
Summary
Background This open-label, first-in-human, phase 1 study evaluated AMG 232, an oral selective MDM2 inhibitor in patients with TP53 wild-type (P53WT), advanced solid tumors or multiple myeloma (MM). Methods In the dose escalation (n = 39), patients with P53WT refractory solid tumors enrolled to receive once-daily AMG 232 (15, 30, 60, 120, 240, 480, and 960 mg) for seven days every 3 weeks (Q3W). In the dose expansion (n = 68), patients with MDM2-amplified (well-differentiated and de-differentiated liposarcomas [WDLPS and DDLPS], glioblastoma multiforme [GBM], or other solid tumors [OST]), MDM2-overexpressing ER+ breast cancer (BC), or MM received AMG 232 at the maximum tolerated dose (MTD). Safety, pharmacokinetics, pharmacodynamics, and efficacy were assessed. Results AMG 232 had acceptable safety up to up to 240 mg. Three patients had dose-limiting toxicities of thrombocytopenia (n = 2) and neutropenia (n = 1). Due to these and other delayed cytopenias, AMG 232 240 mg Q3W was determined as the highest tolerable dose assessed in the dose expansion. Adverse events were typically mild/moderate and included diarrhea, nausea, vomiting, fatigue, decreased appetite, and anemia. AMG 232 plasma concentrations increased dose proportionally. Increases in serum macrophage inhibitor cytokine-1 from baseline were generally dose dependent, indicating p53 pathway activation. Per local review, there were no responses. Stable disease (durability in months) was observed in patients with WDLPS (3.9), OST (3.3), DDLPS (2.0), GBM (1.8), and BC (1.4–2.0). Conclusions In patients with P53WT advanced solid tumors or MM, AMG 232 showed acceptable safety and dose-proportional pharmacokinetics, and stable disease was observed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The tumor suppressor p53 is a key regulator of cell cycle progression and apoptosis [1, 2]. Mouse double minute 2 homolog (MDM2) inhibits p53 activity by acting as an E3 ubiquitin ligase to promote its degradation, by binding and blocking the p53 transcriptional activation domain, and by exporting p53 from the nucleus to the cytoplasm [3, 4]. Among TP53 wild-type (P53WT) solid tumors, MDM2 amplification has been demonstrated in well-differentiated and de-differentiated liposarcomas (WDLPS and DDLPS, respectively) and in glioblastoma multiforme (GBM) [5, 6]. Overexpression of MDM2 protein has been shown in WDLPS and DDLPS, estrogen receptor positive (ER+) breast cancer, and multiple myeloma [7,8,9,10,11]. MDM2 amplification and MDM2 overexpression, which result in p53 inactivation and decreased apoptosis, have been associated with poor outcomes [12,13,14].
Although many tumors harbor non-targetable mutations in TP53, MDM2 has become an attractive therapeutic target in the treatment of TP53 wild-type (P53WT) cancers. Several MDM2 inhibitors are in clinical investigation as monotherapy or combined with other therapies for the treatment of P53WT hematologic malignancies and solid tumors [15,16,17,18]. In clinical studies of MDM2 inhibitors, increases in circulating macrophage inhibitor cytokine-1 (MIC-1) has been used as a pharmacodynamic marker of p53 activation [19,20,21].
AMG 232 is an investigational oral, selective MDM2 inhibitor that restores p53 tumor suppression by blocking the MDM2-p53 interaction with picomolar affinity [22]. In tumor xenograft models, treatment with AMG 232 resulted in tumor growth inhibition and caused regression of MDM2-amplified tumors through the induction of growth arrest and apoptosis [23]. The primary objectives of this open-label, first-in-human, phase 1 study were to assess the safety and tolerability, pharmacokinetics, maximum tolerated dose (MTD), pharmacodynamics, and efficacy of AMG 232 in patients with P53WT solid tumors or multiple myeloma.
Methods
Patients
Patients aged ≥18 years with pathologically-documented, P53WT (per next-generation sequencing) treatment-refractory solid tumors measurable per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 or Macdonald criteria for glioblastoma multiforme (GBM), or progressive multiple myeloma measurable per International Myeloma Working Group (IMWG) response criteria were eligible. Additional eligibility criteria were Eastern Cooperative Oncology Group (ECOG) performance status ≤2; life expectancy >3 months; adequate hematologic (ANC ≥1.5 × 109/L for solid tumors or ≥ 1.0 × 109/L for multiple myeloma; platelet count ≥100 × 109/L for solid tumors or ≥ 75 × 109/L for multiple myeloma; hemoglobin >9 g/dL), renal (estimated glomerular filtration rate ≥ 45 mL/min/1.73 m2), hepatic (AST and AST <2.5 × ULN; ALP <2.0 × ULN; total bilirubin <1.5 × ULN), and coagulation (prothrombin time or partial thromboplastin time < 1.5 × ULN) functions. In the dose expansion, five tumor types were defined: well-differentiated or dedifferentiated liposarcoma; relapsed GBM with MDM2 amplification; estrogen receptor positive (ER+) breast cancer refractory to hormonal treatments; relapsed multiple myeloma progressive after ≥1 prior treatment; other advanced solid tumors with MDM2 amplification. Key exclusion criteria included active or untreated brain metastases; unresolved toxicity from prior anticancer therapy, excluding alopecia; antitumor therapy or major surgery within 28 days of starting study treatment; investigational device or drug within 30 days or 5 half-lives of starting study treatment; liposarcomas with >3 prior approved therapies; multiple myeloma with del (17p) or IgM subtype, non-secretory or hyposecretory disease, lack of ≥25% reduction in M-protein for ≥6 weeks with prior therapy, corticosteroid therapy within 3 weeks of study, POEMS syndrome, or plasma cell leukemia or lymphoplasmacytic lymphoma. Institutional review board approval was obtained for all study procedures. All patients provided informed consent before enrollment.
Study design and treatment
This open-label phase 1 study was conducted at 16 centers (ClinicalTrials.gov, NCT01723020). TP53 mutation status was confirmed by central laboratory assessment. The study was planned with two parts: a 3-part dose escalation (Part 1) and a dose expansion (Part 2). In the dose escalation, multiple-patient cohorts were planned to enroll sequentially (Part 1A) or in parallel (Parts 1B and 1C; 3 + 3 design) and to receive AMG 232 once daily (QD) for 3 days (Part 1B) or 7 days (Part 1C) every 3 weeks (Q3W) at prespecified doses of 15, 30, 60, 120, 240, 480, and 960 mg. Intermediate doses were allowed when deemed appropriate. The dose expansion was planned for patients with MDM2-amplified tumors (group 1; liposarcomas, GBM, and other solid tumors), potentially MDM2-overexpressing tumors (group 2; ER+ metastatic breast cancer), or multiple myeloma (group 3).
Each patient was monitored for 21 days for the occurrence of dose-limiting toxicities (DLTs), defined as febrile neutropenia, neutropenic infection, grade 4 neutropenia lasting >7 days, grade ≥ 3 thrombocytopenia lasting >7 days (solid tumors only), grade 3 thrombocytopenia with grade ≥ 2 bleeding (solid tumors only), grade 3 or 4 thrombocytopenia with grade > 1 bleeding (multiple myeloma only), grade 4 thrombocytopenia (solid tumors only), or grade 4 thrombocytopenia lasting >14 days (multiple myeloma only), or as grade ≥ 3 nausea, vomiting, or diarrhea after support; grade 3 fatigue lasting >7 days; any other grade ≥ 3 adverse event (AE); grade ≥ 3 kidney injury (multiple myeloma only), or treatment-related AEs not returning to grade ≤ 1 (solid tumors only) per Common Terminology Criteria for Adverse Events (CTCAE), version 4.0. The MTD was defined as the maximum dose, at which the probability of a DLT was ≤25% in Part 1A and ≤ 33% in Parts 1B and 1C. Treatment continued until disease progression, intolerable toxicity, or withdrawal of consent.
Study assessments
Safety
AEs were recorded for all enrolled patients.
Pharmacokinetics
Plasma samples for the measurement of AMG 232 pharmacokinetics in Parts 1 and 2 were collected predose and at 1, 3, 5, and 7 h postdose on days 1 and 7 and 24 and 72 h postdose from day 7 of cycle 1; predose on days 1 and 7 of cycle 2; and at the end of study. Plasma AMG 232 levels were measured using a validated high performance liquid chromatography mass spectrometry method [24]. Pharmacokinetic and exposure parameters were estimated, including terminal half-life (tmax), maximum observed plasma concentration (Cmax), area under the concentration-versus-time curve at 24 h (AUC24h), volume of distribution (Vz/F), terminal elimination half-life (t1/2,z), and clearance (CL/F). Non-compartmental analysis was performed using WinNonlin Professional software, version 6.3. Parameters were summarized descriptively.
Circulating MIC-1
In Parts 1 and 2, serum samples for the assessment of circulating macrophage inhibitor cytokine-1 (MIC-1) were collected on the pharmacokinetic sample schedule. Serum MIC-1 concentrations were measured using a validated ELISA (human GDF-15 Quantikine®, R&D Systems Inc.).
Efficacy
Efficacy response was assessed using revised Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1 [25] Macdonald criteria for GBM [26] or International Myeloma Working Group (IMWG) response criteria for multiple myeloma [27].
Statistical analysis
Primary endpoints were the patient incidence of AEs, DLTs, and clinically significant changes in safety assessments; AMG 232 and pharmacokinetic parameters; and the MTD in Part 1. Secondary/exploratory endpoints included tumor response and change in serum MIC-1 level. Data were summarized descriptively. Qualified researchers may request data from Amgen clinical studies. Complete details are available at the following: http://www.amgen.com/datasharing.
Results
Patients
Overall, 107 patients enrolled (dose escalation, n = 39; dose expansion, n = 68). Patients in the dose escalation had a variety of refractory advanced solid tumors, and those in the dose expansion had WDLPS (n = 10), DDLPS (n = 10), GBM (n = 10), other solid tumors (n = 16), breast cancer (n = 12), or multiple myeloma (n = 10; Table 1). Most patients had received 3 or more lines of therapy (dose escalation, 69%; dose expansion, 72%).
AMG 232 was administered to all 39 patients in the dose escalation (15 mg, n = 3; 30 mg, n = 3; 60 mg, n = 4; 120 mg, n = 7; 240 mg, n = 8; 300 mg, n = 4; 360 mg, n = 4; 480 mg, n = 6) and to all 68 patients in the dose expansion (240 mg). The intermediate doses of 300 mg and 360 mg were assessed due to the occurrence of AEs. Reasons for discontinuing treatment across the entire study were disease progression (n = 74), AEs (n = 21), patient request (n = 11), and unknown (n = 1).
Safety and tolerability
DLTs and MTD
Three patients in the dose escalation had DLTs. The first patient (120-mg cohort) with esophageal cancer had a DLT consisting of grade 3 thrombocytopenia on day 15 that worsened to grade 4 on day 17, lasted seven days, and required a platelet transfusion. The second patient (360-mg cohort) with rectal cancer had a DLT consisting of grade 4 thrombocytopenia on day 28. The third patient (480-mg cohort) with head and neck cancer had grade 3 neutropenia on day 22 that delayed treatment in the next cycle and was therefore considered a DLT. Two additional patients in the 300-mg cohort had cytopenias (grade 4 neutropenia; grade 4 thrombocytopenia) outside of the 21-day DLT evaluation window that were considered in the dose escalation decisions. Based on the protocol-specified definition using DLTs incidence, the MTD for AMG 232 for 7 days Q3W was not reached. However, when the DLTs and delayed cytopenias were considered, the highest safe and tolerable dose of AMG 232 was 240 mg, which was the dose evaluated in the dose expansion.
Safety and tolerability in the dose escalation
In the dose escalation, 37 (95%) patients had treatment-emergent AEs (Table 2), most of which were grade 1 or 2. The most common (occurring in ≥20% of patients) treatment-emergent AEs were diarrhea (67%), nausea (59%), vomiting, (51%), fatigue (41%), decreased appetite (39%), thrombocytopenia (36%), anemia (26%), neutropenia (26%), and abdominal pain (21%). Thirty-four (87%) patients in the dose escalation had AEs that were considered by the investigators to be treatment related. The most common (occurring in ≥20% of patients) treatment-related AEs were diarrhea (64%), nausea (51%), vomiting (46%), fatigue (41%), thrombocytopenia (36%), decreased appetite (26%), and neutropenia (21%). Most treatment-related AEs were grade 1 or 2.
Serious AEs occurred in 14 (36%) patients during the dose escalation, including six (15%) whose serious AEs were considered treatment-related and predominantly included gastrointestinal toxicity (Table 2). Overall, eight patients in the dose escalation had AEs resulting in treatment discontinuation: thrombocytopenia (n = 5), neutropenia (n = 2), and febrile neutropenia (n = 1). Four patients in the dose escalation had fatal AEs of disease progression while on study.
Safety and tolerability in the dose expansion
In the 240-mg dose expansion, 67 (99%) patients had treatment-emergent AEs (Table 3), most of which were grade 1 or 2. The most common (occurring in ≥20% of patients) treatment-emergent AEs were diarrhea (72%), nausea (72%), vomiting, (59%), fatigue (53%), decreased appetite (41%), anemia (25%), and thrombocytopenia (24%). Sixty-five (96%) patients in the dose expansion had AEs that were considered by the investigators to be related to treatment with AMG 232. The most common (occurring in ≥20% of patients) treatment-related AEs were diarrhea (68%), nausea (68%), vomiting (47%), fatigue (47%), decreased appetite (41%), and thrombocytopenia (21%). Most treatment-related AEs were grade 1 or 2.
During the dose expansion, 29 (43%) patients had serious AEs, including seven (10%) whose serious AEs were considered treatment-related (Table 3). Overall, 13 (19%) patients in the dose expansion had AEs resulting in treatment discontinuation: vomiting (n = 4), fatigue (n = 2), and nausea, dyspnea, pulmonary embolism, asthenia, malaise, ECOG performance status 4, thrombocytopenia, neutropenia, intestinal adhesion lysis, and oculogyric crisis (n = 1 each). Three patients in the dose expansion had fatal AEs while on study, including two with disease progression and one with physical deterioration.
Pharmacokinetics of AMG 232
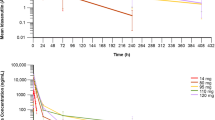
Plasma samples for the evaluation of AMG 232 pharmacokinetics were available for 106 patients. AMG 232 pharmacokinetic profiles for the dose escalation and dose expansion are shown in Fig. 1. Plasma concentrations of AMG 232 increased dose proportionally. The mean AUC accumulation ratio between days 1 and day 7 across all dosing groups in the dose escalation and in the dose expansion was less than 2-fold with the once-daily dosing regimen (Table 4, Table 5). The mean estimated apparent volume of distribution was 615 L across all dosing cohorts. With oral administration, the estimated mean apparent clearance of AMG 232 was 30.2 L/h across groups and varied among individuals.

Mean (± SD) pharmacokinetic profile of AMG 232 following oral administration every 3 weeks in the dose escalation (a) and in the dose expansion (b). DDLPS, dedifferentiated liposarcoma; GBM, glioblastoma multiforme; OST, other solid tumor; WDLPS, well differentiated liposarcoma
AMG 232 pharmacodynamic effects
Thirty-nine patients in the dose escalation had available pre-treatment and post-treatment blood samples for the assessment of serum MIC-1; serum MIC- was not assessed in the dose expansion. From baseline to day 15, increases in serum MIC-1 (post-treatment to pre-treatment ratios) were generally dose-dependent (Fig. 2). Mean serum MIC-1 ratios increased up to day 7 and decreased until cycle 2, suggesting that MIC-1 changes were dependent on AMG 232 exposure.

Mean (± SE) ratio of post-treatment versus pre-treatment serum MIC-1 in the dose escalation
Efficacy
Imaging for the local evaluation of tumor response was available for 38 of 39 patients in the dose escalation and 60 of 68 patients in the dose expansion. One patient in the dose escalation had no postbaseline imaging due to an AE. Eight patients in the dose expansion had no postbaseline imaging due to clinical disease progression (n = 3), AEs (n = 3), and patient request (n = 2). By local evaluation, no objective responses were observed. Per central evaluation, three patients (4%) with WDLPS, squamous cell carcinoma, and breast cancer had unconfirmed partial responses with durations of 2.4, 0.1, and 2.0 months, respectively. In the dose escalation, 31 (80%) patients had stable disease and five (13%) had progressive disease (Fig. 3a). Based on evaluation of non-target lesions, 2 (5%) patients in the dose escalation had non − complete response/non − progressive disease. Overall, 45 (66%) patients in the dose expansion had stable disease and 15 (22%) had progressive disease (Fig. 3b). Stable disease as a best result in the dose expansion was observed among 10 of 10 patients with WDLPS, 7 of 10 with DDLPS, six of 10 with GBM, 10 of 16 with other solid tumors, 10 of 12 with breast cancer, and 5 of 10 with multiple myeloma.

Best change from baseline in the sum of longest diameters of target lesions in the dose escalation (a) and in the dose expansion (b). ACC, adenoid cystic carcinoma; CAC, cholangiocarcinoma; Chondro, chondrosarcoma; DDLPS, dedifferentiated liposarcoma; Esoph, esophageal; GBM, glioblastoma multiforme; GPL, glandular parotis left; Gran, granular cell tumor; Leio, leiomyosarcoma; Mel, melanoma; Neuro, neuroendocrine; NSCLC, non–small-cell lung cancer; OST, other solid tumor; Panc, pancreatic; Pleural, pleural mesothelioma; Saliv, salivary gland; SCCHN, squamous cell carcinoma of the head and neck; SFT, solitary fibrous tumor; STS, soft tissue sarcoma; WDLPS, well differentiated liposarcoma. *Patient had partial response per central review
Duration of stable disease in the dose escalation and across histologies in the dose expansion is summarized in Fig. 4. The median duration of stable disease in the dose escalation was 3.1 months (range, 0.7–22.4 mo). In the dose expansion, the overall median duration of stable disease was 2.0 months (range, 0.5–12.9) overall, 3.9 months (range 1.9–6.1) among patients with WDLPS, 2.0 months (0.9–6.9) among patients with DDLPS, 1.8 months (0.5–11.9) among patients with GBM, 3.3 months (0.9–12.9) among patients with other solid tumors, 2.0 months (0.9–4.0) among patients with ER + PR+ breast cancer, and 1.4 months (0.9–3.8) among those with ER + PR– breast cancer.

Duration of stable disease in the dose escalation (a) and in the dose expansion (b). DDLPS, dedifferentiated liposarcoma; GBM, glioblastoma multiforme; OST, other solid tumor; WDLPS, well differentiated liposarcoma
Discussion
In this first-in-human study, AMG 232 was generally well tolerated up to 240 mg. AMG 232 doses up to 480 mg QD for seven days in a 21-day cycle were assessed; however, the MTD was not reached. Three patients had DLTs, two with thrombocytopenia and one with neutropenia. Based on these DLTs and other cytopenias outside of the DLT evaluation window, AMG 232 240 mg was selected as the highest safe and tolerable dose for evaluation in the dose expansion. In both phases of the study, AMG 232–related AEs were generally mild to moderate, with the most frequently occurring AEs being diarrhea, nausea, vomiting, fatigue, decreased appetite, and anemia. Among the AEs resulting in treatment discontinuation, thrombocytopenia, neutropenia, and gastrointestinal toxicity were the most frequent. These results are consistent with other clinical studies of MDM2 inhibitors, in which myelosuppression and gastrointestinal toxicity have been reported [18, 19, 28,29,30,31,32,33]. In this study, five patients died due to disease progression while on the study.
Following QD treatment for seven days in a 21-day cycle, AMG 232 plasma concentrations increased generally dose proportionally, and across all patients, the mean AUC accumulation ratio between days 1 and 7 was less than 2-fold. Overall, AMG 232 exhibited an acceptable pharmacokinetic profile in this population. Furthermore, dose-dependent increases in serum MIC-1 levels from baseline to day 15 of treatment indicated p53 pathway activation, consistent with previous studies of MDM2 inhibitors [19,20,21].
One of the objectives of the dose expansion was to assess the antitumor activity of AMG 232 among patients not only with P53WT tumors but also among those with MDM2 amplification and MDM2 overexpression, which is common in liposarcomas, GBM, breast cancer, and multiple myeloma [5,6,7,8,9,10,11] Per local evaluation, no objective response were observed. Per central evaluation, 3 patients had partial responses. Overall, stable disease was observed in 45 of 68 (66%) patients in the dose expansion, including all 10 patients with WDLPS, 7 of 10 with DDLPS, 6 of 10 with GBM, 10 of 16 with other solid tumors, 7 of 12 with breast cancer, and 5 of 10 with multiple myeloma. Stable disease was observed among patients with WDLPS (median, 3.9 months; range, 1.9–6.1), which is naturally an indolent disease, followed by other solid tumors (median, 3.3 months; range, 1.9–12.9), DDLPS (median, 2.0 months; range, 1.8–.6.9), GBM (median, 1.8 months; range, 1.4–11.9), breast cancer (median, 2.0 months; range, 1.7–4.0), and multiple myeloma (median, 1.0 months; range, 1.2–2.0). Our results are consistent with those of previous reports demonstrating limited clinical activity with HDM2/MDM2 inhibitors [18, 20, 31, 34,35,36]. Because the development of TP53 mutations may contribute to the development of resistance to treatment with HDM2/MDM2 inhibitors, investigation of HDM2/MDM2 inhibitors in combination with other agents may be appropriate [34].
In conclusion, AMG 232 showed acceptable safety, dose-proportional pharmacokinetics, and on-target activity as monotherapy administered at oral doses up to 240 mg QD on days 1–7 per 3-week cycle in patients with P53WT solid tumors. No responses were observed per local evaluation but 3 patients had unconfirmed partial responses per central evaluation. Stable disease was observed among patients regardless of MDM2 amplification or overexpression. Future evaluation of AMG 232 in hematologic malignancies and solid tumors should be considered.
References
Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88(3):323–331
Levine AJ, Oren M (2009) The first 30 years of p53: growing ever more complex. Nat Rev Cancer 9(10):749–758
Moll UM, Petrenko O (2003) The MDM2-p53 interaction. Mol Cancer Res 1(14):1001–1008
Pei D, Zhang Y, Zheng J (2012) Regulation of p53: a collaboration between Mdm2 and Mdmx. Oncotarget 3(3):228–235
Coindre JM, Pedeutour F, Aurias A (2010) Well-differentiated and dedifferentiated liposarcomas. Virchows Arch 456(2):167–179
Rao SK, Edwards J, Joshi AD, Siu IM, Riggins GJ (2010) A survey of glioblastoma genomic amplifications and deletions. J Neuro-Oncol 96(2):169–179
Binh MB, Sastre-Garau X, Guillou L, de Pinieux G, Terrier P, Lagace R, Aurias A, Hostein I, Coindre JM (2005) MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: a comparative analysis of 559 soft tissue neoplasms with genetic data. Am J Surg Pathol 29(10):1340–1347
McCann AH, Kirley A, Carney DN, Corbally N, Magee HM, Keating G, Dervan PA (1995) Amplification of the MDM2 gene in human breast cancer and its association with MDM2 and p53 protein status. Br J Cancer 71(5):981–985
Konduri SD, Medisetty R, Liu W, Kaipparettu BA, Srivastava P, Brauch H, Fritz P, Swetzig WM, Gardner AE, Khan SA, Das GM (2010) Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc Natl Acad Sci U S A 107(34):15081–15086
Elnenaei MO, Gruszka-Westwood AM, A'Hernt R, Matutes E, Sirohi B, Powles R, Catovsky D (2003) Gene abnormalities in multiple myeloma; the relevance of TP53, MDM2, and CDKN2A. Haematologica 88(5):529–537
Quesnel B, Preudhomme C, Oscier D, Lepelley P, Collyn-d'Hooghe M, Facon T, Zandecki M, Fenaux P (1994) Over-expression of the MDM2 gene is found in some cases of haematological malignancies. Br J Haematol 88(2):415–418
Choschzick M, Heilenkotter U, Lebeau A, Jaenicke F, Terracciano L, Bokemeyer C, Sauter G, Simon R (2010) MDM2 amplification is an independent prognostic feature of node-negative, estrogen receptor-positive early-stage breast cancer. Cancer Biomark 8(2):53–60
Grochola LF, Zeron-Medina J, Meriaux S, Bond GL (2010) Single-nucleotide polymorphisms in the p53 signaling pathway. Cold Spring Harb Perspect Biol 2(5):a001032
Park HS, Park JM, Park S, Cho J, Kim SI, Park BW (2014) Subcellular localization of Mdm2 expression and prognosis of breast cancer. Int J Clin Oncol 19(5):842–851
Tisato V, Voltan R, Gonelli A, Secchiero P, Zauli G (2017) MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol 10(1):133
Cassier PA, Castets M, Belhabri A, Vey N (2017) Targeting apoptosis in acute myeloid leukaemia. Br J Cancer 117(8):1089–1098
McCubrey JA, Lertpiriyapong K, Fitzgerald TL, Martelli AM, Cocco L, Rakus D, Gizak A, Libra M, Cervello M, Montalto G, Yang LV, Abrams SL, Steelman LS (2017) Roles of TP53 in determining therapeutic sensitivity, growth, cellular senescence, invasion and metastasis. Adv Biol Regul 63:32–48
Wagner AJ, Banerji U, Mahipal A, Somaiah N, Hirsch H, Fancourt C, Johnson-Levonas AO, Lam R, Meister AK, Russo G, Knox CD, Rose S, Hong DS (2017) Phase I trial of the human double minute 2 inhibitor MK-8242 in patients with advanced solid tumors. J Clin Oncol 35(12):1304–1311
Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, Heil F, Rueger R, Graves B, Ding M, Geho D, Middleton SA, Vassilev LT, Nichols GL, Bui BN (2012) Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol 13(11):1133–1140
de Jonge M, de Weger VA, Dickson MA, Langenberg M, Le Cesne A, Wagner AJ, Hsu K, Zheng W, Mace S, Tuffal G, Thomas K, Schellens JH: A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in Patients with solid tumours. Eur J Cancer 76(144–151, 2017
Nemunaitis J, Young A, Ejadi S, Miller W, Chen LC, Nichols G, Blotner S, Vazvaei F, Zhi J, Razak A (2018) Effects of posaconazole (a strong CYP3A4 inhibitor), two new tablet formulations, and food on the pharmacokinetics of idasanutlin, an MDM2 antagonist, in patients with advanced solid tumors. Cancer Chemother Pharmacol 81(3):529–537
Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, Canon J, Chen A, Chen X, Chow D, Deignan J, Duquette J, Eksterowicz J, Fisher B, Fox BM, Fu J, Gonzalez AZ, Gonzalez-Lopez De Turiso F, Houze JB, Huang X, Jiang M, Jin L, Kayser F, Liu JJ, Lo MC, Long AM, Lucas B, LR MG, Mcintosh J, Mihalic J, Oliner JD, Osgood T, Peterson ML, Roveto P, Saiki AY, Shaffer P, Toteva M, Wang Y, Wang YC, Wortman S, Yakowec P, Yan X, Ye Q, Yu D, Yu M, Zhao X, Zhou J, Zhu J, Olson SH, Medina JC (2014) Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J Med Chem 57(4):1454–1472
Canon J, Osgood T, Olson SH, Saiki AY, Robertson R, Yu D, Eksterowicz J, Ye Q, Jin L, Chen A, Zhou J, Cordover D, Kaufman S, Kendall R, Oliner JD, Coxon A, Radinsky R (2015) The MDM2 inhibitor AMG 232 demonstrates robust antitumor efficacy and potentiates the activity of p53-inducing cytotoxic agents. Mol Cancer Ther 14(3):649–658
Erba HP, Becker PS, Shami PJ, Grunwald MR, Flesher DL, Zhu M, Rasmussen E, Henary HA, Anderson AA, Wang ES (2019) Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Advances 3(13):1939–1949
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247
Macdonald DR, Cascino TL, Schold SC Jr, Cairncross JG (1990) Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol 8(7):1277–1280
Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, Kumar S, Hillengass J, Kastritis E, Richardson P, Landgren O, Paiva B, Dispenzieri A, Weiss B, LeLeu X, Zweegman S, Lonial S, Rosinol, L, Zamagni E, Jagannath S, Sezer O, Kristinsson SY, Caers J, Usmani SZ, Lahuerta JJ, Johnsen HE, Beksac M, Cavo M, Goldschmidt H, Terpos E, Kyle RA, Anderson KC, Durie BG, Miguel JF: International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 15(12):e538–e548, 2014
Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, Bowen D, Martinelli G, Drummond MW, Vyas P, Kirschbaum M, Iyer SP, Ruvolo V, Gonzalez GM, Huang X, Chen G, Graves B, Blotner S, Bridge P, Jukofsky L, Middleton S, Reckner M, Rueger R, Zhi J, Nichols G, Kojima K (2016) Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Cancer Res 22(4):868–876
Siu LL, Italiano A, Miller WH, Blay JY, Gietema JA, Bang YJ, Mileshkin LR, Hirte HW, Reckner M, Higgins B, Jukofsky L, Blotner S, Zhi J, Middleton S, Nichols GL, Chen LC (2014) Phase 1 dose escalation, food effect, and biomarker study of RG7388, a more potent second-generation MDM2 antagonist. In: Patients (pts) with solid tumors. J Clin Oncol 32(15S):abstr 2535
Kurzrock R, Blay JY, Nguyen BB, Wagner A, Maki RG, Schwartz GK, Patnaik A, Gore L, Wu L, Vassilev LT, Ding M, Geho D, Zhi J, Middleton S, Nichols GL (2012) A phase I study of MDM2 antagonist RG7112 in patients (pts) with relapsed/refractory solid tumors. J Clin Oncol 30(15S):e13600
Ravandi F, Gojo I, Patnaik MM, Minden MD, Kantarjian H, Johnson-Levonas AO, Fancourt C, Lam R, Jones MB, Knox CD, Rose S, Patel PS, Tibes R (2016) A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML). Leuk Res:92–48, 100
Yee K, Martinelli G, Vey N, Dickinson MJ, Seiter K, Assouline S, Drummond MW, Yoon SS, Kasner M, Lee JH, Kelly KR, Blotner S, Higgins B, Middleton S, Nichols G, Chen G, Zhong H, Pierceall WE, Zhi J, Chen LC (2014) Phase 1/1b study of RG7388, a potent MDM2 antagonist, in acute myelogenous leukemia (AML) patients (pts). Blood 124(21):abstr 116
Yee K, Uy G, Assouline S, Britten CD, Zhi J, Blotner S, Pierceall WE, Higgins B, Chen LC (2018) A phase I study of the MDM2 antagonist RO6839921, a pegylated intravenous prodrug of idasanutlin, in patients with AML. Mol Cancer Ther 17(1):A082
Jung J, Lee JS, Dickson MA, Schwartz GK, Le Cesne A, Varga A, Bahleda R, Wagner AJ, Choy E, de Jonge MJ, Light M, Rowley S, Mace S, Watters J (2016) TP53 mutations emerge with HDM2 inhibitor SAR405838 treatment in de-differentiated liposarcoma. Nat Commun 7:12609
Lulla RR, Goldman S, Yamada T, Beattie CW, Bressler L, Pacini M, Pollack IF, Fisher PG, Packer RJ, Dunkel IJ, Dhall G, Wu S, Onar A, Boyett JM, Fouladi M (2016) Phase I trial of p28 (NSC745104), a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in pediatric patients with recurrent or progressive central nervous system tumors: a pediatric brain tumor consortium study. Neuro-Oncology 18(9):1319–1325
Warso MA, Richards JM, Mehta D, Christov K, Schaeffer C, Rae Bressler L, Yamada T, Majumdar D, Kennedy SA, Beattie CW, Das Gupta TK (2013) A first-in-class, first-in-human, phase I trial of p28, a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in patients with advanced solid tumours. Br J Cancer 108(5):1061–1070
Funding
This study was funded by Amgen Inc. Ben Scott, PhD (Scott Medical Communications, LLC), provided medical writing assistance funded by Amgen Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Mrinal M. Gounder declares personal fees and advisory board for Bayer, Epizyme, Karyopharm, Amgen Inc., Daichi, Springworks, Tracon, and GlaxoSmithKline. Jean Yves Blay declares research support and honoraria from Amgen Inc., Roche, and Novartis. Philippe A. Cassier declares research support from Novartis, Astra-Zeneca, Plexxikon, Merck Sharp & Dohme, and Debio; institutional research support from Abbvie, Bayer, Bristol-Myers Squibb, GlaxoSmithKline, Merck Sharp & Dohme, Novartis, Roche, Lilly, and Taiho; honoraria from Amgen Inc., Novartis, and AstraZeneca; and travel from Bristol-Myers Squibb, Roche, and Amgen Inc. Jean-Charles Soria declares employment by MedImmune. Sant Chawla declares honoraria, consultant/advisor, research funding, and speakers’ bureau for Amgen Inc., Roche, Threshold Pharmaceuticals, GlaxoSmithKline, CytRx Corporation, Ignyta, Immune Design, TRACON Pharma, SARC, Karyopharm Therapeutics, and Janssen. Andrew J. Wagner declares research support from Amgen Inc.; personal fees from Eli Lilly, Loxo, and Five Prime Therapeutics; and grants from Merck, Sanofi, Daichi-Sankyo, Plexxikon, Karyopharm, Five Prime Therapeutics, and AADI. David Siegel declares a grant from Celgene; speakers bureau and advisory boards for Amgen Inc., Celgene, Bristol Myers-Squibb, Janssen, Takeda, and Karyopharm; and advisory board for Merck. Haby A. Henary and Erik Rasmussen declare employment and stock ownership in Amgen Inc. Larry Gluck, Richard Frank, Ferry Eskens, Vincent de Weger, and Filip De Vos have no conflicts to disclose.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Financial support
Amgen Inc.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gluck, W.L., Gounder, M.M., Frank, R. et al. Phase 1 study of the MDM2 inhibitor AMG 232 in patients with advanced P53 wild-type solid tumors or multiple myeloma. Invest New Drugs 38, 831–843 (2020). https://doi.org/10.1007/s10637-019-00840-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-019-00840-1