
Abstract
The two murine double minute (MDM) family members MDM2 and MDMX are at the center of an intense clinical assessment as molecular target for the management of cancer. Indeed, the two proteins act as regulators of P53, a well-known key controller of the cell cycle regulation and cell proliferation that, when altered, plays a direct role on cancer development and progression. Several evidence demonstrated that functional aberrations of P53 in tumors are in most cases the consequence of alterations on the MDM2 and MDMX regulatory proteins, in particular in patients with hematological malignancies where TP53 shows a relatively low frequency of mutation while MDM2 and MDMX are frequently found amplified/overexpressed. The pharmacological targeting of these two P53-regulators in order to restore or increase P53 expression and activity represents therefore a strategy for cancer therapy. From the discovery of the Nutlins in 2004, several compounds have been developed and reported with the ability of targeting the P53-MDM2/X axis by inhibiting MDM2 and/or MDMX. From natural compounds up to small molecules and stapled peptides, these MDM2/X pharmacological inhibitors have been extensively studied, revealing different biological features and different rate of efficacy when tested in in vitro and in vivo experimental tumor models. The data/evidence coming from the preclinical experimentation have allowed the identification of the most promising molecules and the setting of clinical studies for their evaluation as monotherapy or in therapeutic combination with conventional chemotherapy or with innovative therapeutic protocols in different tumor settings. Preliminary results have been recently published reporting data about safety, tolerability, potential side effects, and efficacy of such therapeutic approaches. In this light, the aim of this review is to give an updated overview about the state of the art of the clinical evaluation of MDM2/X inhibitor compounds with a special attention to hematological malignancies and to the potential for the management of pediatric cancers.
Similar content being viewed by others
Background
The MDM2/X genes and proteins
The murine double minute 2 (MDM2) gene was originally discovered through screening of a cDNA library of RNA isolated from a spontaneously transformed derivative of mouse 3T3 cells [1]. The characterization of the newly discovered MDM2 gene showed its tumorigenic potential when experimentally overexpressed, a preliminary evidence followed by the full identification and the structural description of a key evolutionarily conserved gene with a crucial role in regulating cellular growth [2, 3]. Human MDM2 gene (HDM2) maps on chromosome 12q14.3–q15 and both expression and functions are tightly regulated at transcriptional level (e.g., presence of different promoters, gene polymorphisms, splicing variants), at translational level and at post-translational level by several cell signals that regulate protein accumulation and activity [4]. The MDM2 gene codifies for an E3 ubiquitin ligase that acts as a powerful inhibitor of the tumor suppressor P53, the master regulator of the cell cycle progression, in a feedback loop that is attracting great interests and attention as a potential target for tumor therapeutic purposes [5]. The tight link between MDM2 and P53 has been recently addressed from Tan and colleagues in an interesting analysis of the evolutionary history of the MDM genes in relation to P53 [6]. In their study, the authors have highlighted a preserved cellular role throughout history of this genes’ family because, as expected, the maintenance of DNA integrity and the ability to respond to DNA damage are inalienable functions from an evolutionary perspective. Of note, the functional relationship between MDM and P53 has been found also in organisms that existed for one billion years, suggesting that these two proteins have evolved together to maintain preserved and controlled synergic functions [6]. However, although the interest on MDM2 rises on its key biological target, MDM2 can also interact with other molecules including upstream regulators (effectors) and downstream proteins (affectors) leading to several P53-independent effects [7].
The tumor suppressor P53 has been defined as “guardian of the genome” in light of its nature, a multifunctional transcription factor that can be activated by several stress signals and which is able to regulate a wide panel of target genes leading to different biological functions, overall aiming at monitoring and controlling cell cycle progression and cellular proliferation state [8,9,10]. The biological impact of any alteration of TP53 gene and/or P53 protein activity can be summarized by the evidence that TP53 knockout mice appear normal but are predisposed to spontaneous development of tumors at young age [11, 12], and that the presence of mutant TP53 allele makes the mice more prone to the development of tumors [13]. In humans, the protein is mutated in most of solid tumors [14] and it has been widely demonstrated that the newly acquired oncogenic functions allow uncontrolled cell survival/proliferation and acquisition of invasive/metastatic potentials [15]. In the context of hematological malignancies, the frequency of mutations of TP53 has a minor impact compared to solid tumors since most types of leukemia express wild-type P53, and the reported functional aberrations of the protein are in most cases the consequence of alterations on P53 regulatory proteins, in particular MDM2 and its paralog MDMX. The MDMX (namely MDM4) gene is located on human chromosome 1q32 and encodes for an MDM2 effector that plays a key role in both modulating P53 and regulating MDM2 activity. This protein was discovered more than 20 years ago as an additional P53-modulating factor characterized by a structural similarity to MDM2 [16], showing a stimulatory effect on the E3 activity of MDM2 without a detectable direct E3 ubiquitin ligase activity of MDMX itself [17]. MDM2 and MDMX are therefore key (non-overlapping) regulators of P53 as demonstrated by some fundamental in vivo preclinical experiments performed in MDM2 and MDMX null mice, showing that both MDM2 −/− and MDMX −/− genetic background are characterized by embryonic lethality and that the lethality can be completely rescued by the loss of P53 [18,19,20]. In particular, several studies have explored the developmental effects of the loss of MDM2 or MDMX, highlighting that they are highly tissue-specific and, although associated with uncontrolled activation of P53 target genes, the level of alteration of P53 do not correlate to the severity of the observed tissue-specific morphological alterations [21]. Both MDM2 and MDMX are frequently found altered in human cancers (Fig. 1). The MDM2 gene is overexpressed, mainly by gene amplification, in several solid cancers [22] including in situ and invasive breast adenocarcinomas [23], sarcomas (either common bone and soft tissue forms) [24,25,26], esophageal cancer [27], and endometrial stromal tumor [28]. It is of interest that, while amplification of the MDM2 gene is a major mechanism of MDM2 overexpression, MDM2 gene amplification and mutation of P53 are commonly mutually exclusive events [26]. Moreover, genetic variants of the MDM2 gene such as single nucleotide polymorphisms affect the basal levels of MDM2 and altering P53 mono- and poly-ubiquitination [29,30,31]. In the same fashion, MDMX can be also found amplified or overexpressed in a number of types of tumors including glioblastomas [32], retinoblastomas [33], as well as sarcomas, and breast cancers [22]. Amplification of MDM2 and amplification/overexpression of MDMX together with alterations of P53 in the context of hematological malignancies are summarized in Table 1.

Expression levels of MDM2 and MDMX in human cancers. Graphical representation of the expression levels of MDM2 (upper panel) and MDMX (lower panel) in human cancers obtained from studies collected in cBioPortal For Cancer Genomics (www.cbioportal.org). The picture shows RNA values of patients expressing the wild-type or mutated genes. TCGA The Cancer Genome Atlas Network
The MDM2/X axis with P53
In normal conditions, to maintain an appropriate biological outcome, cellular homeostasis is set to preserve low P53 levels and avoid excessive P53 accumulation with a concomitant and unnecessary activation of the P53-mediated pathways. Accumulation and activation of P53 are strictly regulated processes under the control of several regulatory signals and molecular mediators [34, 35]. Intracellular P53 is maintained low mainly thanks to the short half-life of the protein that is subjected to constant proteasome-dependent degradation. This is a reversible process allowing, when necessary, the rapid interruption of the protein degradation with an increase of protein levels to react and respond to specific signals of cellular stress and damage. Ubiquitination-mediated proteasome degradation is the critical process involved in regulating cellular accumulation and activity of P53 [36]. Nonetheless, ubiquitin-independent mechanisms are not excluded [37].
Ubiquitin-mediated protein degradation is a three-step process involving at least the E1 (Ub-activating) enzyme, the E2 (Ub-conjugating) enzyme, and E3 (Ub protein) ligase. A number of E-ligases able to mediate P53 ubiquitination have been identified so far, and the list includes, within the others, molecules such as COP1, CHIP, ARF-BP1, MKRN1, and several members of the TRIM protein family [36, 38,39,40,41,42,43,44]. Nonetheless, the best-validated ubiquitin ligase for P53 is MDM2, a protein that exerts its negative regulatory activity at different levels. The binding of the MDM2 amino-terminal domain to P53 is sufficient to inhibit the transcriptional activity of P53 affecting both the P53-mediated cell cycle arrest and the apoptosis functions [45]. Moreover, the presence of a nuclear export signal into MDM2 allows, after the binding of P53, the export from the nucleus to the cytoplasm of the protein complex [46]. This process has a dual effect: the prevention of P53 binding to the DNA precluding its activity as transcription factor and the degradation of P53 by cytoplasmic proteasomes [47,48,49]. The proteasome-mediated P53 degradation occurs via engagement of the really interesting new gene (RING) domain of MDM2 (i.e. really interesting new gene domain, the one that retains the ligase activity) to the N-terminal transactivation domain of P53, with the transfer of ubiquitin primarily into six lysine residues of the C-regulatory region of P53, the main sites of ubiquitin ligation [50, 51]. However, although MDM2 ubiquitin ligase activity is mandatory for P53 processing, MDM2 can catalyze both mono-ubiquitination (conjugation with an ubiquitin monomer at one or multiple sites) and poly-ubiquitination (conjugation with a polymeric ubiquitin chain) of P53 in a dosage-dependent manner [52]. In particular, in the presence of low levels of MDM2, it is likely that P53 undergoes to MDM2-mediated mono-ubiquitination and cytoplasmic translocation, while in the presence of stressed conditions MDM2 activities are high and there is a preferential poly-ubiquitination of P53 leading to degradation in the nucleus [52].
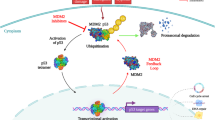
As transcriptional factor, P53 binds the promoter region of MDM2 and regulates the protein expression in an autoregulatory feedback loop that is critical in maintaining the appropriate balance between the levels of both MDM2 and P53 proteins (Fig. 2) [53]. Briefly, in normal conditions, P53 acts as transcription factor of the MDM2 gene and induces expression of MDM2 protein that binds P53 and induces in turn its degradation. The overall result is a tight and constant balance of the two proteins’ levels [53]. The autoregulatory loop between P53 and MDM2 crosses with MDMX that is able to interact with both P53 and MDM2. In particular, MDMX binds the N-terminal transactivation domain of P53 and represses P53 transcriptional activity without a direct E3 ligase activity [16], and differently from MDM2, the absence of P53-responsive elements in the MDMX structure makes MDMX not susceptible of P53-mediated regulation. On the other side, the presence of a RING domain allows MDMX to cross-reacts with MDM2 creating RING-mediated heterodimers [54, 55]. In the form of heterodimer with MDMX, MDM2 is more stable and exhibits a higher efficacy in downregulating P53 [56]. The MDMX-mediated regulation of MDM2 and the implications for P53 regulation have been recently addressed [57]. Overall, it is widely accepted that MDMX and MDM2 are two non-overlapping inhibitors of P53 [58], but it has been demonstrated that they can act downregulating P53 through the generation of heterodimers in normal conditions but also through a positive regulation of P53 after DNA damage [59]. In particular, the switch between negative and positive regulators occurs following the phosphorylation on serine residues that changes the conformation of the proteins re-directing the E3 ubiquitin ligase activity of MDM2 away from P53 and toward MDMX and MDM2 itself [59].

Schematic representation of the autoregulatory loop between P53, MDM2, and MDMX and pharmacological inhibitors. The picture shows the main molecular links between P53, MDM2, and MDMX together with the MDM2 and MDMX inhibitors under clinical evaluation and the key compounds used in clinical studies for combination therapy
Overall, the deep understanding of the MDM2/MDMX-P53 axis achieved in the last years has provided the biological rationale for the design of innovative chemical and synthesis procedures to develop molecules able to inhibit this interaction and restore the antitumor activity of P53. Crystal structure studies of P53 in complex with the MDM proteins have highlighted that three amino acids, Phe19, Trp23, and Leu26, play a key role in the MDM2-P53 interaction, differently from the MDMX-P53 interaction that must be targeted independently from MDM2, opening the doors to the identification of several classes of antitumor agents with different clinical potential [60, 61]. From a clinical perspective, the predominant role of MDM2 and MDMX in regulating P53 through the P53-MDM2/X axis has led to several reports in this field in the last few years; however, suggestions about new compounds and therapeutic combinations are coming from preclinical studies, and novel compounds have recently entered into clinical trials.
In this light, the aim of this review is to give an updated overview of the impact of the newly discovered compounds and to revise and comment the recent suggestions coming from their clinical evaluation as monotherapy or in combination with conventional chemotherapy and innovative therapeutic combinations, with particular attention to their potential for hematological malignancies and pediatric cancers.
Targeting the MDM2/X-P53 axis for therapeutic purposes
In the past few years, several molecules have been developed in order to release P53 from the control of MDM2 and restore its oncosuppressor activity. This goal has been obtained through different “molecular strategies” aiming at (i) blocking MDM2 expression, (ii) blocking the physical interaction between MDM2 and P53, (iii) modulating the E3 ubiquitin ligase activity of MDM2, and (iv) targeting the MDM2-P53 (protein–protein) complex. Insight on structural studies and chemical/technological procedures involved in the design of successful MDM2/X inhibitors and processes in understanding their potential are beyond of the purposes of the present reviews, and the details can be found in several recent publications [62,63,64]. Here, we will focus on the compounds that are now under clinical evaluation (and summarized in Table 2) to discuss their clinical potential and the recent results coming from their assessment.
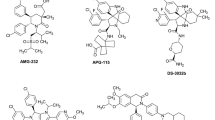
The cis-imidazolines “Nutlins” compounds: RG7112 and RG7388 (Idasanutlin)
The RG7112 (RO5045337) molecule from Roche has been the first MDM2 inhibitor undergoing clinical evaluation. This molecule derives from the optimization of the original molecules belonging to the Nutlins, a group of small molecules (Nutlin-1, Nutlin-2, Nutlin-3) acting by preventing the binding of MDM2 to P53. At the structural levels, the Nutlins are cis-imidazoline analogs discovered by Vassilev and colleagues in 2004 as compounds able to bind MDM2 in the P53-binding pocket with subsequent P53 accumulation, P53 pathways activation with the induction of cell cycle arrest, and apoptosis and growth inhibition in cancer cells and human tumor-xenografted nude mice [65]. In addition to P53 activation, it has been also reported that Nutlin-3a (the most active and potent enantiomer of the Nutlins’ family) was able to induce P53-independent mechanisms of cell death by enhancing the stability of P73, a member of the P53 family which is also regulated by MDM2 and that shows pro-apoptotic activities [66]. Actually, the interaction with P73 is not the only P53-independent effect of MDM2, since additional regulatory targets of MDM2 have been reported such as pRb [67] and E2F/DP [68]. In fact, it has been shown that antagonism of MDM2 by Nutlin-3a in cells with mutant P53 enhances chemo-sensitivity in an E2F1-dependent fashion, providing a therapeutic advantage also in tumors characterized by mutant P53 in combination with chemotherapy [69]. In terms of preclinical evaluation, it has been widely demonstrated in both in vitro and in vivo tumors models that Nutlin-3a is a powerful antitumor agent able to target different types of solid tumors, such as breast cancer, melanoma, retinoblastoma, prostate cancer, and lymphoma, as well as hematological malignancies. In particular, in the context of melanoma, important results have been achieved by using Nutlin-3 both as single treatment, revealing a complete inhibition of tumor cells invasiveness [70], and in combination with other molecules, such as inhibitors of Aurora A kinase that are synergized with Nutlin-3 by promoting immune-mediated tumor clearance in patient-derived xenograft models [71]. Also in the context of breast cancer in recent years, the use of Nutlin-3 was preclinically validated in combination with (i) carboplatin, in a humanized orthotropic breast-to-lung metastatic model [72] and (ii) paclitaxel, against aggressive diseases such as triple-negative and Numb-deficient human breast cancers [73, 74]. In particular, the combination Nutlin-3 plus paclitaxel in patient-derived xenograft (PDX) models showed persistent tumor growth inhibition and prevention of cancer stem-cell-driven tumor relapse after removal of chemotherapy [74]. In the context of hematological malignancies, our group has significantly contributed to the demonstration and characterization of the potential of Nutlin-3 in preclinical models based on established cell lines and patient-derived cells of acute myeloid leukemia (AML) and of chronic lymphocytic leukemia (CLL). In these settings, the efficacy of Nutlin-3 has been related to activation of P53 apoptotic pathway [75, 76] and modulation of a specific panel of genes and proteins related to cell survival [77,78,79,80], apoptosis [81, 82], and cell cycle progression [83]. Other groups validated the cytotoxic preclinical effects of Nutlin-3 in different hematological malignancies. In particular, significant results were obtained in the field of acute lymphoblastic leukemia (ALL), demonstrating the efficacy of the activation of P53 pathway and subsequent cell cycle block and apoptosis induction in aggressive childhood leukemia cells in vitro and in mice xenograft [84, 85] as well as reduction of cell viability, involving also autophagy, in adult ALL samples [86]. Our group has also significantly contributed to the demonstration of a synergistic activity of Nutlin-3 in different leukemic models, irrespectively of the P53 status: (i) with the multi-kinase inhibitors Dasatinib and Sorafenib in B cell chronic lymphocytic leukemia (B-CLL) and AML models via inhibition of the Akt pathway by inducing apoptosis/autophagy [87, 88]; (ii) with nanoparticles engineered with rituximab thus targeting CD20+ cells enhancing antibody-dependent cellular cytotoxicity and increasing survival in B-leukemic xenografted mice [89, 90]; (iii) with sodium dichloroacetate via increased expression of the P53-target genes MDM2, PUMA, TIGAR, and CDKN1A [91]; and (iv) with ibrutinib via inhibition of the BCR signaling and MAPK/PI3K pro-survival pathways [92]. The functional activation of P53 pathway after ex vivo Nutlin-3 treatment was proposed by Pozzo et al. as assay to detect P53 dysfunction in B-CLL patient samples [93]. The first published results on the clinical potential of RG7112 have been obtained in a trial of the European Community (EudraCT number: 2009-015522-10) investigating the pharmacodynamics of RG7112 in patients with MDM2-amplified liposarcoma [94]. Although reporting an overall feasibility of RG7112-mediated inhibition of MDM2 and P53 activation in this type of tumor in vivo, the trial highlighted several clinical adverse events related to the drug treatment including hematological toxicity, making the long-term treatment with RG7112 a challenge [94]. A phase I study on patients with advanced solid tumors has assessed the dose and treatments protocols showing that although RG7112 was generally well tolerated, the more effective schedule in terms of adequate P53 tumor activation was anyhow associated to increased hematological toxicities [95]. RG7112 has been clinically evaluated also in a phase I study clinical study involving different types of hematological malignancies including relapsed/refractory AML, ALL, chronic myeloid leukemia (CML) as well as CLL, and small cell lymphocytic leukemia (sCLL). RG7112 showed sufficient clinical activity to lead to P53 stabilization and transcriptional activation of P53 target genes in extremely poor prognosis (relapsed/refractory) AML patients and in CLL/sCLL patients [96]. In terms of potential molecular biomarkers, treatment with RG7112 led to increased expression of several P53 target genes including BAX, PUMA, FDXR, MDM2, ZMAT3, FAS, TNFRSF10B, CDKN1A, and TP53NP1 in circulating P53 wild-type leukemic cells following different kinetics of activation [96]. In AML, RG7112 administration showed clinical activity as monotherapy, particularly in relapsed/refractory AML with some patient reaching complete remission and hematopoietic recovery allowing subsequent transplantation [96], as well as by combining RG7112 with cytarabine, suggesting that combinations with other therapeutic agents might result in a synergic effect [97].
The evidence of hematotoxicity following treatment with RG7112 was not surprising since MDM2 plays a role in normal hematopoiesis. Indeed, preclinical studies highlighted that MDM2 antagonists induce major hematopoietic defects and inhibition of megakaryopoiesis that may account for the significant marrow suppression of both leukemic and normal progenitors in patients following the treatment [98]. The high doses required to obtain a clinical effect together with the impact of toxicities and complications (i.e., neutropenia, thrombocytopenia including sepsis and hemorrhage) and gastrointestinal toxicity due to the loss of enterocytes attributable to RG7112 administration have prompted the development of a more potent and selective compound of the Nutlins’ family named RG7388 (RO5503781) known as idasanutlin [99]. Idasanutlin is a second-generation MDM2 inhibitor able to induce the expected biological effects at concentrations that are significantly lower than those required by RG7112, both in in vitro and in vivo experimental models [99]. The higher efficacy and the less degree of side effects compared to the original compound have therefore speed up the setting of several clinical trials (Table 2). A recent phase Ib study on relapsed-refractory AML patients has assessed safety and pharmacokinetics of escalating doses of idasanutlin as single agent or in combination with cytarabine, showing a well tolerability of the treatment coupled with clinical response and with the possibility of achieve a good control of the gastrointestinal complications [100]. Moreover, analysis of MDM2 protein expression showed that the levels of MDM2 expression in leukemic blasts are associated to idasanutlin clinical response and might therefore represent a useful biomarker to identify those AML patients who likely will have advantage from idasanutlin treatment [100]. Different idasanutlin-based combined approaches are under evaluation at the clinical level. Among these, the combinations with obinutuzumab and venetoclax might potentially have a significant impact in the context of hematological malignancies. Obinutuzumab is a novel glycoengineered type II anti-C20 monoclonal antibody characterized by enhanced antibody-dependent cellular cytotoxicity and direct cell death compared to rituximab and representing therefore a significant clinical improvement for the treatment of B cell malignancies [101]. Recently, the antitumor potential of the therapeutic combination based on the caspase-independent cell death and antibody-dependent cellular cytotoxicity triggered by obinutuzumab together with the caspase-dependent apoptosis mediated by idasanutlin has been evaluated in in vitro and in vivo models of lymphoma, reporting an enhanced P53 wild-type cell death without signs of reciprocal interference between the two pharmacological compounds [102]. Venetoclax (ABT-199) is a selective oral small molecular BCL-2 inhibitor that has been tested at the preclinical level in combination with idasanutlin in P53 wild-type AML models, showing indeed a synergistic antitumor activity compared with the respective single-agent treatments [103]. Therefore, these studies provided the proof of concept for the clinical evaluation of such therapeutic combinations.
The spirooxindole MI-77301 (SAR405838)
The small-molecule MI-77301 from Sanofi is a spirooxindole-based compound that mimics the three P53 key aminoacidic residues but shows also the ability of establishing additional interaction with an overall a higher binding affinity. After optimization of the two phenyl rings, the newly derived molecule was characterized by an improved affinity to MDM2 leading to an optimized protein binding and enhanced biological activities together with improved in vivo pharmacokinetic features. In particular, the preclinical evaluation of the antitumor activity of MI-77301, also referred as SAR405838, revealed a good chemical stability of the compound and the capacity of inducing cell growth inhibition up to cell cycle arrest and/or apoptosis in a P53-dependent manner in several tumor cell lines (i.e., osteosarcoma, acute leukemia, prostate and colon cancer cells) [104]. In in vivo xenograft models, SAR405838 was able to induce a significant antitumor activity in different types of tumors characterized by P53 wild-type status but lacking MDM2 amplification and a complete and persistent tumor regression in 100% of mice bearing osteosarcoma cancer cells with wild-type P53 and amplified MDM2 [104]. At the molecular level, the pharmacological treatment was able to induce upregulation of MDM2 and P21 in all cancer cell lines and PUMA expression was correlated with apoptosis induction and tumor regression suggesting a direct role in mediating SAR405838 biological effects [104]. A clinical study evaluated the combined treatment of SAR405838 with pimasertib (a small-molecule inhibitor of MEK1 and MEK2) in patients with advanced solid cancer. Although the study is terminated, no results have been published so far. Results from a second clinical trial assessing SAR405838 safety, tolerability, pharmacokinetics, and biological activity in patients with advanced cancer, including solid tumors and lymphoma, for which no further effective standard treatment is available, have been recently published. The pharmacological treatment resulted to be acceptable in terms of safety, and a dose-dependent MIC-1 modulation following P53 pathway activation was observed suggesting that, although a limited activity was observed with the single-agent schedule, SAR405838 might have a potential in combination regimens [105].
The MK-8242 compound (SCH-900242)
MK-8242, also known as SCH-900242, is a compound from Merck pharmaceutic described as a potent, orally bioavailable, small-molecule inhibitor of the MDM2-P53 protein–protein interaction able to induce growth arrest and cell death at IC50 value as low as 20 nM [106]. Two phase I clinical trials have been launched by Merck to evaluate MK-8242 alone in patients with advanced solid tumors and MK-8242 either alone or in combination with cytarabine in AML patients (Table 2). In the context of AML, the study aimed at evaluating safety, tolerability, and pharmacokinetics in adult patients with refractory or recurrent AML together with the evaluation of the response rate and duration of response after treatment [106]. The results of the study highlighted adequate safety and no significant adverse effects, even though the maximum dose tolerated and the recommended phase 2 dose were not established for the better tolerated 7-day on/14-day off dosing schedule as well as the single-agent tolerance since doses above 300 mg (administered twice a day) were not tested [106]. In terms of toxicity, one patient reported a grade 3, drug-related adverse event of atrial fibrillation while the most common side effects were in line with those described with other MDM2 inhibitors including in particular gastrointestinal and hematological adverse events that are dose dependent and mostly less than grade 3 resulting therefore overall well handily. In terms of efficacy assessment, the single-agent clinical effectiveness was limited, probably for the advanced disease state of the patients and the limited number of treatments performed. Moreover, although one of the clinical aims of the study was the evaluation of the combined treatment with chemotherapy (cytarabine), the study terminated before the complete enrollment of this arm of the study. The results of the second study performed in patients with advanced/refractory solid tumors characterized by wild-type P53 status have been recently reported [107]. The 47 patients that received MK-8242 monotherapy (doses ranging from 60 to 500 mg) developed gastrointestinal/hematologic toxicity in line with the previous study. Doses above 300 mg were able to activate P53 pathway, as demonstrated by the expression of the P53 target gene pleckstrin homology-like domain family A member 3 (PHLD3A) which is a biomarker of P53 transcriptional activity, supporting the designation of 400 mg as the recommended phase II dose. The clinical activity was however less impressive than expected showing three patients with liposarcoma with a partial response, 31 patients with stable disease, and eight with progressive disease for a total of 27 patients with liposarcoma with a median progression-free survival of 237 days [107]. These partially satisfying results could be due to previous chemotherapy that might have selected more resistant tumors, and perhaps the achieved P53 activation might have been insufficient to induce significant cell death [107]. Nonetheless, the median progression-free survival appears encouraging, in particular in patients with liposarcoma.
New compounds recently entered in clinical evaluation
In the last few years, additional compounds and small-molecule inhibitors of MDM2/X have entered in clinical evaluation.
AMG232 is a piperidinone-derived compound from Amgen acting as a potent and selective inhibitor of the MDM2−P53 interaction with notable pharmacokinetic properties and in vivo antitumor activity in xenograft models [108]. The preclinical pharmacological characterization showed a high potential of this MDM2 inhibitor compared to other MDM2 inhibitors including RG7112, SAR299155, and RG7388, in blocking tumor cell proliferation and inducing tumor cell apoptosis in a large panel of tumor cell lines in a P53-dependent manner [109]. As expected, the degree of response to the treatment was variable within the different cell lines both in vitro and in vivo, but interestingly showing antitumor activity also in P53 wild-type tumors carrying different genetic aberrations such as KRAS-mutant and BRAF-mutant. Treatment was associated with P53 stabilization and induction of MDM2, P21, and PUMA expression [109]. Moreover, there was an improved antitumor activity associated with P53 increase and p21 induction when used in combination with cytotoxic agents [109]. In addition, in combination with radiation therapy, the compound showed a more potent antitumor activity coupled to antiangiogenesis potential compared to the single treatments, leading to accumulation of several molecular mediators of senescence/autophagy/apoptosis such as FoxM1, ULK-1, DRAM, and BAX [110]. AMG232 is currently under clinical evaluation for the treatment of solid tumors, melanoma, myeloma, and AML leukemia as single treatment or in combination therapies (Table 2).
The CGM097 compound is a substituted dihydroisoquinolinone derivative from Novartis designed to mimic three key hydrophobic interactions made by P53 residues with Phe19, Trp23, and Leu26 in the MDM2 pocket and acting as potent and selective MDM2 inhibitor [111]. The compound is in phase I study as single agent (dose escalation and a dose expansion) in patients with advanced solid tumors characterized by wild-type P53 that undergo standard therapies, but have progressed and in patients with no standard therapy available (Table 2). In the context of hematological malignancies, CGM097 has been evaluated in vitro, showing a potent and selective effect of inhibition of cell proliferation/viability of wild-type P53 primary AML cells and AML cell lines and in vivo antitumor effects in xenograft models, providing the rational for the testing of CGM097 in AML patients [112]. Finally, CGM097 was preclinically evaluated in 24 B cell acute lymphoblastic leukemia patient-derived xenograft (PDX) including hypodiploid, near haploid, mixed lineage leukemia (MLL)-rearranged, CRLF2-rearranged, and BCR-ABL models of the PDX repository (PRoXe; www.proxe.org) showing the ability to induce a significantly improved survival of tumor-bearing mice [113, 114].
The DS3032b compound is an inhibitor of the P53-MDM2 interaction developed by Daiichi Sankyo that has reached the clinical assessment in 2013, and it is now under evaluation in three studies in patients affected by different types of tumors including AML, ALL, CML, MDS (id: NCT02319369), advanced solid tumors or lymphomas (id: NCT01877382), and relapsed/refractory multiple myeloma patients (id: NCT02579824). Recent preliminary results have been presented at the Annual American Society of Clinical Oncology in 2016 on data collected from 31 of 34 patients with solid P53 wild-type tumors (mainly liposarcoma) enrolled in part 1 study. In terms of drug-related adverse effects, gastrointestinal and hematological events were reported with thrombocytopenia or with neutropenia. In terms of efficacy, none of the patients under evaluation had an objective response but the 77% of patients had stable disease with the better outcome in tumors with aberrant MDM2 signaling and wild-type P53 [115]. In December 2016, at the 58th Annual Meeting of the American Society of Hematology Daiichi Sankyo has also announced preliminary safety and efficacy data from the phase I study of DS-3032b in the treatment of hematological malignancies. The dose escalation study in 38 patients enrolled with relapsed/refractory AML or high-risk myelodisplastic syndrome (MDS) has established the maximum tolerated dose at 160 mg once a day for 21 days in a 28-day cycle [116]. In terms of clinical efficacy, the authors report a reduction of bone marrow blasts at the end of the first cycle in 15 out of 38 patients and complete remission in two patients with relapsed/refractory AML [116]. One patient with high-risk MDS achieved marrow complete remission with platelet improvement for 4 months. The three patients (two with AML and one with MDS) showing complete response developed TP53 gene mutation while receiving treatment, suggesting that a combination therapy approach might be more appropriate [116].
HDM201 from Novartis is an imidazopyrrolidinone scaffold-based inhibitor of the P53-MDM2 protein–protein interaction with superior characteristics in terms of in vitro activity/selectivity and of in vivo features of oral bioavailability, pharmacokinetic, and pharmacodynamic profiles as assessed in animals [117, 118]. In particular, the optimized interactions of HDM201 with MDM2 protein are responsible for the increased stabilization of the complex leading to a higher potency of the molecule [119]. In xenograft tumor models, HDM201administration following either a daily low-dose schedule or once at a high-dose schedule induced a differential response; although, the single high-dose schedule lead to rapid and significant induction of P53-dependent PUMA expression and apoptosis together with robust and sustained tumor regression, though the two regimens had an overall comparable long-term efficacy [120]. There are three ongoing clinical studies aimed to assess and compare different schedules of HDM201 in patients with advanced P53 wild-type tumors (Table 2). Some preliminary results obtained from these trials have been recently reported [121]. The 74 patients receiving HDM201 following two regimens reported common grade 3/4 adverse events including anemia, neutropenia, and thrombocytopenia. Gastrointestinal toxicity was common but not dose limiting (mainly nausea) while hematological toxicity appears to be regimen-dependent and at late onset. Clinical benefit was observed but no further details are available so far [121].
Stapled peptides (i.e., stabilized alpha-helical peptides) are currently rising interest as strategy to target protein–protein interactions. At the chemical level, by virtue of the role of the alpha helix in binding and modulating protein–protein interactions, there is the possibility of “mimicking” the α-helices at the binding interface of two proteins to competitively inhibit their contact [122]. As far as the interaction between P53 and MDM2/MDMX is concern, several reports suggest that MDM2 antagonists might not be effective in tumors overexpressing MDMX, highlighting the potential of a concomitant and simultaneous targeting of the two oncosuppressor that have led to the development of “dual inhibitors” of MDM2 and MDMX for cancer therapy [123, 124]. In this light, the stapled peptide ALRN-6924 from Aileron Therapeutics is the first clinical drug candidate that binds, in an equipotent way, and inhibits the two P53 suppressor proteins MDM2 and MDMX. This molecule is now under clinical evaluation in two studies with advanced hematological (id: NCT02909972) and solid malignancies (id: NCT02264613) characterized by wild-type P53 status.
Perspectives for pediatric malignancies
The progresses made by recent medicine have led to the achievement of a high rate of overall survival across pediatric cancer patients. The reported 5-year survival is nearly the 80% for many childhood tumors, and it reaches the 90% in the case of pediatric ALL, with a mortality persistently decreasing [125, 126]. However, this outstanding result has left somehow pediatric cancers excluded from the assessment of newly derived therapeutic molecules. The gap between adult and pediatric tumors consideration is well described by the evidence that, although from 1948 to 2003 the FDA approved the use of 120 new cancer drugs, only 30 have been used in pediatric patients [127]. Anyhow, there are still childhood cancers difficult to treat and, as in the case of pediatric ALL, the occurrence of genetic alterations and the development of relapse disease make the clinical outcome less favorable by using the current standard care, pressing the need of new therapeutic options [128]. In addition, therapy-related late adverse effects in cancer survivors, ranging from infertility and cardiotoxicity up to second cancers development, are becoming key issues for therapy-related risk evaluation and therapy decision [129]. With the aim to establish an international platform in order to identify more effective treatments for children with cancer, a pediatric preclinical testing program (PPTC) has been established. Its aim is to provide reliable preclinical in vivo data using genomically characterized patient-derived xenograft lines in order to identify agents to move forward in pediatric clinical trials (NCI PPTC, www.ncipptc.org). The panel of models accessible through the Consortium for testing drug candidates includes tumor cell lines and xenograft models with different features among which most are patient-derived xenografts (PDX) models obtained by direct implant of tumors in mice [130]. A key feature of pediatric cancers is the low frequency of P53 mutations compared to adult tumors, and it is therefore not surprising that the Nutlins compounds exhibited both in vitro and in vivo antitumor activity against several types of pediatric tumors including solid and hematological malignancies [131]. In the same fashion, the preliminary evaluation of the Nutlin-3-derived compound RG7112, tested in vitro against a panel of 23 pediatric cancer cell lines and in vivo in xenograft models, reported regressions in solid tumors from different histotypes including medulloblastoma, alveolar rhabdomyosarcoma, Wilms, rhabdoid, and Ewing sarcoma xenografts [132]. Moreover, with regard to hematological malignancies, RG7112 showed a significant antitumor activity against ALL xenografts including B cell precursor acute lymphoblastic leukemia (BCP-ALL), T cell acute lymphoblastic leukemia (T-ALL), and MLL-ALL with the most impressive effect reported on infant MLL-rearranged xenograft when RG7112 was used either alone or in combination with established drugs such as vincristine, dexamethasone, and l-asparaginase [85, 132]. In line with previous evidence, in these models, the RG7112-mediated effects led to the induction of P53-dependent cell cycle arrest and apoptosis with the activation of pro-apoptotic targets, such as PUMA, and downregulation of the anti-apoptotic protein surviving supporting the use of this Nutlin-derived molecule in pediatric cancer management [85]. In the pediatric context, while carcinomas are very rare (1.5% of malignancies), the embryonal tumors represent more than one-fourth of all malignant diseases, with neuroblastoma being the most common extracranial solid tumor of childhood [133]. Although patients are generally responsive to treatments, children with high-risk neuroblastoma have also a high risk of relapse and show high rate of mortality. Since P53 is usually unaltered in this type of tumor and genetic alterations of TP53 are rare (in contrast with aberrations of MDM2 that are instead quite common), the use of MDM2 inhibitors to target the P14(ARF)-MDM2-TP53 key axis in neuroblastoma is an appealing therapeutic strategy [134]. In this line, it has been shown that Nutlin 3 is able to induce P53 accumulation with concomitant decrease of proliferation and increased apoptosis in neuroblastoma cells when used alone and in synergy with chemotherapeutic drugs [135]. Moreover, synergistic antitumor activity in neuroblastoma has been demonstrated in different therapeutic combinations such as the one based on Nutlin-3 plus bevacizumab to enhance apoptosis and target tumor angiogenesis [136]. Of note, a sensitizing activity to genotoxic drugs of Nutlin 3 has also been reported in P53-null neuroblastoma cells via upregulation of TAp73 and activation of E2F1, highlighting the P53-independent mechanisms of actions of this inhibitor [137]. The potential therapeutic benefit for neuroblastoma has been evaluated in phase II-like trials with recent MDM2 inhibitors that have entered in clinical evaluation for adult cancers. In this line, Lu and colleagues have reported a P53-mediated apoptotic effect of MI-773 in a P53 wild-type neuroblastoma model with stabilizing effect on P53, both in vitro showing similar efficacy as RG7388 and Nutlin-3 and in an in vivo orthotropic model when the compound was used as single agent. Moreover, when used in combination with doxorubicin, there was a significant a sensitizing activity toward chemotherapy supporting the rational of combination therapies to overcome chemo-resistance [138]. In the context of the PPTP, a second phase II-like trial has been performed to test the potential of MK-8242 as single agent in several tumor models [139]. Kang and co-workers reported sensitivity of different P53 wild-type pediatric tumor cell lines to MK-8242 treatment with an IC50 that was around sixfold lower than the one reported for RG7112 in the same tumor models. The antitumor activity evaluated in vivo highlighted a differential response among the different P53 wild-type solid tumor xenografts, with an overall delay (twofold or greater) in time to event in the treated mice compared to controls, with the exception of the osteosarcoma xenografts that were indeed characterized by a very low P53 expression [139]. The in vivo evaluation of a hematological panel highlighted MLL-rearranged lines as the best responders to MK-8242 treatment, while the ALL-non-MLL models showed only partial responses with no complete remission [139]. Finally, results from a randomized phase II-like trial assessing the MDM2 inhibitor CGM097 in a large and well-characterized leukemia and lymphoma PDX repository (Public Repository of Xenografts, PRoXe; www.proxe.org) have been recently published by Townsend and colleagues [140]. In their work, the authors aimed at testing CGM097 in mice xenografted with adult and pediatric P53 wild-type B-ALL. Overall, treatment with CGM097 led to survival benefit only on wild-type P53 PDX with an increased median survival of 44 days compared to controls; although, heterogeneity of response to CGM097 was observed within the different xenograft models. This study provides the proof of concept for the clinical testing of CGM097 in pediatric B-ALL patients including those that received previous extensive chemotherapeutic treatments [140].
Conclusions
The use of MDM2/X pharmacological inhibitors to activate the P53 pathway and challenge cancer is an appealing and fruitful therapeutic strategy, particularly for the management of hematological malignancies that show low levels of TP53 mutations. The ongoing clinical trials are testing mainly inhibitors of MDM2 while chemical compounds targeting MDMX did not advanced to the clinical phase yet. However, there is great effort at the preclinical levels to design successful MDMX inhibitors and there are great expectations on the results of the clinical study with the first double MDM2-MDMX inhibitor ALRN-6924. Overall, the increasing number of newly derived compounds recently entered in clinical trials and the preliminary results coming from these studies are encouraging. As for many drugs, also this class of compounds will definitely benefit of the combination with a second pharmacological strategy, ranging from conventional cytotoxic chemotherapy up to novel small-molecule inhibitors (Fig. 2), allowing a synergic effect in targeting both the P53 wild-type cancer cells as well as the P53-mutated clones. However, several aspects need to be handled in the future studies. The drug-related toxicity is one of the main issue since the simple adjustment of schedule treatments does not appear sufficient to achieve an adequate control of the gastrointestinal, hematologic, and cardiac adverse effects reported when these inhibitors are used either as monotherapy or in combination with standard chemotherapeutic drugs that might “synergize” also in this aspect [98]. An attractive option could be the “cyclotherapy,” designed to perform a conditioning treatment with low doses of P53 activators to induce cell cycle arrest in normal cells with a cytostatic effect that will be protected from the toxicity of conventional drugs targeting the S/M phases of the cell cycle [141]. Another concern is the development of drug resistance that may impair the clinical potential of these compounds. Resistance to treatment occurs through the development of P53 mutant-clones and as result of other molecular defects and/or altered expression of molecules, such as MDMX, that can confer resistance to MDM2 inhibitors. Hoffman-Luca and colleagues have recently addressed the mechanisms of acquired resistance focusing on the MDM2 inhibitor SAR405838. By using an osteosarcoma cell line (MDM2 amplified and TP53 wild-type) the authors highlighted a difference between the resistance acquired in vitro and the one acquired in in vivo xenograft models [142]. In fact, while the in vitro pharmacological treatment led to the selection of resistant sublines characterized by the development of P53 mutations in the DNA binding domain, the resistant xenografted tumors regrown after drug treatment showed only a partial loss of ex vivo sensitivity to the compound and retained wild-type TP53. The different responses and the different mechanisms of acquired resistance in the two models have obviously important implications in terms of drug development and preclinical assessment. The key point appears to be the “pressure” of the drug that is different in the two models: steady and persistent in vitro while variable in vivo with pharmacokinetic-dependent fluctuations. As expected in the first model, the tumor cells need to develop inactivating TP53 mutations to survive, while in vivo structural mutations might be sufficient to confer the required advantage to the cells to survive [142]. The evidence of acquisition of somatic TP53 mutations and selection of mutated clones is in line with the preclinical results obtained with other MDM2 inhibitors [143, 144] that provide insight into the mechanisms of acquired resistance useful also to assess the potential of combined/sequential therapies between MDM2 inhibitors and other small molecules such ad Bcl-2 inhibitors [145]. The demonstration of the onset of pharmacological resistance to an MDM2 inhibitor at the clinical level has been recently reported by Jung and colleagues [146]. Data were collected from the phase I trial (NCT01636479) with patients affected by de-differentiated/MDM2 amplified liposarcoma under SAR405838 treatment [105]. TP53 mutations appeared during treatment as shown by cell-free DNA analysis and the frequency of mutations increased during the time with a correlation with tumor mass [146]. In conclusion, in the next future, the deep elucidation of the mechanisms underlying the onset of acquired resistance in preclinical models and in long-term follow-up-treated patients will be crucial. In the pediatric context, the phase II-like trials that mimic human randomized clinical trials definitely represent a critical step to gain insight on the potential of MDM2/X inhibitors. These preclinical platforms have already allowed the evaluation of the antitumor activity of several compounds. In addition, they have highlighted key aspects that need to be addressed in the next future including the gap between in vitro activity and in vivo efficacy and the high doses required in the xenograft models that would imply very high doses in patients [140]. Finally, a personalized approach would definitely optimized the use of this therapeutic strategy. To this end, the TP53/MDM2 and MDMX status need to be coupled with new biomarkers that might help in selecting those patients that will benefit of such therapeutic approaches either as monotherapy or as combination treatments.
Abbreviations
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- B-CLL:
-
B cell chronic lymphocytic leukemia
- BCP-ALL:
-
B cell precursor acute lymphoblastic leukemia
- CLL:
-
Chronic lymphocytic leukemia
- CML:
-
Chronic myeloid leukemia
- MDM2:
-
Murine double minute-2
- MDMX:
-
Murine double minute-X
- MDS:
-
Myelodysplastic syndrome
- MLL:
-
Mixed lineage leukemia
- PDX:
-
Patient-derived xenograft
- PHLDA3:
-
Pleckstrin homology-like domain, family A, member 3
- PPTP:
-
Pediatric preclinical testing program
- PRoXe:
-
Public repository of xenografts
- RING:
-
Really interesting new gene
- sCLL:
-
Small cell lymphocytic leukemia
- T-ALL:
-
T cell acute lymphoblastic leukemia
- Ub:
-
Ubiquitin
References
Cahilly-Snyder L, Yang-Feng T, Francke U, George DL. Molecular analysis and chromosomal mapping of amplified genes isolated from a transformed mouse 3T3 cell line. Somat Cell Mol Genet. 1987;13:235–44.
Fakharzadeh SS, Trusko SP, George DL. Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. EMBO J. 1991;10:1565–9.
Fakharzadeh SS, Rosenblum-Vos L, Murphy M, Hoffman EK, George DL. Structure and organization of amplified DNA on double minutes containing the mdm2 oncogene. Genomics. 1993;15:283–90.
Zhao Y, Yu H, Hu W. The regulation of MDM2 oncogene and its impact on human cancers. Acta Biochim Biophys Sin Shanghai. 2014;46:180–9.
Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 2010;20:299–309.
Tan BX, Liew HP, Chua JS, Ghadessy FJ, Tan YS, Lane DP, Coffill CR. Anatomy of Mdm2 and Mdm4 in evolution. J Mol Cell Biol. 2017;9:3–15.
Iwakuma T, Lozano G. MDM2, an introduction. Mol Cancer Res. 2003;1:993–1000.
Kastan MB, Canman CE, Leonard CJ. P53, cell cycle control and apoptosis: implications for cancer. Cancer Metastasis Rev. 1995;14:3–15.
Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979–87.
Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9:862–73.
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery Jr CA, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21.
Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7.
Jackson JG, Lozano G. The mutant p53 mouse as a pre-clinical model. Oncogene. 2013;32:4325–30.
Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9.
Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15:2–8.
Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, van Ham RC, van der Houven van Oordt W, Hateboer G, van der Eb AJ, Jochemsen AG. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996;15:5349–57.
Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc Natl Acad Sci U S A. 2003;100:12009–14.
Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–8.
Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–6.
Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92–5.
Moyer SM, Larsson CA, Lozano G. Mdm proteins: critical regulators of embry ogenesis and homeostasis. J Mol Cell Biol. 2017;9:16–25.
Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–23.
Baliou E, Nonni A, Keramopoulos D, Ragos V, Tsiambas E, Patsouris E, Pavlakis K. Deregulation of p53-MDM2 auto-regulatory pathway in breast carcinoma. J BUON. 2016;21:1099–103.
Panagopoulos I, Bjerkehagen B, Gorunova L, Berner JM, Boye K, Heim S. Several fusion genes identified by whole transcriptome sequencing in a spindle cell sarcoma with rearrangements of chromosome arm 12q and MDM2 amplification. Int J Oncol. 2014;45:1829–36.
Ware PL, Snow AN, Gvalani M, Pettenati MJ, Qasem SA. MDM2 copy numbers in well-differentiated and dedifferentiated liposarcoma: characterizing progression to high-grade tumors. Am J Clin Pathol. 2014;141:334–41.
Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res. 1998;26:3453–9.
Michalk M, Meinrath J, Kunstlinger H, Koitzsch U, Drebber U, Merkelbach-Bruse S, Bollschweiler E, Kloth M, Hartmann W, Holscher A, et al. MDM2 gene amplification in esophageal carcinoma. Oncol Rep. 2016;35:2223–7.
Schoolmeester JK, Sciallis AP, Greipp PT, Hodge JC, Dal Cin P, Keeney GL, Nucci MR. Analysis of MDM2 amplification in 43 endometrial stromal tumors: a potential diagnostic pitfall. Int J Gynecol Pathol. 2015;34:576–83.
Steinman HA, Burstein E, Lengner C, Gosselin J, Pihan G, Duckett CS, Jones SN. An alternative splice form of Mdm2 induces p53-independent cell growth and tumorigenesis. J Biol Chem. 2004;279:4877–86.
Oliner JD, Saiki AY, Caenepeel S. The Role of MDM2 Amplification and Overexpression in Tumorigenesis. Cold Spring Harb Perspect Med. 2016;6:a026336.
Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591–602.
Riemenschneider MJ, Buschges R, Wolter M, Reifenberger J, Bostrom J, Kraus JA, Schlegel U, Reifenberger G. Amplification and overexpression of the MDM4 (MDMX) gene from 1q32 in a subset of malignant gliomas without TP53 mutation or MDM2 amplification. Cancer Res. 1999;59:6091–6.
Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, Teunisse A, Lam S, Ramos Y, Mohan A, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–6.
Chao CC. Mechanisms of p53 degradation. Clin Chim Acta. 2015;438:139–47.
Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 2009;1:a000950.
Pant V, Lozano G. Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev. 2014;28:1739–51.
Tsvetkov P, Reuven N, Shaul Y. Ubiquitin-independent p53 proteasomal degradation. Cell Death Differ. 2010;17:103–8.
Allton K, Jain AK, Herz HM, Tsai WW, Jung SY, Qin J, Bergmann A, Johnson RL, Barton MC. Trim24 targets endogenous p53 for degradation. Proc Natl Acad Sci U S A. 2009;106:11612–6.
Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, O’Rourke K, Koeppen H, Dixit VM. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature. 2004;429:86–92.
Esser C, Scheffner M, Hohfeld J. The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J Biol Chem. 2005;280:27443–8.
Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005;121:1071–83.
Lee EW, Lee MS, Camus S, Ghim J, Yang MR, Oh W, Ha NC, Lane DP, Song J. Differential regulation of p53 and p21 by MKRN1 E3 ligase controls cell cycle arrest and apoptosis. EMBO J. 2009;28:2100–13.
Elabd S, Meroni G, Blattner C. TRIMming p53’s anticancer activity. Oncogene. 2016;35:5577–84.
Love IM, Grossman SR. It takes 15 to tango: making sense of the many ubiquitin ligases of p53. Genes Cancer. 2012;3:249–63.
Chen J, Wu X, Lin J, Levine AJ. mdm-2 inhibits the G1 arrest and apoptosis functions of the p53 tumor suppressor protein. Mol Cell Biol. 1996;16:2445–52.
Freedman DA, Levine AJ. Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol Cell Biol. 1998;18:7288–93.
Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303.
Jain AK, Barton MC. Making sense of ubiquitin ligases that regulate p53. Cancer Biol Ther. 2010;10:665–72.
Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9.
Rodriguez MS, Desterro JM, Lain S, Lane DP, Hay RT. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol Cell Biol. 2000;20:8458–67.
Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–45.
Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–5.
Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–32.
Sharp DA, Kratowicz SA, Sank MJ, George DL. Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J Biol Chem. 1999;274:38189–96.
Tanimura S, Ohtsuka S, Mitsui K, Shirouzu K, Yoshimura A, Ohtsubo M. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 1999;447:5–9.
Leslie PL, Ke H, Zhang Y. The MDM2 RING domain and central acidic domain play distinct roles in MDM2 protein homodimerization and MDM2-MDMX protein heterodimerization. J Biol Chem. 2015;290:12941–50.
Wang X, Jiang X. Mdm2 and MdmX partner to regulate p53. FEBS Lett. 2012;586:1390–6.
Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG, Parant J, Lozano G, Yuan ZM. Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem. 2002;277:19251–4.
Medina-Medina I, Garcia-Beltran P, de la Mora-de la Mora I, Oria-Hernandez J, Millot G, Fahraeus R, Reyes-Vivas H, Sampedro JG, Olivares-Illana V. Allosteric interactions by p53 mRNA govern HDM2 E3 ubiquitin ligase specificity under different conditions. Mol Cell Biol. 2016;36:2195–205.
Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–53.
Popowicz GM, Czarna A, Holak TA. Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle. 2008;7:2441–3.
Estrada-Ortiz N, Neochoritis CG, Domling A. How to design a successful p53-MDM2/X interaction inhibitor: a thorough overview based on crystal structures. ChemMedChem. 2016;11:757–72.
Lemos A, Leao M, Soares J, Palmeira A, Pinto M, Saraiva L, Sousa ME. Medicinal chemistry strategies to disrupt the p53-MDM2/MDMX interaction. Med Res Rev. 2016;36:789–844.
Riedinger C, McDonnell JM. Inhibitors of MDM2 and MDMX: a structural perspective. Future Med Chem. 2009;1:1075–94.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8.
Lau LM, Nugent JK, Zhao X, Irwin MS. HDM2 antagonist Nutlin-3 disrupts p73-HDM2 binding and enhances p73 function. Oncogene. 2008;27:997–1003.
Xiao ZX, Chen J, Levine AJ, Modjtahedi N, Xing J, Sellers WR, Livingston DM. Interaction between the retinoblastoma protein and the oncoprotein MDM2. Nature. 1995;375:694–8.
Zhang Z, Wang H, Li M, Rayburn ER, Agrawal S, Zhang R. Stabilization of E2F1 protein by MDM2 through the E2F1 ubiquitination pathway. Oncogene. 2005;24:7238–47.
Ambrosini G, Sambol EB, Carvajal D, Vassilev LT, Singer S, Schwartz GK. Mouse double minute antagonist Nutlin-3a enhances chemotherapy-induced apoptosis in cancer cells with mutant p53 by activating E2F1. Oncogene. 2007;26:3473–81.
Worrall C, Suleymanova N, Crudden C, Trocoli Drakensjo I, Candrea E, Nedelcu D, Takahashi SI, Girnita L, Girnita A. Unbalancing p53/Mdm2/IGF-1R axis by Mdm2 activation restrains the IGF-1-dependent invasive phenotype of skin melanoma. Oncogene. 2017;36:3274–86.
Vilgelm AE, Pawlikowski JS, Liu Y, Hawkins OE, Davis TA, Smith J, Weller KP, Horton LW, McClain CM, Ayers GD, et al. Mdm2 and aurora kinase a inhibitors synergize to block melanoma growth by driving apoptosis and immune clearance of tumor cells. Cancer Res. 2015;75:181–93.
Tonsing-Carter E, Bailey BJ, Saadatzadeh MR, Ding J, Wang H, Sinn AL, Peterman KM, Spragins TK, Silver JM, Sprouse AA, et al. Potentiation of carboplatin-mediated DNA damage by the Mdm2 modulator Nutlin-3a in a humanized orthotopic breast-to-lung metastatic model. Mol Cancer Ther. 2015;14:2850–63.
Wali VB, Langdon CG, Held MA, Platt JT, Patwardhan GA, Safonov A, Aktas B, Pusztai L, Stern DF, Hatzis C. Systematic drug screening identifies tractable targeted combination therapies in triple-negative breast cancer. Cancer Res. 2017;77:566–78.
Tosoni D, Pambianco S, Ekalle Soppo B, Zecchini S, Bertalot G, Pruneri G, Viale G, Di Fiore PP, Pece S. Pre-clinical validation of a selective anti-cancer stem cell therapy for Numb-deficient human breast cancers. EMBO Mol Med. 2017;9:655–71.
Secchiero P, Barbarotto E, Tiribelli M, Zerbinati C, di Iasio MG, Gonelli A, Cavazzini F, Campioni D, Fanin R, Cuneo A, Zauli G. Functional integrity of the p53-mediated apoptotic pathway induced by the nongenotoxic agent nutlin-3 in B-cell chronic lymphocytic leukemia (B-CLL). Blood. 2006;107:4122–9.
Athanasakis E, Melloni E, Rigolin GM, Agnoletto C, Voltan R, Vozzi D, Piscianz E, Segat L, Dal Monego S, Cuneo A, et al. The p53 transcriptional pathway is preserved in ATMmutated and NOTCH1mutated chronic lymphocytic leukemias. Oncotarget. 2014;5:12635–45.
Secchiero P, Melloni E, di Iasio MG, Tiribelli M, Rimondi E, Corallini F, Gattei V, Zauli G. Nutlin-3 up-regulates the expression of Notch1 in both myeloid and lymphoid leukemic cells, as part of a negative feedback antiapoptotic mechanism. Blood. 2009;113:4300–8.
Secchiero P, Voltan R, di Iasio MG, Melloni E, Tiribelli M, Zauli G. The oncogene DEK promotes leukemic cell survival and is downregulated by both Nutlin-3 and chlorambucil in B-chronic lymphocytic leukemic cells. Clin Cancer Res. 2010;16:1824–33.
Voltan R, di Iasio MG, Bosco R, Valeri N, Pekarski Y, Tiribelli M, Secchiero P, Zauli G. Nutlin-3 downregulates the expression of the oncogene TCL1 in primary B chronic lymphocytic leukemic cells. Clin Cancer Res. 2011;17:5649–55.
Tisato V, Norcio A, Celeghini C, Milani D, Gonelli A, Secchiero P. Upregulation of SOCS-1 by Nutlin-3 in acute myeloid leukemia cells but not in primary normal cells. Clinics (Sao Paulo). 2014;69:68–74.
Secchiero P, di Iasio MG, Melloni E, Voltan R, Celeghini C, Tiribelli M, Dal Bo M, Gattei V, Zauli G. The expression levels of the pro-apoptotic XAF-1 gene modulate the cytotoxic response to Nutlin-3 in B chronic lymphocytic leukemia. Leukemia. 2010;24:480–3.
Voltan R, Secchiero P, Corallini F, Zauli G. Selective induction of TP53I3/p53-inducible gene 3 (PIG3) in myeloid leukemic cells, but not in normal cells, by Nutlin-3. Mol Carcinog. 2014;53:498–504.
Zauli G, Voltan R, di Iasio MG, Bosco R, Melloni E, Sana ME, Secchiero P. miR-34a induces the downregulation of both E2F1 and B-Myb oncogenes in leukemic cells. Clin Cancer Res. 2011;17:2712–24.
Kaindl U, Morak M, Portsmouth C, Mecklenbrauker A, Kauer M, Zeginigg M, Attarbaschi A, Haas OA, Panzer-Grumayer R. Blocking ETV6/RUNX1-induced MDM2 overexpression by Nutlin-3 reactivates p53 signaling in childhood leukemia. Leukemia. 2014;28:600–8.
Richmond J, Carol H, Evans K, High L, Mendomo A, Robbins A, Meyer C, Venn NC, Marschalek R, Henderson M, et al. Effective targeting of the P53-MDM2 axis in preclinical models of infant MLL-rearranged acute lymphoblastic leukemia. Clin Cancer Res. 2015;21:1395–405.
Borthakur G, Duvvuri S, Ruvolo V, Tripathi DN, Piya S, Burks J, Jacamo R, Kojima K, Ruvolo P, Fueyo-Margareto J, et al. MDM2 inhibitor, Nutlin 3a, induces p53 dependent autophagy in acute leukemia by AMP kinase activation. PLoS One. 2015;10:e0139254.
Zauli G, Voltan R, Bosco R, Melloni E, Marmiroli S, Rigolin GM, Cuneo A, Secchiero P. Dasatinib plus Nutlin-3 shows synergistic antileukemic activity in both p53 wild-type and p53 mutated B chronic lymphocytic leukemias by inhibiting the Akt pathway. Clin Cancer Res. 2011;17:762–70.
Zauli G, Celeghini C, Melloni E, Voltan R, Ongari M, Tiribelli M, di Iasio MG, Lanza F, Secchiero P. The sorafenib plus nutlin-3 combination promotes synergistic cytotoxicity in acute myeloid leukemic cells irrespectively of FLT3 and p53 status. Haematologica. 2012;97:1722–30.
Voltan R, Secchiero P, Ruozi B, Caruso L, Forni F, Palomba M, Zauli G, Vandelli MA. Nanoparticles loaded with Nutlin-3 display cytotoxicity towards p53(wild-type) JVM-2 but not towards p53(mutated) BJAB leukemic cells. Curr Med Chem. 2013;20:2712–22.
Voltan R, Secchiero P, Ruozi B, Forni F, Agostinis C, Caruso L, Vandelli MA, Zauli G. Nanoparticles engineered with rituximab and loaded with Nutlin-3 show promising therapeutic activity in B-leukemic xenografts. Clin Cancer Res. 2013;19:3871–80.
Agnoletto C, Melloni E, Casciano F, Rigolin GM, Rimondi E, Celeghini C, Brunelli L, Cuneo A, Secchiero P, Zauli G. Sodium dichloroacetate exhibits anti-leukemic activity in B-chronic lymphocytic leukemia (B-CLL) and synergizes with the p53 activator Nutlin-3. Oncotarget. 2014;5:4347–60.
Voltan R, Rimondi E, Melloni E, Rigolin GM, Casciano F, Arcidiacono MV, Celeghini C, Cuneo A, Zauli G, Secchiero P. Ibrutinib synergizes with MDM-2 inhibitors in promoting cytotoxicity in B chronic lymphocytic leukemia. Oncotarget. 2016;7:70623–38.
Pozzo F, Dal Bo M, Peragine N, Bomben R, Zucchetto A, Rossi F, Degan M, Rossi D, Chiarenza A, Grossi A, et al. Detection of TP53 dysfunction in chronic lymphocytic leukemia by an in vitro functional assay based on TP53 activation by the non-genotoxic drug Nutlin-3: a proposal for clinical application. J Hematol Oncol. 2013;6:83.
Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, Heil F, Rueger R, Graves B, Ding M, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012;13:1133–40.
Patnaik A, Tolcher A, Beeram M, Nemunaitis J, Weiss GJ, Bhalla K, Agrawal M, Nichols G, Middleton S, Beryozkina A, et al. Clinical pharmacology characterization of RG7112, an MDM2 antagonist, in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2015;76:587–95.
Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, Bowen D, Martinelli G, Drummond MW, Vyas P, et al. Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Cancer Res. 2016;22:868–76.
Yee K, Martinelli G, Assouline S, Kasner M, Vey N, Kelly KR, Mark WD, Dennis M, Seiter K, Blotner S, et al. Phase 1b study of the MDM2 antagonist RG7112 in combination with 2 doses/schedules of cytarabine. Blood. 2013;122.
Mahfoudhi E, Lordier L, Marty C, Pan J, Roy A, Roy L, Rameau P, Abbes S, Debili N, Raslova H, et al. P53 activation inhibits all types of hematopoietic progenitors and all stages of megakaryopoiesis. Oncotarget. 2016;7:31980–92.
Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, Chu XJ, Bartkovitz D, Podlaski F, Janson C, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem. 2013;56:5979–83.
Reis B, Jukofsky L, Chen G, Martinelli G, Zhong H, So WV, Dickinson MJ, Drummond M, Assouline S, Hashemyan M, et al. Acute myeloid leukemia patients’ clinical response to idasanutlin (RG7388) is associated with pre-treatment MDM2 protein expression in leukemic blasts. Haematologica. 2016;101:e185–8.
Farhadfar N, Litzow MR. New monoclonal antibodies for the treatment of acute lymphoblastic leukemia. Leuk Res. 2016;49:13–21.
Herting F, Herter S, Friess T, Muth G, Bacac M, Sulcova J, Umana P, Dangl M, Klein C. Antitumour activity of the glycoengineered type II anti-CD20 antibody obinutuzumab (GA101) in combination with the MDM2-selective antagonist idasanutlin (RG7388). Eur J Haematol. 2016;97:461–70.
Lehmann C, Friess T, Birzele F, Kiialainen A, Dangl M. Superior anti-tumor activity of the MDM2 antagonist idasanutlin and the Bcl-2 inhibitor venetoclax in p53 wild-type acute myeloid leukemia models. J Hematol Oncol. 2016;9:50.
Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barriere C, Stuckey JA, Meagher JL, Bai L, Liu L, et al. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014;74:5855–65.
de Jonge M, de Weger VA, Dickson MA, Langenberg M, Le Cesne A, Wagner AJ, Hsu K, Zheng W, Mace S, Tuffal G, et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur J Cancer. 2017;76:144–51.
Ravandi F, Gojo I, Patnaik MM, Minden MD, Kantarjian H, Johnson-Levonas AO, Fancourt C, Lam R, Jones MB, Knox CD, et al. A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML). Leuk Res. 2016;48:92–100.
Wagner AJ, Banerji U, Mahipal A, Somaiah N, Hirsch H, Fancourt C, Johnson-Levonas AO, Lam R, Meister AK, Russo G, et al. Phase I trial of the human double minute 2 inhibitor MK-8242 in patients with advanced solid tumors. J Clin Oncol. 2017;35:1304–11.
Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, Canon J, Chen A, Chen X, Chow D, et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J Med Chem. 2014;57:1454–72.
Canon J, Osgood T, Olson SH, Saiki AY, Robertson R, Yu D, Eksterowicz J, Ye Q, Jin L, Chen A, et al. The MDM2 inhibitor AMG 232 demonstrates robust antitumor efficacy and potentiates the activity of p53-inducing cytotoxic agents. Mol Cancer Ther. 2015;14:649–58.
Werner LR, Huang S, Francis DM, Armstrong EA, Ma F, Li C, Iyer G, Canon J, Harari PM. Small molecule inhibition of MDM2-p53 interaction augments radiation response in human tumors. Mol Cancer Ther. 2015;14:1994–2003.
Holzer P, Masuya K, Furet P, Kallen J, Valat-Stachyra T, Ferretti S, Berghausen J, Bouisset-Leonard M, Buschmann N, Pissot-Soldermann C, et al. Discovery of a dihydroisoquinolinone derivative (NVP-CGM097): a highly potent and selective MDM2 inhibitor undergoing phase 1 clinical trials in p53wt tumors. J Med Chem. 2015;58:6348–58.
Weisberg E, Halilovic E, Cooke VG, Nonami A, Ren T, Sanda T, Simkin I, Yuan J, Antonakos B, Barys L, et al. Inhibition of wild-type p53-expressing aml by the novel small molecule HDM2 inhibitor CGM097. Mol Cancer Ther. 2015;14:2249–59.
Townsend EC, DeSouza T, Murakami MA, Montero J, Stevenson K, Christie AL, Christodolou AN, Vojinovic U, Kopp N, Barzaghi-Rinaudo P, et al. The MDM2 inhibitor NVP-CGM097 is highly active in a randomized preclinical trial of B-cell acute lymphoblastic leukemia patient derived xenografts. Blood. 2015;126:797.
Townsend EC, Murakami MA, Christodoulou A, Christie AL, Koster J, DeSouza TA, Morgan EA, Kallgren SP, Liu H, Wu SC, et al. The public repository of xenografts enables discovery and randomized phase II-like trials in mice. Cancer Cell. 2016;29:574–86.
Mrinal MG, Todd Michael B, Gary KS, Tyler M, Richard DC, Saeheum S, Prasanna K, Roohi G, Oleg Z, Michael MR, et al. A phase 1 study of the MDM2 inhibitor DS-3032b in patients (pts) with advanced solid tumors and lymphomas. J Clin Oncol. 2016;34:2581.
DiNardo CD, Rosenthal J, Andreeff M, Zernovak O, Kumar P, Gajee R, Chen S, Rosen M, Song S, Kochan J, et al. Phase 1 dose escalation study of MDM2 inhibitor DS-3032b in patients with hematological malignancies—preliminary results. Blood. 2016;128:593.
Furet P, Masuya K, Kallen J, Stachyra-Valat T, Ruetz S, Guagnano V, Holzer P, Mah R, Stutz S, Vaupel A, et al. Discovery of a novel class of highly potent inhibitors of the p53-MDM2 interaction by structure-based design starting from a conformational argument. Bioorg Med Chem Lett. 2016;26:4837–41.
Holzer P, Chène P, Ferretti S, Furet P, Gabriel T, Gruenenfelder B, Guagnano V, Hofmann F, Kallen J, Mah R, et al. Abstract 4855: discovery of NVP-HDM201—first disclosure of a next-generation Mdm2 inhibitor with superior characteristics. Cancer Res. 2016;76:4855.
Stachyra-Valat T, Baysang F, D’Alessandro A-C, Dirk E, Furet P, Guagnano V, Kallen J, Leder L, Mah R, Masuya K, et al. Abstract 1239: NVP-HDM201: biochemical and biophysical profile of a novel highly potent and selective PPI inhibitor of p53-Mdm2. Cancer Res. 2016;76:1239.
Ferretti S, Rebmann R, Berger M, Santacroce F, Albrecht G, Pollehn K, Sterker D, Wartmann M, Hueber A, Wiesmann M, et al. Abstract 1224: insights into the mechanism of action of NVP-HDM201, a differentiated and versatile next-eneration small-molecule inhibitor of Mdm2, under evaluation in phase I clinical trials. Cancer Res. 2016;76:1224.
Hyman D, Chatterjee M, Langenberg MHG, Lin CC, Suárez C, Tai D, Cassier P, Yamamoto N, De Weger VA, Jeay S, et al. Dose- and regimen-finding phase I study of NVP-HDM201 in patients (pts) with TP53 wild-type (wt) advanced tumors. Eur J Cancer. 2016;69:S128-9.
Iyer VV. A review of stapled peptides and small molecules to inhibit protein-protein interactions in cancer. Curr Med Chem. 2016;23:3025–43.
Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, Olson KA, Kesavan K, Gangurde P, Mukherjee A, et al. Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci U S A. 2013;110:E3445–54.
Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, Kung AL, Wahl GM, Walensky LD. A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell. 2010;18:411–22.
Smith MA, Altekruse SF, Adamson PC, Reaman GH, Seibel NL. Declining childhood and adolescent cancer mortality. Cancer. 2014;120:2497–506.
Pritchard-Jones K, Hargrave D. Declining childhood and adolescent cancer mortality: great progress but still much to be done. Cancer. 2014;120:2388–91.
Adamson PC, Houghton PJ, Perilongo G, Pritchard-Jones K. Drug discovery in paediatric oncology: roadblocks to progress. Nat Rev Clin Oncol. 2014;11:732–9.
Ding LW, Sun QY, Tan KT, Chien W, Thippeswamy AM, Eng Juh Yeoh A, Kawamata N, Nagata Y, Xiao JF, Loh XY, et al. Mutational landscape of pediatric acute lymphoblastic leukemia. Cancer Res. 2017;77:390–400.
Armenian SH, Robison LL. Childhood cancer survivorship: an update on evolving paradigms for understanding pathogenesis and screening for therapy-related late effects. Curr Opin Pediatr. 2013;25:16–22.
Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, Gorlick R, Kolb EA, Zhang W, Lock R, et al. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer. 2007;49:928–40.
Van Maerken T, Rihani A, Van Goethem A, De Paepe A, Speleman F, Vandesompele J. Pharmacologic activation of wild-type p53 by nutlin therapy in childhood cancer. Cancer Lett. 2014;344:157–65.
Carol H, Reynolds CP, Kang MH, Keir ST, Maris JM, Gorlick R, Kolb EA, Billups CA, Geier B, Kurmasheva RT, et al. Initial testing of the MDM2 inhibitor RG7112 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2013;60:633–41.
Kaatsch P. Epidemiology of childhood cancer. Cancer Treat Rev. 2010;36:277–85.
Van Maerken T, Vandesompele J, Rihani A, De Paepe A, Speleman F. Escape from p53-mediated tumor surveillance in neuroblastoma: switching off the p14(ARF)-MDM2-p53 axis. Cell Death Differ. 2009;16:1563–72.
Barbieri E, Mehta P, Chen Z, Zhang L, Slack A, Berg S, Shohet JM. MDM2 inhibition sensitizes neuroblastoma to chemotherapy-induced apoptotic cell death. Mol Cancer Ther. 2006;5:2358–65.
Patterson DM, Gao D, Trahan DN, Johnson BA, Ludwig A, Barbieri E, Chen Z, Diaz-Miron J, Vassilev L, Shohet JM, et al. Effect of MDM2 and vascular endothelial growth factor inhibition on tumor angiogenesis and metastasis in neuroblastoma. Angiogenesis. 2011;14:255–66.
Peirce SK, Findley HW. The MDM2 antagonist nutlin-3 sensitizes p53-null neuroblastoma cells to doxorubicin via E2F1 and TAp73. Int J Oncol. 2009;34:1395–402.
Lu J, Guan S, Zhao Y, Yu Y, Wang Y, Shi Y, Mao X, Yang KL, Sun W, Xu X, et al. Novel MDM2 inhibitor SAR405838 (MI-773) induces p53-mediated apoptosis in neuroblastoma. Oncotarget. 2016;7:82757–69.
Kang MH, Reynolds CP, Kolb EA, Gorlick R, Carol H, Lock R, Keir ST, Maris JM, Wu J, Lyalin D, et al. Initial testing (stage 1) of MK-8242-A novel MDM2 inhibitor-by the pediatric preclinical testing program. Pediatr Blood Cancer. 2016;63:1744–52.
Townsend EC, Murakami MA, Christodoulou A, Christie AL, Koster J, DeSouza TA, Morgan EA, Kallgren SP, Liu H, Wu SC, et al. The public repository of xenografts enables discovery and randomized phase II-like trials in mice. Cancer Cell. 2016;30:183.
Rao B, Lain S, Thompson AM. p53-Based cyclotherapy: exploiting the ‘guardian of the genome’ to protect normal cells from cytotoxic therapy. Br J Cancer. 2013;109:2954–8.
Hoffman-Luca CG, Yang CY, Lu J, Ziazadeh D, McEachern D, Debussche L, Wang S. Significant differences in the development of acquired resistance to the MDM2 inhibitor SAR405838 between in vitro and in vivo drug treatment. PLoS One. 2015;10:e0128807.
Aziz MH, Shen H, Maki CG. Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene. 2011;30:4678–86.
Jones RJ, Bjorklund CC, Baladandayuthapani V, Kuhn DJ, Orlowski RZ. Drug resistance to inhibitors of the human double minute-2 E3 ligase is mediated by point mutations of p53, but can be overcome with the p53 targeting agent RITA. Mol Cancer Ther. 2012;11:2243–53.
Hoffman-Luca CG, Ziazadeh D, McEachern D, Zhao Y, Sun W, Debussche L, Wang S. Elucidation of acquired resistance to Bcl-2 and MDM2 inhibitors in acute leukemia in vitro and in vivo. Clin Cancer Res. 2015;21:2558–68.
Jung J, Lee JS, Dickson MA, Schwartz GK, Le Cesne A, Varga A, Bahleda R, Wagner AJ, Choy E, de Jonge MJ, et al. TP53 mutations emerge with HDM2 inhibitor SAR405838 treatment in de-differentiated liposarcoma. Nat Commun. 2016;7:12609.
Han X, Garcia-Manero G, McDonnell TJ, Lozano G, Medeiros LJ, Xiao L, Rosner G, Nguyen M, Fernandez M, Valentin-Vega YA, et al. HDM4 (HDMX) is widely expressed in adult pre-B acute lymphoblastic leukemia and is a potential therapeutic target. Mod Pathol. 2007;20:54–62.
Hendy OM, Elghannam DM, El-Sharnouby JA, Goda EF, El-Ashry R, Al-Tonbary Y. Frequency and prognostic significance of murine double minute protein-2 overexpression and p53 gene mutations in childhood acute lymphoblastic leukemia. Hematology. 2009;14:335–40.
Moorman AV, Harrison CJ, Buck GA, Richards SM, Secker-Walker LM, Martineau M, Vance GH, Cherry AM, Higgins RR, Fielding AK, et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood. 2007;109:3189–97.
Stengel A, Kern W, Haferlach T, Meggendorfer M, Fasan A, Haferlach C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia. 2017;31:705–11.
Falk IJ, Willander K, Chaireti R, Lund J, Nahi H, Hermanson M, Green H, Lotfi K, Soderkvist P. TP53 mutations and MDM2(SNP309) identify subgroups of AML patients with impaired outcome. Eur J Haematol. 2015;94:355–62.
Li L, Tan Y, Chen X, Xu Z, Yang S, Ren F, Guo H, Wang X, Chen Y, Li G, Wang H. MDM4 overexpressed in acute myeloid leukemia patients with complex karyotype and wild-type TP53. PLoS One. 2014;9:e113088.
Long J, Parkin B, Ouillette P, Bixby D, Shedden K, Erba H, Wang S, Malek SN. Multiple distinct molecular mechanisms influence sensitivity and resistance to MDM2 inhibitors in adult acute myelogenous leukemia. Blood. 2010;116:71–80.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21.
Quintas-Cardama A, Hu C, Qutub A, Qiu YH, Zhang X, Post SM, Zhang N, Coombes K, Kornblau SM. p53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia. 2017;31:1296-305.
Welch JS, Petti AA, Miller CA, Fronick CC, O’Laughlin M, Fulton RS, Wilson RK, Baty JD, Duncavage EJ, Tandon B, et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med. 2016;375:2023–36.
Forero-Castro M, Robledo C, Lumbreras E, Benito R, Hernandez-Sanchez JM, Hernandez-Sanchez M, Garcia JL, Corchete-Sanchez LA, Tormo M, Barba P, et al. The presence of genomic imbalances is associated with poor outcome in patients with burkitt lymphoma treated with dose-intensive chemotherapy including rituximab. Br J Haematol. 2016;172:428–38.
Leventaki V, Rodic V, Tripp SR, Bayerl MG, Perkins SL, Barnette P, Schiffman JD, Miles RR. TP53 pathway analysis in paediatric Burkitt lymphoma reveals increased MDM4 expression as the only TP53 pathway abnormality detected in a subset of cases. Br J Haematol. 2012;158:763–71.
Tessoulin B, Eveillard M, Lok A, Chiron D, Moreau P, Amiot M, Moreau-Aubry A, Le Gouill S, Pellat-Deceunynck C. p53 dysregulation in B-cell malignancies: More than a single gene in the pathway to hell. Blood Rev. 2017. (Epub ahead of print).
Baliakas P, Hadzidimitriou A, Sutton LA, Rossi D, Minga E, Villamor N, Larrayoz M, Kminkova J, Agathangelidis A, Davis Z, et al. Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia. 2015;29:329–36.
Hurtado AM, Chen-Liang TH, Przychodzen B, Hamedi C, Munoz-Ballester J, Dienes B, Garcia-Malo MD, Anton AI, de Arriba F, Teruel-Montoya R, et al. Prognostic signature and clonality pattern of recurrently mutated genes in inactive chronic lymphocytic leukemia. Blood Cancer J. 2015;5:e342.
Liu L, Fan L, Fang C, Zou ZJ, Yang S, Zhang LN, Li JY, Xu W. S-MDM4 mRNA overexpression indicates a poor prognosis and marks a potential therapeutic target in chronic lymphocytic leukemia. Cancer Sci. 2012;103:2056–63.
Nadeu F, Delgado J, Royo C, Baumann T, Stankovic T, Pinyol M, Jares P, Navarro A, Martin-Garcia D, Bea S, et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood. 2016;127:2122–30.
Stilgenbauer S, Schnaiter A, Paschka P, Zenz T, Rossi M, Dohner K, Buhler A, Bottcher S, Ritgen M, Kneba M, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014;123:3247–54.
Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, Dawson KJ, Iorio F, Nik-Zainal S, Bignell GR, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997.
Carballo-Zarate AA, Medeiros LJ, Fang L, Shah JJ, Weber DM, Thomas SK, Manasanch EE, Hao S, Shen Q, Orlowski RZ, et al. Additional-structural-chromosomal aberrations are associated with inferior clinical outcome in patients with hyperdiploid multiple myeloma: a single-institution experience. Mod Pathol. 2017;30:843–53.
Elnenaei MO, Gruszka-Westwood AM, A’Hernt R, Matutes E, Sirohi B, Powles R, Catovsky D. Gene abnormalities in multiple myeloma: the relevance of TP53, MDM2, and CDKN2A. Haematologica. 2003;88:529–37.
Kryukov F, Nemec P, Radova L, Kryukova E, Okubote S, Minarik J, Stefanikova Z, Pour L, Hajek R. Centrosome associated genes pattern for risk sub-stratification in multiple myeloma. J Transl Med. 2016;14:150.
Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, Sougnez C, Knoechel B, Gould J, Saksena G, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25:91–101.
Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, Proszek PZ, Johnson DC, Kaiser MF, Melchor L, et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol. 2015;33:3911–20.
Fan C, Wei J, Yuan C, Wang X, Jiang C, Zhou C, Yang M. The functional TP53 rs1042522 and MDM4 rs4245739 genetic variants contribute to non-Hodgkin lymphoma risk. PLoS One. 2014;9:e107047.
Hartmann E, Fernandez V, Stoecklein H, Hernandez L, Campo E, Rosenwald A. Increased MDM2 expression is associated with inferior survival in mantle-cell lymphoma, but not related to the MDM2 SNP309. Haematologica. 2007;92:574–5.
Kridel R, Meissner B, Rogic S, Boyle M, Telenius A, Woolcock B, Gunawardana J, Jenkins C, Cochrane C, Ben-Neriah S, et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood. 2012;119:1963–71.
Salido M, Baro C, Oscier D, Stamatopoulos K, Dierlamm J, Matutes E, Traverse-Glehen A, Berger F, Felman P, Thieblemont C, et al. Cytogenetic aberrations and their prognostic value in a series of 330 splenic marginal zone B-cell lymphomas: a multicenter study of the Splenic B-Cell Lymphoma Group. Blood. 2010;116:1479–88.
Xu-Monette ZY, Medeiros LJ, Li Y, Orlowski RZ, Andreeff M, Bueso-Ramos CE, Greiner TC, McDonnell TJ, Young KH. Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood. 2012;119:3668–83.
Xu-Monette ZY, Moller MB, Tzankov A, Montes-Moreno S, Hu W, Manyam GC, Kristensen L, Fan L, Visco C, Dybkaer K, et al. MDM2 phenotypic and genotypic profiling, respective to TP53 genetic status, in diffuse large B-cell lymphoma patients treated with rituximab-CHOP immunochemotherapy: a report from the International DLBCL Rituximab-CHOP Consortium Program. Blood. 2013;122:2630–40.
Young KH, Leroy K, Moller MB, Colleoni GW, Sanchez-Beato M, Kerbauy FR, Haioun C, Eickhoff JC, Young AH, Gaulard P, et al. Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: an international collaborative study. Blood. 2008;112:3088–98.
Braggio E, Keats JJ, Leleu X, Van Wier S, Jimenez-Zepeda VH, Valdez R, Schop RF, Price-Troska T, Henderson K, Sacco A, et al. Identification of copy number abnormalities and inactivating mutations in two negative regulators of nuclear factor-kappaB signaling pathways in Waldenstrom’s macroglobulinemia. Cancer Res. 2009;69:3579–88.
Chang H, Samiee S, Li D, Patterson B, Chen CI, Stewart AK. Analysis of IgH translocations, chromosome 13q14 and 17p13.1(p53) deletions by fluorescence in situ hybridization in Waldenstrom’s macroglobulinemia: a single center study of 22 cases. Leukemia. 2004;18:1160–2.
Hunter ZR, Xu L, Yang G, Zhou Y, Liu X, Cao Y, Manning RJ, Tripsas C, Patterson CJ, Sheehy P, Treon SP. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123:1637–46.
Nguyen-Khac F, Lambert J, Chapiro E, Grelier A, Mould S, Barin C, Daudignon A, Gachard N, Struski S, Henry C, et al. Chromosomal aberrations and their prognostic value in a series of 174 untreated patients with Waldenstrom’s macroglobulinemia. Haematologica. 2013;98:649–54.
Poulain S, Braggio E, Roumier C, Aijjou R, Broucqsault N, Galiegue-Zouitina S, Manier S, Soenen V, Nibourel O, Duthilleul P, et al. High-throughput genomic analysis in Waldenstrom’s macroglobulinemia. Clin Lymphoma Myeloma Leuk. 2011;11:106–8.
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
VT and RV carried out the primary literature search; VT and RV drafted the manuscript; AG performed the search literature and carried out the revision process; and PS and GZ participated in discussing, revising and editing the manuscript. All authors read and approved final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tisato, V., Voltan, R., Gonelli, A. et al. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol 10, 133 (2017). https://doi.org/10.1186/s13045-017-0500-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-017-0500-5