogen bond standard deviation for the hydrogen bonds of the main chain. In our study, a parameter value of 0.7A° was found (Fig. 3E). Moreover, the G-factor that measures the overall normality of the protein structure was found to be 0.2 (Fig. 3F).
Molecular docking
Docking analysis is mainly performed to determine the interaction between the protein molecule and the ligand. A binding site describes the interaction between the protein macromolecule and the ligand, and bioactive compounds with lowest binding energy represent the most significant interaction. In the present study, compounds of Ashwagandha (Withanoside IV, Withanoside V, Withanolide A, Withanolide B, and Sitoindoside IX) and two control drugs (Gentamicin and Doxycycline hydrochloride) were docked against bovine NLRP9. The results of binding affinities and inhibition constant (Ki) obtained are shown in Table 3.
In the present study, Withanolide B showed the highest binding affinity score of −10.5 kcal/mol followed by Sitoindoside IX (−10.3 kcal/mol). Similarly, Withanolide A produced the least binding energy of −8.7 kcal/mol. Among the drugs, Doxycycline hydrochloride showed the highest binding energy of −10.3 kcal/mol, whereas Gentamicin had the lowest energy of −8.1 kcal/mol.
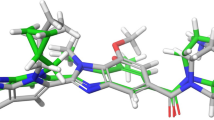
In Fig. 4 green balls and sticks display hydrogen bonds, violet balls and sticks represent hydrophobic bonds (Pi-Pi/Pi-sigma/amide-Pi interactions), pink balls and sticks display hydrophobic interactions (Pi-alkyl/alkyl interaction stacking), gold balls and sticks represent hydrophobic bonds (Pi-sulfur), and white balls and sticks display carbon-hydrogen bonds.


2D molecular interaction of NLRP9 with (A) Withanoside IV (B) Withanoside V (C) Withanolide A (D) Withanolide B (E) Sitoindoside IX (F) Gentamicin (G) Doxycycline hydrochloride
In the present study, docking of NLRP9 with Withanoside IV created hydrogen bonds (ILE613, ILE617), van der Waals interaction (HIS604, VAL605, PRO607, GLU609 CYS612, SER614, ASN615, THR616, and LYS619), and alkyl (LYS579). The docking of Withanoside V with NLRP9 formed hydrogen bonds (HIS604, SER614, LYS619, and ARG645), van der Waals interaction (GLU603, GLU609), and alkyl (PRO607) residues respectively. In the present study, Withanolide A docked with bovine NLRP9 formed hydrogen bond (CYS116, THR115, GLY159, ILE160, GLY161, LYS162, THR163, THR164, HIS449) and van der waals interaction (PRO115, LEU117, PHE123, TYR124, VAL119, THR128, PRO157, GLU158, PHE302, TYR310, PRO341, PHE342). Also, Withanolide B formed conventional hydrogen bonds with LYS579, HIS604, and van der Waals interaction (GLY577, VAL605, THR616, GLU618, and LYS619) and carbon-hydrogen bond (THR578). Similarly, Sitoindoside IX formed hydrogen bond (HIS604, THR616, and LYS619) and van der Waals interaction (LYS579, VAL605, PRO607, ILE613, SER614, ASN615, ILE617, and GLU618).
Furthermore, with drugs like Gentamicin, NLRP9 formed hydrogen bonds (ARG285, HIS604, SER614, LYS619) and van der Waals interaction (LYS281, GLY577, THR578, LYS579, GLU580, PRO607, THR616). Finally, the docking of Doxycycline hydrochloride with NLRP9 formed conventional hydrogen bonds (LYS579, GLU580, LYS619), van der Waals interaction (GLU577, THR578, HIS604, PRO607, GLU609, SER614), carbon hydrogen bond (THR578, SER614), Pi-Pi stacked (HIS604), and Pi-alkyl (PRO607) residues respectively.
Molecular simulation
The model was a single one and had one chain with 899 amino acids. In the present study, during the dynamic simulation process, the radius of gyration (Rg) and RMSD values were used to calculate structure deviation that determines whether the protein suffered any distortion or maintained its conformation during the 100-ns duration The Rg that measures protein compactness was found to be 34.77A° (Fig. 5A). The trajectory RMSD values were found to be 0.003 to 0.005A° (Fig. 5B). The RMSD values per residue for the 899 residue chain varied and were very low with the highest value being 0.01A° during the 100-ns period (Fig. 5C). Bfactor (atomic fluctuation) per residue and RMSD analysis provide insights about the flexibility of the structure and identify regions of the structure having more mobility and areas that are fixed during the whole simulation. In this study, the B-factor per residue varied, and the values were very small (Fig. 5D). The 5-residue running average hydrophobicity value is shown in Fig. 5E. Moreover the hydrogen bonds in the protein chain are represented in Fig. 5F. The MDWeb simulation results are shown in Fig. 5 respectively.

MDWeb simulation analysis: (A) Radius of gyration. (B) Trajectory RMSD. (C) RMSD values per residue. (D) BF values per residue. (E) Hydrophobicity index. (F) H-bond interactions
Protein Contacts Atlas
In this study, at the atomic levels, the protein contact visualization was studied using Protein Contacts Atlas tool. The nodes in the complex network represent the nodes that finally denote nucleic acids or proteins. The links between the nodes in the chain subunits represent the interaction surfaces. The chord plots of NLRP9 protein show the visualization of protein contacts at the secondary level (Fig. 6A). Similarly, the atomic neighborhood of the selected residues or ligands is represented by an asteroid plot (ligand/residue centric view). The asteroid plot of NLRP9 protein is shown in Fig. 6B. In the present study, the scatter plots of NLRP9 protein are shown in Fig. 6C respectively. The scatter plots give insights into the quantitative characteristics per residue.

Protein contact atlas of bovine NLRP9 protein: (A) Chord plot. (B) Asteroid plot. (C) Scatter plot
Electrostatic potential
In the binding of molecules to proteins, electrostatic interactions are one of the most important guiding forces. The electrostatic interactions play a pivotal role in protein stability and proper folding. In the present study, the Delphi web server read 14279 as the number of atom coordinates with 6581 as the total number of atomic charges and many dielectric boundary points (155,227) respectively. The total grid energy was 56,261.76 kT, corrected reaction field energy (−7237.99 kT), and coulombic energy (−78,747.26 kT). The electrostatic potential map generated by the Delphi tool is shown in Fig. 7A–B.

Delphi electrostatic energy visualization. (A) Surface view. (B) Potential map in cube format wherein red and blue lines correspond to negative and positive polarity
STRING
In the present study, the protein network of bovine NLRP9 was studied using the STRING database. The specific lines of evidence are represented by line colors linking the protein nodes that are involved in creating functional association whereas the confidence is denoted by the distance between nodes as recognized by the Bayesian scoring system (Fig. 8A). Across the huge transcriptomic and proteomic expression datasets, the co-expression channel mainly is dependent on a gene by gene correlation tests. In bovine animals, the observed gene co-expression is depicted in Fig. 8B. The co-expression method describes the functional association whereas the color intensity in the triangle-matrices represents the confidence aspect of two proteins that are known to be functionally associated keeping in view the whole data expression of the organism. Moreover, the similarities in proteins were provided by gene co-occurrence that provided computing scores. The color intensity describes the degree of similarity between proteins in different organisms as shown in Fig. 8C.


STRING analysis. (A) Network interaction. (B) Gene co-expression. (C) Gene co-occurrence
MoDEL
In the present study, the results of principal component analysis (PCA) generated by the MoDEL web server are shown in Fig. 9 and Table 4.

PCA by MoDEL server. (A) Eigenvalues of CA atoms. (B) Eigenvalues of all atoms. (C) SASA. (D) Cross-correlation network. (E) Displacement projections (CA atoms). (F) Fraction histogram
In the present study, the eigenvalues for CA atoms and all atoms are shown in Fig. 9A, B respectively. The total variance for CA atoms was found to be 477.61A° and 14,590.7A° for all atoms (Table 4). In the present study, solvent accessible surface area (SASA) was 19,508.5±160.5 (all residues), 9339.5±149.7 (polar), and 10,169.1±111.9 (apolar) residues respectively (Fig. 9C). The cross-correlation network relationship of the residues is shown in Fig. 9D. wherein the covariance matrix describes the coupling between residue pairs, i.e., red (correlated), blue (anti-correlated), and white (uncorrelated) respectively. The displacement projection of the first 5 eigenvalues for CA and all atoms are shown in Table 4. Fig. 9D represents the displacement projection for CA atoms. In the present study, Fig. 9E histogram shows the fraction projection of CA atoms of the protein backbone.
Discussion
The role of NLRP9 in host innate immunity and other inflammatory disorders has been found. In the process of drug discovery, a significant interest of examination lies in the identification, synthesis, and purification of natural molecules [41. ]. The drug-like characteristics of bioactives were evaluated by the five parameters rule of Lipinski, and these properties are particularly essential as they are associated with dissolution and intestinal permeability [42,43,44].
Bioactive compounds with less non-toxic nature, diverse phytoconstituents, and bioavailability have been screened and used for their therapeutic ability against various diseases [45,46,47]. In the Indian Ayurvedic system of medicine, Withania somnifera is one of the most valuable medicinal plants that possess a diverse range of phytochemicals like Withaferin A, steroidal lactones, and alkaloids of Withanolide series, Withanolides (II, III, IV, V) and few other compounds [48]. Computational approaches of docking provide insights into protein’s interaction with drug molecules. The docking algorithms generate the binding pose of compounds and scoring properties help to rank them [49,50,51]. Numerous studies have reported beneficial properties of active constituents of Ashwagandha against genital disease caused by Herpes Simplex virus, inhibit Bursal infectious viral disease, and possess anti-influenza activity against chikungunya [52,53,54. ]. Studies have reported antioxidant, anti-inflammatory, immunomodulatory, anticancer, anxiolytic, antioxidant, neuroprotective, cardiovascular, and antidepressant properties in extracts of Ashwagandha [30, 55,56,57].
Doxycycline hydrochloride is a second-generation broad-spectrum antibiotic that is effective against bacterial (Streptococcus pneumoniae, Mycoplasma influenza), viral (vesicular stomatitis virus, chikungunya virus, dengue virus), COVID-19 infections [58,59,60,61]. Our analysis of molecular docking approaches revealed that Withanolide B and doxycycline hydrochloride have the potential to target inflammatory disease proteins and could act as potential inhibitors.
Molecular dynamics provides insights into molecular processes of molecules and rely on empirical potentials. Rg value measures the compactness of protein structure and a protein molecule with a stable Rg value depicts higher compactness. The complete Rg analysis predicts ligand binding to the particular protein that renders it to become more compact and flexible. In the present study, values for RMSD were found to be very small. During the simulation process, a stable value for RMSD determines a good and stable convergence [62]. RMSD analysis provides information about deviation by comparing reference protein structure with the corresponding molecule [63]. High values of RMSD could be associated with significant instability. B-factor and RMSD values identify the flexible areas and the loop regions in the protein molecule. B-factors are associated with the packing of atoms in a protein molecule, and simple algorithms have been developed to determine them depending on the 3D structure of the protein [64, 65] and multiple picosecond MD simulations [66]. B-factors help to investigate the flexibility of protein and differentiate between contacts of crystal packing from the physiological sites of protein-protein binding [67, 68 ]. A greater number of hydrogen bonds determines a stable and significant interaction.
Screening a large number of compounds to estimate the exact scoring function requires much time, and usually, the docking scores and the experimental binding affinities show no correlation. Henceforth, docking results are improved by employing post-processing approaches [69, 70. ].
The contact pattern is important as it helps to evaluate the function and tertiary structure of the protein. Chord plots describe the non-covalent contacts in the secondary structures with edges representing protein contacts and arc the secondary structure. The number of residues in the secondary structure is directly proportional to the size of the arc and simultaneously the thickness of the chord to contact number. The circular arrangement with the residues in the first shell represents non-covalent interactions. Asteroid plots are most significant as they provide insights about structure-based drug designing and engineering of proteins and depict the effect of mutations. The residues in the second shell are represented by larger concentric circles with residues in the nearby neighborhood not interacting with the ligands directly. The larger concentric circles represent the second shell residues that include neighbors of every contacting residue that do not contact the ligand directly. The number of atomic contacts depicts the node size in the inner and outer concentric circles. Scatter plots represent the values for two variables for each chain residue. The residues in the chain indicate the points that describe the value of variables with x and y coordinates. In the scatter plot, the residue properties (network centrality, degree of residue extent, and complex solvent accessible area) of each of the residues are plotted concerning one another. It is important for residues in the outlier areas that may be associated with the structure, stability, and function of the protein [71, 72].
In the binding of small molecules to proteins, electrostatic interactions are particularly important [73]. In the recognition of ligands, electrostatic interactions (stable or transient) along with hydrophobic association and shape complementarity that define enthalpic contribution play a significant role. Many computational approaches have been developed that provide vital insights about the biological and network pathways primarily based on genomic or other datasets with promising results. In system biology, protein-protein interactions (PPI) play an important role in evaluating target protein function and molecule’s drug ability. A PPI network is generally a heterogenous protein network connected by edges that represent interactions. In such graphs, nodes denote proteins, and the adjacent nodes joined by edges represent the physically interacting proteins. The basis for PPI, folding, and assembly of proteins is provided by the non-covalent interactions between the side chain residues [74]. Within the cell, the function of proteins can be inferred by the study of PPIs. Revealing information about protein-protein interactions helps to discover new drug targets [75]. In the present study, associated eigenvalues represent motion stiffness, and its value is proportional to the energy required to deform protein structure. Therefore, the lower the eigenvalues, the easier will be the deformation. The variance of every normal mode is related inversely to its eigenvalue. PCA reduces the complexity of high-dimensional data while retaining original patterns and trends.
Conclusion
The molecular docking and simulation approach was applied to natural compounds of Withania somnifera to reveal their potential against the bovine inflammatory NLRP9 protein. Our study suggested that natural bioactives of Withania somnifera that follow Lipinski’s rule of five could be developed as potent inhibitors against bovine NLRP9. The drug-likeliness and toxicities of the bioactives are important in the drug-development process. Molecular modeling predicted the quality and correctness of protein structure with 96.23% of the residues lying in the Ramachandran favored region. The total energy deviation of bovine NLRP9 protein was evaluated by determining Z-score. Molecular simulation studies revealed structure deviation, protein compactness, stability, mobility, and hydrophobicity. Protein Contacts Atlas provided insights into visualization analysis of non-covalent contacts in a protein structure. The assessment of electrostatic interactions through in silico approaches helps to determine the energy of protein-ligand complexes and thus is important in virtual drug design and screening. In living organisms, protein-protein interactions perform a significant function in the cellular systems. The behavior of protein motion can be easily assessed by PCA based on eigenvalues. The present study demonstrates that this herb can serve as an alternative to treat inflammatory conditions in addition to currently available procedures. However, in vivo and in vitro studies are further required to validate the role of this compound to combat inflammatory states. Thus, based on the observations, we can conclude that bioactives of Withania somnifera within the acceptable range of drug-likeliness can prove to be a potent and efficacious molecule that could interact with inflammatory proteins and can be further repurposed as a drug candidate.
Data availability
In this study, all the data generated has been published in this article.
References
Zhang Y, Liang C (2016) Innate recognition of microbial-derived signals in immunity and inflammation. Sci China Life Sci 59(12):1210–1217
Abe T, Marutani Y, Shoji I (2019) Cytosolic DNA-sensing immune response and viral infection. Microbiol Immunol 63(2):51–64
Fitzgerald KA, Kagan JC (2020) Toll-like receptors and the control of immunity. Cell 180(6):1044–1066
Fritz JH, Ferrero RL, Philpott DJ, Girardin SE (2006) Nod-like proteins in immunity, inflammation and disease. Nat Immunol 7(12):1250–1257
Meylan E, Tschopp J, Karin M (2006) Intracellular pattern recognition receptors in the host response. Nature 442(7098):39–44
Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM (2019) Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol 40(11):1035–1052
Rathinam VA, Fitzgerald KA (2016) Inflammasome complexes: emerging mechanisms and effector functions. Cell 165(4):792–800
Bauernfeind F, Hornung V (2013) Of inflammasomes and pathogens–sensing of microbes by the inflammasome. EMBO Mol Med 5:814–826
Man SM, Kanneganti TD (2016) Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol 16(1):7–21
Jones JD, Vance RE, Dangl JL (2016) Intracellular innate immune surveillance devices in plants and animals. Science 354(6316):aaf6395
Lamkanfi M, Dixit VM (2012) Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol 28:137–161
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140(6):805–820
Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13(6):397–411
Davis BK, Wen H, Ting JP (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29:707–735
Kufer TA, Sansonetti PJ (2011) NLR functions beyond pathogen recognition. Nat Immunol 12(2):121–128
Peng H, Zhang W, Xiao T, Zhang Y (2014) Expression patterns of Nlrp9a, Nlrp9b and Nlrp9c during mouse development. Biologia. 69(1):107–112
Jiang Y, Xie M, Chen W, Talbot R, Maddox JF, Faraut T, Dalrymple BP (2014) The sheep genome illuminates biology of the rumen and lipid metabolism. Science 344(6188):1168–1173
Calvo JH, Chantepie L, Serrano M, Sarto MP, Iguacel LP, Jiménez MÁ, Lahoz B (2020) A new allele in the BMP15 gene (FecXRA) that affects prolificacy co-segregates with FecXR and FecXGR in Rasa aragonesa sheep. Theriogenology 144:107–111
Amoushahi M, Steffensen LL, Galieva A, Agger J, Heuck A, Siupka P et al (2020) Maternally contributed Nlrp9b expressed in human and mouse ovarian follicles contributes to early murine preimplantation development. J Assist Reprod Genet 37(6):1355–1365
Zhu S, Ding S, Wang P, Wei Z, Pan W, Palm NW, Flavell RA (2017) Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546(7660):667–670
Fernandez MV, Budde J, Del-Aguila JL, Ibanez L, Deming Y, Harari O et al (2018) Evaluation of gene-based family-based methods to detect novel genes associated with familial late onset alzheimer disease. Front Neurosci 12:209
Yanling Q, Xiaoning C, Fei B, Liyun F, Huizhong H, Daqing S (2018) Inhibition of NLRP9b attenuates acute lung injury through suppressing inflammation, apoptosis and oxidative stress in murine and cell models. Biochem Biophys Res Commun 503(2):436–443
Gil-Varea E, Urcelay E, Vilarino-Guell C, Costa C, Midaglia L, Matesanz F et al (2018) Exome sequencing study in patients with multiple sclerosis reveals variants associated with disease course. J Neuroinflammation 15(1):265
Castano-Rodriguez N, Kaakoush NO, Goh KL, Fock KM, Mitchell HM (2014) The NOD-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer: a case-control study and gene expression analyses. PloS One 9(6):e98899
Bai R, Yao C, Zhong Z, Ge J, Bai Z, Ye X et al (2021) Discovery of natural antiinflammatory alkaloids: potential leads for the drug discovery for the treatment of inflammation. Eur J Med Chem 213:113165
Borquaye LS, Darko G, Laryea MK, Roberts V, Boateng R, Gasu EN et al (2017) Anti-Inflammatory activities of extracts from Oliva sp., Patella Rustica, and Littorina littorea collected from Ghana’s coastal shorelines. Cogent Biol 3:1364063
Saleem S, Muhammad G, Hussain MA, Altaf M, Bukhari SNA (2020) Withania somnifera L.: Insights into the phytochemical profile, therapeutic potential, clinical trials, and future prospective. Iran J Basic Med Sci 23(12):1501
Priyanka G, Anil Kumar B, Lakshman M, Manvitha V, Kala Kumar B (2020) Adaptogenic and immunomodulatory activity of Ashwagandha root extract: an experimental study in an equine model. Front Vet Sci 7:541112
Bano A, Sharma N, Dhaliwal HS et al (2015a) A systematic and comprehensive review on Withania somnifera (L.) Dunal—an Indian ginseng. Br J Pharmaceut Res 7:63–75
Farooqui AA, Farooqui T, Madan A, Ong JHJ, Ong WY (2018) Ayurvedic medicine for the treatment of dementia: mechanistic aspects. Evid Based Complement Alternat Med:2018
Kim SH, Singh KB, Hahm ER, Lokeshwar BL, Singh SV (2020) Withania somnifera root extract inhibits fatty acid synthesis in prostate cancer cells. J Tradit Complement Med 10(3):188–197
Maheswari P, Harish S, Navaneethan M, Muthamizhchelvan C, Ponnusamy S, Hayakawa Y (2020) Bio-modified TiO2 nanoparticles with Withania somnifera, Eclipta prostrata and Glycyrrhiza glabra for anticancer and antibacterial applications. Mater Sci Eng C 108:110457
Birla H, Keswani C, Rai SN, Singh SS, Zahra W, Dilnashin H, Singh SP (2019) Neuroprotective effects of Withania somnifera in BPA induced-cognitive dysfunction and oxidative stress in mice. Behav Brain Funct 15(1):1–9
Yang H, Lou C, Sun L, Li J, Cai Y, Wang Z, Tang Y (2019) admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 35(6):1067–1069
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7(1):1–13
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 26(2):283–291
Kleywegt GJ, Jones TA (1996) Phi/psi-chology: Ramachandran revisited. Structure 4(12):1395–1400
Tallei TE, Tumilaar SG, Niode NJ, Kepel BJ, Idroes R, Effendi Y, Emran TB (2020) Potential of plant bioactive compounds as SARS-CoV-2 main protease (Mpro) and spike (S) glycoprotein inhibitors: a molecular docking study. Scientifica 2020
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461
Li L, Li CA, Sarkar S, Zhang J, Witham S, Zhang Z, Wang L, Smith N, Petukh M, Alexov E (2012) DelPhi: a comprehensive suite for DelPhi software and associated resources. Bmc Biophys 5:1
Mahmud S, Paul GK, Afroze M, Islam S, Gupt SBR, Razu MH, Simal-Gandara J (2021) Efficacy of phytochemicals derived from Avicennia officinalis for the management of COVID-19: a combined in silico and biochemical study. Molecules 26(8):2210
Rehman MU, Ali A, Ansar R, Arafah A, Imtiyaz Z, Wani TA et al (2022) In Silico molecular docking and dynamic analysis of natural compounds against major non-structural proteins of SARS-COV-2. J Biomol Struct Dyn:1–17
Malla BA, Ali A, Maqbool I, Dar NA, Ahmad SB, Alsaffar RM, Rehman MU (2022) Insights into molecular docking and dynamics to reveal therapeutic potential of natural compounds against P53 protein. J Biomol Struct Dyn:1–20
Lipinski CA (2004) Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol 1(4):337–341
Ali A, Malla BA, Manzoor Z, Bashir SM, Dar MA, Ganie SA, Mir MUR (2022) Immunogenetic disorders: treatment with phytomedicines. Clinical Applications of Immunogenetics. Academic Press, pp 89–130
Alsaffar RM, Ali A, Rashid SM, Ahmad SB, Alkholifi FK, Kawoosa MS, Rehman MU (2023) Zerumbone protects rats from collagen-induced arthritis by inhibiting oxidative outbursts and inflammatory cytokine levels. ACS Omega
Bhardwaj VK, Singh R, Sharma J, Rajendran V, Purohit R, Kumar S (2020) Identification of bioactive molecules from Tea plant as SARS-CoV-2 main protease inhibitors. J Biomol Struct Dyn:1–10
Tong X, Zhang H, Timmermann BN (2011) Chlorinated Withanolides from Withania somnifera. Phytochemistry Lett 4(4):411–414
Du X, Li Y, Xia YL, Ai SM, Liang J, Sang P, Ji XL, Liu SQ (2016) Insights into protein–ligand interactions: mechanisms, models, and methods. Int J Mol Sci 17(2):144
Kairys V, Baranauskiene L, Kazlauskiene M, Matulis D, Kazlauskas E (2019) Binding affinity in drug design: experimental and computational techniques. Expert Opin Drug Discovery 14(8):755–768
Vuignier K, Schappler J, Veuthey JL, Carrupt PA, Martel S (2010) Drug-protein binding: a critical review of analytical tools. Anal Bioanal Chem 398(1):53–66
Jain J, Narayanan V, Chaturvedi S, Pai S, Sunil S (2018) In vivo evaluation of Withania somnifera-based Indian traditional formulation (Amukkara Choornam), against Chikungunya virus-induced morbidity and arthralgia. J Evid-Based Integr Med 23:2156587218757661
Cai Z, Zhang G, Tang B, Liu Y, Fu X, Zhang X (2015) Promising anti-influenza properties of active constituent of Withania somnifera ayurvedic herb in targeting neuraminidase of H1N1 influenza: Computational study. Cell Biochem Biophys 72(3):727–739
Ganguly B, Umapathi V, Rastogi SK (2018) Nitric oxide induced by Indian ginseng root extract inhibits Infectious Bursal Disease virus in chicken embryo fibroblasts in vitro. J Anim Sci Technol 60:2
Bano A, Sharma N, Dhaliwal HS et al (2015b) A systematic and comprehensive review on Withania somnifera (L.) Dunal—an Indian ginseng. Br J Pharmaceut Res 7:63–75
Kulkarni SK, Dhir A (2008) Withania somnifera: an Indian ginseng. Progress in neuro-psychopharmacology & biological psychiatry 32(5):1093–1105
Mirjalili MH, Moyano E, Bonfill M, Cusido RM, Palazón J (2009) Steroidal lactones from Withania somnifera, an ancient plant for novel medicine. Molecules (Basel, Switzerland), 14(7):2373–2393
Cazalis J, Bodet C, Gagnon G, Grenier D (2008) Doxycycline reduces lipopolysaccharide-induced inflammatory mediator secretion in macrophage and ex vivo human whole blood models. J Periodontol 79(9):1762–1768
Wu J, Maoqiang L, Fan H, Zhenyu B, Qifang H, Xuepeng W, Liulong Z (2016) Rutin attenuates neuroinflammation in spinal cord injury rats. J Surg Res 203(2):331–337
Rothan HA, Mohamed Z, Paydar M, Rahman NA, Yusof R (2014) Inhibitory effect of doxycycline against dengue virus replication in vitro. Arch Virol 159(4):711–718
Gendrot M, Andreani J, Jardot P, Hutter S, Delandre O, Boxberger M, Pradines B (2020) In vitro antiviral activity of doxycycline against SARS-CoV-2. Molecules 25(21):5064
Melvin LS, Welling U, Kandel Y, Levinson ZA, Taoka H, Stock HJ, Demmerle W (2022) Applying stochastic simulation to study defect formation in EUV photoresists. Jpn J Appl Phys 61(SD):SD1030
Schreiner W, Karch R, Knapp B, Ilieva N (2012) Relaxation estimation of RMSD in molecular dynamics immunosimulations. Comput Math Methods Med 2012:173521
Yin H, Li YZ, Li ML (2011) On the relation between residue flexibility and residue interactions in proteins. Protein Pept Lett 18:450–456
Weiss MS (2007) On the interrelationship between atomic displacement parameters (ADPs) and coordinates in protein structures. Acta Crystallogr D63:1235–1242
Pang YP (2016) Use of multiple picosecond high-mass molecular dynamics simulations to predict crystallographic B-factors of folded globular proteins. Heliyon 2:e00161
Vihinen M, Torkkila E, Riikonen P (1994) Accuracy of protein flexibility predictions. Proteins 19:141–149
Liu Q, Li Z, Li J (2014) Use B-factor related features for accurate classification between protein binding interfaces and crystal packing contacts. BMC Bioinf 15:S3
Quiroga R, Villarreal MA (2016) Vinardo: a scoring function based on Autodock Vina improves scoring, docking, and virtual screening. PloS One 11(5):e0155183
Rastelli G, Pinzi L (2019) Refinement and rescoring of virtual screening results. Front Chem 7:498
Mendes E, Lokan C, Harrison R, Triggs C (2005) A replicated comparison of cross-company and within-company effort estimation models using the isbsg database. In 11th IEEE International Software Metrics Symposium (METRICS'05). IEEE, p 10
del Sol A, Fujihashi H, Amoros D, Nussinov R (2006) Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Mol Syst Biol 2(1):2006–0019
Li R, Zhu S (2020) NLRP6 inflammasome. Molecular aspects of medicine 76:100859
Ofran Y, Rost B (2003) Predicted protein–protein interaction sites from local sequence information. FEBS Lett 544(1-3):236–239
Pedamallu CS, Posfai J (2010) Open source tool for prediction of genome wide protein-protein interaction network based on ortholog information. Source Code Biol Med 5(1):1–6
Funding
The authors are thankful to researchers supporting project number (RSPD2023R722), King Saud University, Riyadh Saudi Arabia for funding this research.
Author information
Authors and Affiliations
Contributions
Conceptualization, A.A., methodology, A.A., M.U.R., S.B.A., software, A.A., M.U.R, validation, A.A., A. K, S.M., T.M.M., formal analysis, A.A., M.U.R., investigation, A.A., M.U.R, writing—original draft preparation, A.A., M.U.R., G.J., A.A., I.M., writing—review and editing., A.A., M.U.R., A.K., visualization., A.A., supervision A.A., M.U.R. All authors have read and agreed to the published manuscript version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ali, A., Mir, G. ., Ayaz, A. et al. In silico analysis and molecular docking studies of natural compounds of Withania somnifera against bovine NLRP9. J Mol Model 29, 171 (2023). https://doi.org/10.1007/s00894-023-05570-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-023-05570-z