
Abstract
For dehydration of CaC2O4·H2O and thermal dissociation of CaCO3 carried out in Mettler Toledo TGA/SDTA-851e/STARe thermobalance similar experimental conditions was applied: 9–10 heating rates, q = 0.2, 0.5, 1, 2, 3, 6, 12, 24, 30, and 36 K min−1, for sample mass 10 mg, in nitrogen atmosphere (100 ml min−1) and in Al2O3 crucibles (70 μl). There were analyzed changes of typical TGA quantities, i.e., T, TG and DTG in the form of the relative rate of reaction/process intended to be analyzed on-line by formula (10). For comparative purposes, the relationship between experimental and equilibrium conversion degrees was used (for \( P = P^{{\ominus}} \)). It was found that the solid phase decomposition proceeds in quasi-equilibrium state and enthalpy of reaction is easily “obscured” by activation energy. For small stoichiometric coefficients on gas phase side (here: ν = 1) discussed decomposition processes have typical features of phenomena analyzable by known thermokinetic methods.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the discussion on thermokinetic analysis of reaction/process of thermal decomposition of compounds undergoing destruction with observable weight loss:
(where the stoichiometric coefficient ν is sum of coefficients for gaseous products) an important problem is description of thermodynamic conditions under which experiments are carried out. Classic ‘single kinetic triplet’ f(α) or g(α)-E-A applies to thermokinetic conditions, not necessarily equilibrium. In all conditions, due to temperature (T) and pressure (P), appropriate laws determine equilibrium degree of conversion according to:
-
(1)
modified van ‘t Hoff’s isobar for thermal dissociation of chemical compounds in solid state [1–5],
- (2)
Reaction course in accordance with these laws causes that its determinant are thermodynamic quantities expressed by:
instead of ‘single kinetic triplet’.
It is proved that reaction enthalpy (Δr H)—as an average quantity—is independent of temperature (T) and pressure (P), when P < 3 MPa [8].
In practice, in TGA analysis, the most commonly used is experimental degree of conversion (α) at a given temperature, which must be lower than equilibrium one (αeq) [9–11], but not always so.
A combination of both rights with regard to conditions (T, P) can be carried out using the basic relations for constant thermodynamic equilibrium of reaction:
where constant K for reaction (1) is expressed in the following manner:
In Eq. 4 one substitutes in accordance with Refs. [1–4]:
Using the similar procedure in Eq. 3 one obtains:
Identical results were presented in works [6, 7].
Finally, formula (6) is expressed as follows:
According to Eq. 7 intensive collection of gaseous products producing vacuum (\( P^{{\ominus}} > P \)) moves equilibrium line α eq over the course deriving from isobaric conditions (4) together with (5).
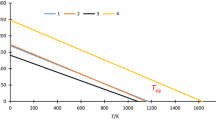
It is common to carry out studies on thermal processes just in discussed way—the example is presented in Fig. 1 quoted from [12].

Scope of the work
Basing on the analysis of thermal decomposition of two model chemical compounds (CaC2O4·H2O and CaCO3), often used as test substances, in inert atmosphere in dynamic conditions at different heating rates (q), own assessment of thermodynamic conditions of decomposition using the relative rate of reaction/process has been presented.
Basing on three-parametric equation [5], the relative rate of reaction/process was proposed for dynamic conditions [13], which results from considerations on the equilibrium course of chemical reaction of solid phase dissociation, when \( P = P^{{\ominus}} \). It means that the term \( r = - {{{\text{d}}\ln \alpha } \mathord{\left/ {\vphantom {{{\text{d}}\ln \alpha } {{\text{d}}\left( {{1 \mathord{\left/ {\vphantom {1 T}} \right. \kern-\nulldelimiterspace} T}} \right)}}} \right. \kern-\nulldelimiterspace} {{\text{d}}\left( {{1 \mathord{\left/ {\vphantom {1 T}} \right. \kern-\nulldelimiterspace} T}} \right)}} \) versus T should be constant, i.e., r = r eq = const. (r in K).
Therefore there was the study carried out, in which it was assumed that relationships r versus T will be analyzed without any data selection procedure in order to analyze graphic images obtained in dynamic conditions, electronically recording the course of thermal dissociation of selected two chemical compounds in the relation: mass versus temperature.
Assumptions
Assuming in Eq. 7 \( P^{{\ominus}} = P, \) after differentiation towards temperature one obtains:
This result can be presented (by analogy), using equilibrium relative rate of reaction/process in the following way:
Equation 9 represents straight line parallel to temperature axis (T), limited by perpendicular at the point with coordinates [0, T eq] (e.g., see Fig. 3 in [14]).
It was assumed that experimental data can be directly measured by TGA [13, 15]:
where m i is initial mass (mg), DTG (mg K−1) and TG (mg).
The relative rate of reaction/process is linear relationship of correlated coefficients a 1 and a 2 [13]:
Coefficients a 1 and a 2 originate from equations:
(A) [5]:
or
(B) [16]:
Further considerations follow from comparison of temperature of maximum reaction rate T m determined experimentally and calculated by formula (14).
According to Ref. [13] temperature of flex point (T fp) in version [7] (formula (1–8) is identified with the T m:
Another relationship is of thermodynamic-correlation nature and interconnects a 1 and a 2 by temperature T r [16]:
According to Ref. [14], temperature T r is represented by slope of straight lines (11) and (9). To some extent this is the analogue of isokinetic temperature, because (in theory) r = const. Analysis of experimental data consists in determination of linear relationships (11), while the relative rate of reaction/process is determined by formula (10). Next, formulas in forms (14) and (15) were used.
Each analysis of compounds (CaC2O4·H2O and CaCO3) was preceded by determination of activation energy according to modified Kissinger law in version [17]:
to confirm whether own data are within the range of data presented in literature.
The considerations were carried out in three areas: thermokinetic, thermodynamic, and relative rate. This approach follows from the fact that such considerations prevail in such order in literature.
The basic element of analysed thermoanalytical results consists in evaluation, whether the relative rate of reaction/process r calculated according to Eq. 10 versus T presents relationship (9), of which graphical image is rectangle area bounded by temperature T eq, or linear relationship (11) with a negative slope, expressed by coefficient a 2.
Calcium oxalate monohydrate
Thermokinetics
Thermal decomposition of CaC2O4·H2O, especially at the initial stage of the process (dehydration) is still featured in both newer literature [18–23] and own works [13, 17, 24]. An example of single kinetic triplet according to Ref. [21] is expressed as:
and according to Ref. [17]:
and from own results according to Eq. 16 was obtained:
i.e., E = 89.05 kJ mol−1, lnA = 15.845 (A in s−1).
Thermodynamics
Equation 9 is plotted on the basis of thermodynamic data for standard conditions:
-
T = 298.15 K and P ≈ 0.1 MPa for dehydration according to Ref. [18]:
-
ΔH = 36.4 kJ mol−1, (according to Ref. [25] enthalpy of vaporization of water in 298.15 K is 44.0 kJ mol−1)
-
ΔG = 2.2 kJ mol−1,
-
ΔC p = −10.2 J (K mol)−1—originates from summing up values: 298 C p, Table 1 in [18] and entropy in these conditions:
-
ΔS = 0.1147 kJ (K mol)−1 (acc. [26]: ΔS = 0.156 kJ (K mol)−1)
From these data, equilibrium temperature T eq (also called temperature of inversion) was determined iteratively from Gibbs–Helmholtz equation and Kirchhoff’s law: when \( \Updelta G \, = \, 0\quad {\text{then}}\quad T = T_{\text{eq}} \) and
next: T eq = 317.4 K
which is to all intents and purposes consistent with the formula:
From the calculations follows the average enthalpy of reaction in the temperature range of 298–318 K: Δr H = 36.3 kJ mol−1. Figure 2 shows relationship between degree of conversion and temperature for ten heating rates, and Fig. 3—between the relative rate of reaction/process and temperature according to Eq. 10, giving the coefficient of determination (r 2) with equilibrium line (9) limited by straight line T eq = 318 K (45 °C) for selected heating rates (q = 0.2, 3, and 30 K min−1).

Ten experimental alpha (α)–temperature (T) curves for dehydration of calcium oxalate monohydrate in nitrogen, obtained at different heating rates: 0.2, 0.5, 1, 2, 3, 6, 12, 18, 24, and 30 K min−1

Relationship between the relative rate of reaction (r) and temperature (T) for dehydration of calcium oxalate monohydrate (3 of the 10 heating rates). Marked linear relationships (11) concerns selected experimental points
Equilibrium relationship is as follows:
According to Eqs. 9 and 19 one obtains:
Equation 19 was determined on the basis of thermodynamic data [18] in standard conditions, T = 298 K, and thus, referring to Eq. 5, equilibrium conversion degree (for ν = 1) in natural way concerns this reference condition. Under these considerations, Eq. 19 should be regarded as acceptable estimation, assuming that in considered temperature range thermodynamic quantities are close to standard ones.
The difference can be attributed to slightly differing values of entropy. The issue, what may be surprising here, is the fact that this is thermodynamic or maximum value at given temperature and at atmospheric pressure P = 0.1 MPa. This does not mean that at T = 298 K, the decomposition occurs and is detectable, because in the way here may be conditions of kinetic nature, e.g., very slow evaporation or atmospheric equilibrium. For this reaction, Δr H ≈ 36 kJ/mol is determined by the activation energy E = 80 kJ/mol (or higher) and by the heat of vaporization of water, more than 40 kJ/mol.
In turns, Fig. 4 shows experimental results that show straight lines (11). From Fig. 4 follows that each straight line (11) together with (20) intersect at a wide temperature range and not only at determined temperature T r = 390.2 K. With the exception of q = 0.2 and 1 K min−1, each straight line (for q = 0.5, 2, 3, 6, 12, 18, 24, and 30 K min−1) intersects at higher temperature, i.e., 399 K.

Graphical presentation of linear relation r = a 1–a 2 T for dehydration of calcium oxalate monohydrate
Relative rate
Table 1 presents calculated coefficients of Eq. 11 and temperature of maximum reaction rate T m determined experimentally and calculated by Eq. 14—Fig. 5 compares the two temperatures toward q.

Relationship between the temperature of the maximal rate of reaction (T m) and heating rate (q) for dehydration of calcium oxalate monohydrate
According to Fig. 2 experimental degrees of conversion α satisfy inequality αeq > α for all heating rates q, and according to Kissinger law (as well as in version (16)) temperatures of maximum reaction rate T m increase with increasing heating rate q. However, from Fig. 3 follows that virtually all the points showing the relative rate of reaction r in terms of temperature are outside the rectangle (20). It demonstrates relative dynamic reaction course for small heating rates (according to Table 1) for q = 0.2, 0.5, 1 K min−1, moderated course (q = 1 and 3 K min−1) and close to equilibrium, when a 2 < 100 for high heating rates (q ≥ 6 K min−1). Relativity of dynamics here means a reference to temperature, as well to maximum thermal rate (dα/d T) obtained at low heating rates (q), as to temperature ranges, in which conversion degree achieve the end of reaction (α → 1).
However, courses similar to the parallel (a 2 = small) are greater in value than the expression r eq = Δr H/νR = 4366.13 K and closer to the ratio E/R ≅ 10000 K.
Characteristic temperatures in these analyses are of very important control meaning, namely: T eq and T m, T f as well as T r, which are determined experimentally. For large heating rates, thermal dissociation approaches to quasi-equilibrium with a large temperature shift of the final reaction temperature T f in relation to T eq (T f > T eq). Instead, for small heating rates high values of coefficient a 2 are observed, what means that even for the lowest heating rate, often regarded as a pseudo-isothermal (q = 0.2 K min−1), occurs the largest deformation of the equilibrium distribution according to Eq. 7 by factor \( \left( {T/T_{\text{f}} } \right)^{{a_{2} }} \) (Eq. 13), which gradually reaches value of 1 for a 2 = 0 [16].
According to Fig. 5 and Table 1 the lower coefficient a 2, the differences between fixed temperature T m calculated by Eq. 14 become very large—only for large values of a 2, so for very small heating rates formula (14) leads to values compatible with experiment.
Establishing the meaning of temperature T r , for the data set in Table 1, Eq. 15 was determined:
what leads to T r = 390.2 K.
Again, one may also note that constant term in Eq. 21 is much higher than the one resulting from enthalpy (r eq = 4366.13 K)—the same observation is included in Ref. [16].
Calcium carbonate (calcite)
Thermokinetics
Calcium carbonate was within the frame of the project described in the report [12]—the results were analyzed and discussed further in subsequent works [27–29], and then in Ref. [16] in terms of significance of Eqs. (12–13). Results of studies on thermal dissociation of CaCO3 discussed at the ICTAC Conference [12, 27–29], also summarized in Tables 1 and 2 in Ref. [16], were used applying Kissinger law in version (16).
It was obtained:
-
for decomposition in nitrogen: E = 201.0 kJ mol−1, ln A = 13.46 (A in s−1) (r 2 = 0.9865, sl = 0.00001), according to Ref. [12] similar values was obtained by H. O. Desseyn: ln A = 13.58 (A in s−1) and E = 199 kJ mol−1, but only in this one case),
-
for decomposition in vacuum: E = 111.9 kJ mol−1, ln A = 6.05 (A in s−1) (r 2 = 0.9435, sl = 0.00122)—in Ref. [12] several pairs are similar.
Next, own data analyzed according to Eq. 16 lead to equation:
Relationship between conversion degree and temperature at variable heating rates and on the background of equilibrium conversion degree is presented in Fig. 6.

Nine experimental alpha (α)–temperature (T) curves for decomposition of calcium carbonate in nitrogen, obtained at different heating rates: 0.2; 0.5; 1; 3; 6; 12; 24; 30 and 36 K min−1
Thermodynamics
Equilibrium relationship for calcite was taken from Ref. [16] (Δr H = 174.9 kJ mol−1):
that is:
Compared to the ICTAC data presented in Fig. 1, results of these studies (Fig. 6) point to an even greater shift of experimental curves of conversion degree α to the left in relation to the equilibrium curve (7) for \( P^{{\ominus}}\,=\,P \) , i.e. αeq < α.
Relative rate
In Ref. [16], it was found that a 2 coefficients are: for decomposition in nitrogen a 2 = 208 → 63 and for heating rate q = 1–25 K min−1 (Table 1 in Ref. [16]), but they are much lower in case of decomposition in vacuum: a 2 = 661–317, q = 1.8 to 10 K min−1 (Table 2 in Ref. [16]).
Figure 7 presents—similarly as in Fig. 3—relationships of the relative rate of reaction/process according to Eq. 10, giving determination coefficient (r 2) together with the equilibrium line (23) limited by T eq = 1162.7 K. Table 2 compiles determined coefficients of linear relationship (11) and temperature T m (experimental) calculated by Eq. 14.

Relationship between the relative rate of reaction (r) and temperature (T) for decomposition of calcium carbonate. Marked relationships (11) concerns selected experimental points, for which (r 2) reached the highest value (even for r 2 = 0.2232, sl = 0.05, together with increase of q: r 2 → 1)
In Fig. 7 was indicated temperature T r following from Eq. 25, because it relates to small values of a 2 and thus exposes range closer to equilibrium. Figure 8 presents again data from Fig. 7, where straight lines (11) present experimental results.

Graphical presentation of linear relation r = a 1 − a 2 T for decomposition of calcium carbonate
Coefficients a 1 and a 2 satisfy linear relationship (Fig. 9):
which determines T r = 1043.4 K. Previous analyses given in Ref. [16] set different coefficients of relationship (11):

Relation between coefficients: a 1 versus a 2 for decomposition of calcium carbonate
From Fig. 8 follows that each straight line (11) together with (24) intersect at a wide temperature range, and not only at determined temperature T r. We note again that constant term in Eq. 25 is higher than the one resulting from enthalpy (r eq = 21039.6 K).
Referring to Table 2, it was again established that for small values of a 2 experimental temperature T m is much lower than the one calculated by Eq. 14. Observed thermal dissociation of CaCO3 can be defined as proceeding under quasi-equilibrium conditions in relation to anticipated range of characteristic temperature according to Eq. 7 for \( P^{{\ominus }} = P \). Maybe this is the reason of observing more chaotic arrangement of points of Fig. 8 in comparison to Fig. 4. Approximately, when a 2 → 0, then Eq. 13 simplifies to relationship known as temperature criterion [24, 30]: ln α = const. − E/RT, then Δr H → E.
Discussion
The results of investigations on thermal dissociation of two chemical substances frequently used as test compounds (standards) in thermal analysis:
-
dehydration of CaC2O4·H2O,
-
thermal decomposition of CaCO3 (calcite).
The investigations were carried out under identical test conditions. Previously, trial and error method was used for selection of experimental conditions—mainly heating rates, nitrogen flow, crucibles—in order to a 2 ratio was greater than 100. According to Tables 1 and 2 in case of calcite, it was manage to get it completely, but for dehydration of oxalate—only in a large extent.
Observing the experimental conversion degrees α toward temperature profile T, one may conclude that both compounds behave differently in relation to the equilibrium curves:
-
dehydration proceeds under curve resulting from relationship (19), α < α eq ,
-
thermal dissociation of carbonate proceeds above relationship (22), α > α eq .
In detailed studies, the relative rate of reaction/process was explicitly determined from experimental data according to the formula (10), i.e., using T, TG, DTG, and m i—deliberately it was not used the option of smoothing by Savitzky–Golay method [31], which very useful for studies of complex organic mixtures such as coal tar pitches [15].
In the range of 0 < α < 0.05–0.1 one always must take into account the occurrence of oscillations arising from expression appearing in formula (10) by the ratio −DTG/TG ∝ dα/αdT, i.e., one is close to an indeterminate form 0/0 and using filters of type [31] does not help much here.
What is puzzling, however, is how are observed such differences between temperatures of maximum reaction rate, determined experimentally and calculated by Eq. 14. Substituting empirical relationship (15) to the formula (14) yields:
For a 2 = 0 (and small values):
when a 2 is large, then according to Ref. [16]
what, on the basis of this studies, means approaching to compliance of both temperatures T m and T eq.
Returning to the Eq. 28 is easy to notice that one obtains this formula, if both Eq. 8 and condition dαeq/dT = 0 are used, thus this is the maximum occurring far beyond the scale where αeq ⋙ 1 (compare with Ref. [32]). For these reasons, the differences [Δr H/2νR − (T m)exp] must be very large, and they decreases, when a 2 = large.
Thus, one may note that the quasi-equilibrium conditions prevent accurate determination of enthalpy of reaction Δr H, because dynamic conditions directly dictate relationships dominated by activation energy E. Thermal analyses of complex organic compounds in the form of salts containing anion halogens (Cl, Br and J), as described by Błażejowski et al. [3, 4, 33–36], behave differently due to the formation of a significant volume of gas (ν ≥ 2). These compounds are characterized by thermal decomposition, combined with the total volatilization of gaseous products. From this comparison, one may even get the feeling that the presence of solid phase (ν = 1) inhibits the process of destruction and it is necessary helping it through physical occurrence (vacuum, intense collection of gaseous products, volatilization).
Conclusions
-
1.
On the basis of dehydration of calcium oxalate monohydrate and thermal dissociation of calcium carbonate (calcite) in specially selected conditions, it was found that the reactions proceed in a quasi-equilibrium conditions in relation to modified van ‘t Hoff’s isobars expressed by Eq. 7 when \( P^{{\ominus}} = P \). In the case of CaCO3 were observed small values of coefficient a 2 (maximum a 2 = 88.65), and the experimental curves of conversion degree toward temperature profile satisfy the relation α > αeq, what means an intense collection of gaseous product from reaction zone and, in consequence, T eq > T f . In the case of dehydration of CaC2O4·H2O were observed very intensive courses for small heating rates q = 0.2 to 1 K min−1, what also translates to deformation equilibrium curve (7) by significant factor \( (T/T_{f} )^{{a_{2} }} \) (Eq. 13), but at higher heating rates also importance of a 2 decreases. The reference to equilibrium decomposition is consistent with expectations, because α < αeq.
-
2.
Quasi-equilibrium conditions prevent precise determination of enthalpy of reaction (Δr H), because dynamic conditions impose relationships dominated by activation energy (E). Despite the very significant correlation between coefficients a 1 and a 2 (see Refs. [37–40]), the constant term in Eq. 15 is however greater than the expression (Δ r H/νR). For two reaction analysed in current paper it was not obtained values of a 2 = 0 and at the same time T f = T eq. Temperature T r, which is an analogue of isokinetic temperature according to Arrhenius law (here: r = r eq = const.), may be determined by correlation—Figs. 4 and 8 indicate that the effect of convergence in one point (for one coordinate) is blurred. It should, however, be borne in mind that similar situation also concerns isokinetic temperature in isokinetic/compensation effect—for example in Ref. [41].
-
3.
Formula (14)—characteristic for the model of relative rate of reaction/process (12, 13) and (11) becomes less important, when a 2 = 0 or a 2 = small for calculation of T m.
-
4.
In the case of dehydration of CaC2O4·H2O at low heating rates q = 0.2–1 K min−1 is observed high dynamics of decomposition determined by coefficient a 2. It disappears at higher heating rates, approaching values close to but higher than r eq = 4366.13 K (formula (20)), but significantly shifted above equilibrium temperature above T f ⋙ T eq. This effect is related to phase transformation of the reaction product—water that goes into the gas phase with a variable rate depending on the time and temperature. At lower temperature, but during the very long time (q = 0.2 K min−1), there are more favorable conditions for the evacuation of water from solid surface. In a very short time (high heating rate q ≥ 6 K min−1) high temperatures are necessary.
-
5.
Formula (10) is very simple and enables on-line analysis, and in particular its linear relationship with temperature (11) becomes clearer with increasing heating rate q.
Experimental methodology
Thermal decomposition of calcium oxalate monohydrate CaC2O4·H2O (Aldrich, Cat. No. 28,984-1) and calcium carbonate CaCO3 (Mettler—Test Sample) was carried out using Mettler Toledo TGA/SDTA-851e/STARe, for weight samples of 10 mg, in atmosphere of nitrogen (100 ml min−1), in Al2O3 crucible (70 μl), for 10 heating rates: q = 0.2; 0.5; 1; 2; 3; 6; 12; 18; 24 and 30 K min−1 (in the case of CaC2O4·H2O) and for 9 heating rates: q = 0.2; 0.5; 1; 3; 6; 12; 24; 30 and 36 K min−1 (in case of CaCO3).
Temperatures indicated in the text by T relate to temperature of the reacting sample.
In relation to ICTAC research [12, 27–29] the scope of research was broadened on both lower (q = 0.2 K min−1—it is treated as pseudo-isothermal conditions) and higher heating rates (q = 36 K min−1).
Abbreviations
- α:
-
Conversion degree, 0 ≤ α ≤ 1
- αeq :
-
Equilibrium conversion degree, 0 ≤ αeq ≤ 1, P = const
- α eq :
-
Equilibrium conversion degree, 0 ≤ α eq ≤ 1, T, P = var
- a 0, a 1 and a 2 :
-
Coefficients of three-parametric equation, acc. to Eq. 12
- A :
-
Pre-exponential factor in Arrhenius equation, s−1
- ΔC p :
-
Heat capacity of reaction, J (K mol)−1
- E :
-
Activation energy, J mol−1
- f(α) and g(α):
-
Kinetic functions toward conversion degree α
- ∆r H :
-
Average reaction enthalpy, J mol−1
- ∆G :
-
Free enthalpy, J mol−1
- ∆S :
-
Entropy, J (K mol)−1
- K :
-
Thermodynamic equilibrium constant
- m i :
-
Initial mass of sample, mg
- P :
-
Pressure, Pa
- \( P^{{\ominus}} \) :
-
Standard pressure, \( P^{{\ominus}} \cong 0.1\;{\text{MPa}} \)
- q :
-
Heating rate, K min−1
- r :
-
Relative rate of reaction/process, K
- R :
-
Universal gas constant, R = 8,314 J (K mol)−1
- T :
-
Temperature, K
- T r :
-
Temperature–slope in Eq. 15, K
- T eq :
-
Equilibrium temperature for αeq = 1, \( P^{{\ominus}} \approx 0.1\;{\text{MPa}} \), K
- ν:
-
Stoichiometric coefficient
- r 2 :
-
Determination coefficient for linear function, 0 ≤ r 2 ≤ 1
- sl:
-
Significance level
- 298:
-
Standard state
- α:
-
Equilibrium conversion degree
- eq:
-
Equilibrium state
- exp:
-
Experimental
- f:
-
Final state
- fp:
-
Flex point
- i:
-
Initial state
- m:
-
Point of the maximal rate of reaction/process
- r:
-
Reaction
References
Kowalewska E, Błażejowski J. Thermochemical properties of H2SnCl6 complexes. Part I. Thermal behaviour of primary n-alkylammonium hexachlorostannates. Thermochim Acta. 1986;101:271–89.
Janiak T, Błażejowski J. Thermal features, thermochemistry and kinetics of the thermal dissociation of hexachlorostannates of aromatic mono-amines. Thermochim Acta. 1989;156:27–43.
Janiak T, Błażejowski J. Thermal features, thermolysis and thermochemistry of hexachlorostannates of some mononitrogen aromatic bases. Thermochim Acta. 1990;157:137–54.
Dokurno P, Łubkowski J, Błażejowski J. Thermal properties, thermolysis and thermochemistry of alkanaminium iodides. Thermochim Acta. 1990;165:31–48.
Mianowski A. Thermal dissociation in dynamic conditions by modeling thermogravimetric curves using the logarithm of conversion degree. J Therm Anal Calorim. 2000;59:747–62.
Mianowski A, Bigda R. Thermodynamic interpretation of three-parametric equation; Part I. New from of equation. J Therm Anal Calorim. 2003;74:423–32.
Mianowski A. Consequences of Holba-Šesták equation. J Therm Anal Calorim. 2009;96:507–13.
Szarawara J, Piotrowski J. Theoretical foundations of chemical technology. Warszawa: WN; 2010. p. 151–156 (in Polish).
Šesták J. Heat, Thermal analysis and society. Czech Republic: Nucleus HK; 2004. p. 210.
Holba P, Šesták J. Kinetics with regard to the equilibrium processes studied by nonisothermal technique. Z für Phys Chemie Neue Folge. 1972;80:1–20.
Czarnecki J, Šesták J. Practical thermogravimetry. J Therm Anal Calorim. 2000;60:759–78.
Brown ME, Maciejewski M, Vyazovkin S, Nomen R, Sempere J, Burnham A, Opfermann J, Strey R, Anderson HL, Kemmler A, Keuleers R, Janssens J, Desseyn HO, Chao-Rui L, Tang TB, Roduit B, Málek J, Mitsuhashi T. Computational aspects of kinetic analysis: Part A: the ICTAC kinetics project-data, methods and results. Thermochim Acta. 2000;355:125–43.
Mianowski A. Analysis of the thermokinetics under dynamic conditions by relative rate of thermal decomposition. J Therm Anal Calorim. 2001;63:765–76.
Mianowski A, Bigda R. Thermodynamic interpretation of three-parametric equation: Part II: the relative rate of reaction. J Therm Anal Calorim. 2003;74:433–42.
Mianowski A, Robak Z, Bigda R, Łabojko G. Relative rate of reaction/process under dynamic conditions as the effect of TG/DTG curves transformation in thermal analysis. Przemysł Chemiczny. 2010;89:780–5. (in Polish).
Mianowski A, Baraniec I. Three-parametric equation in evaluation of thermal dissociation of reference compound. J Therm Anal Calorim. 2009;96:179–87.
Mianowski A, Bigda R. The Kissinger law and isokinetic effect: part II: experimental analysis. J Therm Anal Calorim. 2004;75:355–72.
Rak J, Skurski P, Gutowski M, Błażejowski J. Thermodynamics of the thermal decomposition of calcium oxalate monohydrate examined theoretically. J Therm Anal. 1995;43:239–46.
Kutaish N, Aggarwal P, Dollimore D. Thermal analysis of calcium oxalate samples obtained by various preparative routes. Thermochim Acta. 1997;297:131–7.
Gao Z, Amasaki I, Nakada M. A description of kinetics of thermal decomposition of calcium oxalate monohydrate by means of the accommodated Rn model. Thermochim Acta. 2002;385:95–103.
Liqing L, Donghua C. Application of iso-temperature method of multiple rate to kinetic analysis. J Therm Anal Calorim. 2004;78:283–93.
Frost RL, Weier ML. Thermal treatment of whewellite—thermal analysis and Raman spectroscopic study. Thermochim Acta. 2004;409:79–85.
Vlaev L, Nedelchev N, Gyurova K, Zagorcheva M. A comparative study of non-isothermal kinetics of decomposition of calcium oxalate monohydrate. J Anal Appl Pyrolysis. 2008;81:253–62.
Mianowski A, Radko T. The possibility of identification of activation energy by means of the temperature criterion. Thermochim Acta. 1994;247:389–405.
Barin I. Thermochemical data of pure substances, Vol. 1. Weinheim: VCH Verlagsgesellschaft; 1989. p. 649–50.
Latimer WM, Schutz PW, Hicks JFG Jr. The heat capacity and entropy of calcium oxalate from 19 to 300° absolute. The entropy and free energy of oxalate ion. J Am Chem Soc. 1933;55:971–5.
Maciejewski M. Computational aspects of kinetic analysis: part B: the ICTAC Kinetics Project—the decomposition kinetics of calcium carbonate revisited, or some tips on survival in the kinetic minefield. Thermochim Acta. 2000;355:145–54.
Burnham AK. Computational aspects of kinetic analysis: part D: the ICTAC kinetics project-multi-thermal-history model-fitting methods and their relation to isoconversional methods. Thermochim Acta. 2000;355:165–70.
Roduit B. Computational aspects of kinetic analysis. part E: the ICTAC kinetics Project—numerical techniques and kinetics of solid state processes. Thermochim Acta. 2000;355:171–80.
Ortega A. The incorrectness of the temperature criterion. Thermochim Acta. 1996;276:189–98.
Savitzky A, Golay MJE. Smoothing and differentiation of data by simplified least-squares procedures. Anal Chem. 1964;36:1627–39.
Mianowski A. The Kissinger law and isokinetic effect. J Therm Anal Calorim. 2003;74:953–73.
Błażejowski J. Thermal properties of amine hydrochlorides. Part I. Thermolysis of primary n-alkylammonium chlorides. Thermochim Acta. 1983;68:233–60.
Łubkowski J, Błażejowski J. Thermal properties and thermochemistry of alkanaminium bromides. Thermochim Acta. 1990;157:259–77.
Thanh HV, Gruzdiewa L, Rak J, Błażejowski J. Thermal behaviour and thermochemistry of hexachlorozirconates of mononitrogen aromatic bases. Thermochim Acta. 1993;230:269–92.
Janiak T, Rak J, Błażejowski J. Thermal features and thermochemistry of hexachlorohafnates of nitrogen aromatic bases. Theoretical studies on the geometry and thermochemistry of HfCl6 2−. J Therm Anal. 1995;43:231–7.
Mianowski A, Siudyga T. Thermal analysis of polyolefin and liquid paraffin mixtures. J Therm Anal Calorim. 2003;74:623–30.
Mianowski A, Błażewicz S, Robak Z. Analysis of the carbonization and formation of coal tar pitch mesophase under dynamic conditions. Carbon. 2003;41:2413–24.
Mianowski A, Bigda R, Zymla V. Study on kinetics of combustion of brick-shaped carbonaceous materials. J Therm Anal Calorim. 2006;84:563–74.
Mianowski A, Siudyga T. Influence of sample preparation on thermal decomposition of wasted polyolefins-oil mixtures. J Therm Anal Calorim. 2008;92:543–52.
Owczarek M, Mianowski A. The influence of oxygen upon reactivity of chars in the presence of steam. In: Chemical technology at the turn of century. Gliwice: SKKTChem, Silesian University of Technology; 2000. p. 535–538.
Acknowledgements
We feel grateful to Dr. Wojciech Balcerowiak from ICSO “Blachownia” (Kędzierzyn-Koźle, Poland) for providing the results of the investigations for a series of chemical compounds and thermal decomposition of calcium carbonate and calcium oxalate monohydrate.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Mianowski, A., Baraniec-Mazurek, I. & Bigda, R. Some remarks on equilibrium state in dynamic condition. J Therm Anal Calorim 107, 1155–1165 (2012). https://doi.org/10.1007/s10973-011-1909-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-011-1909-7