
Abstract
Background and aim
To describe current diagnostic and therapeutic strategies in organic acidurias (OADs) and to evaluate their impact on the disease course allowing harmonisation.
Methods
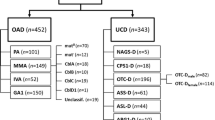
Datasets of 567 OAD patients from the E-IMD registry were analysed. The sample includes patients with methylmalonic (MMA, n = 164), propionic (PA, n = 144) and isovaleric aciduria (IVA, n = 83), and glutaric aciduria type 1 (GA1, n = 176). Statistical analysis included description and recursive partitioning of diagnostic and therapeutic strategies, and odds ratios (OR) for health outcome parameters. For some analyses, symptomatic patients were divided into those presenting with first symptoms during (i.e. early onset, EO) or after the newborn period (i.e. late onset, LO).
Results
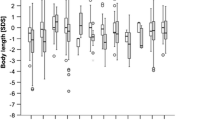
Patients identified by newborn screening (NBS) had a significantly lower median age of diagnosis (8 days) compared to the LO group (363 days, p < 0.001], but not compared to the EO group. Of all OAD patients 71 % remained asymptomatic until day 8. Patients with cobalamin-nonresponsive MMA (MMA-Cbl−) and GA1 identified by NBS were less likely to have movement disorders than those diagnosed by selective screening (MMA-Cbl−: 10 % versus 39 %, p = 0.002; GA1: 26 % versus 73 %, p < 0.001). For other OADs, the clinical benefit of NBS was less clear. Reported age-adjusted intake of natural protein and calories was significantly higher in LO patients than in EO patients reflecting different disease severities. Variable drug combinations, ranging from 12 in MMA-Cbl− to two in isovaleric aciduria, were used for maintenance treatment. The effects of specific metabolic treatment strategies on the health outcomes remain unclear because of the strong influences of age at onset (EO versus LO), diagnostic mode (NBS versus selective screening), and the various treatment combinations used.
Conclusions
NBS is an effective intervention to reduce time until diagnosis especially for LO patients and to prevent irreversible cerebral damage in GA1 and MMA-Cbl−. Huge diversity of therapeutic interventions hampers our understanding of optimal treatment.

Similar content being viewed by others

Change history
12 December 2017
Due to an unfortunate error during the typesetting process, the collaborators were presented incorrectly.
Abbreviations
- AAM(s):
-
Amino acid mixture(s)
- E-IMD:
-
European registry and network for intoxication type metabolic diseases
- EO:
-
Early onset (i.e. onset of first symptoms during the newborn period)
- GA1:
-
Glutaric aciduria type 1
- HRF:
-
High-risk family screening
- IVA:
-
Isovaleric aciduria
- IVD:
-
Isovaleryl-CoA dehydrogenase
- LO:
-
Late onset (i.e. onset of first symptoms after the newborn period)
- MMA:
-
Methylmalonic aciduria (isolated forms)
- MMA-Cbl+ :
-
Cobalamin-responsive methylmalonic aciduria
- MMA-Cbl− :
-
Cobalamin-nonresponsive methylmalonic aciduria
- NBS:
-
Newborn screening
- OAD(s):
-
Organic aciduria(s)
- OR:
-
Odds ratio
- PA:
-
Propionic aciduria
- Q1:
-
First quartile
- Q3:
-
Third quartile
- WHO:
-
World Health Organisation
References
Baumgartner MR, Hörster F, Dionisi-Vici C et al (2014) Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis 9:130
Bijarnia S, Wiley V, Carpenter K, Christodoulou J, Ellaway CJ, Wilcken B (2008) Glutaric aciduria type I: outcome following detection by newborn screening. J Inherit Metab Dis 31:503–507
Boneh A, Beauchamp M, Humphrey M, Watkins J, Peters H, Yaplito-Lee J (2008) Newborn screening for glutaric aciduria type I in Victoria: treatment and outcome. Mol Genet Metab 94:287–291
De Baulny HO, Benoist JF, Rigal O, Touati G, Rabier D, Saudubray JM (2005) Methylmalonic and propionic acidaemias: management and outcome. J Inherit Metab Dis 28:415–423
Dionisi-Vici et al (2006) 'Classical' organic acidurias, propionic aciduria, methylmalonic aciduria and isovaleric aciduria: long-term outcome and effects of expanded newborn screening using tandem mass spectrometry. J Inherit Metab Dis 29:383–389
Ensenauer R, Vockley J, Willard JM et al (2004) A common mutation is associated with a mild, potentially asymptomatic phenotype in patients with isovaleric acidemia diagnosed by newborn screening. Am J Hum Genet 75:1136–1142
Evans S, Alroqaiba N, Daly A, Neville C, Davies P, Macdonald A (2012) Feeding difficulties in children with inherited metabolic disorders: a pilot study. J Hum Nutr Diet 25:209–216
FAO (2001) Human energy requirements: Report of a Joint FAO/WHO/UNU Expert Consultation. FAO Report Series, No. 1. FAO, Rome
Garbade SF, Greenberg CR, Demirkol M et al (2014) Unravelling the complex MRI pattern in glutaric aciduria type I using statistical models-a cohort study in 180 patients. J Inherit Metab Dis 37:763–773
Grünert SC, Müllerleile S, de Silva L et al (2012a) Propionic acidemia: neonatal versus selective metabolic screening. J Inherit Metab Dis 35:41–49
Grünert SC, Wendel U, Lindner M et al (2012b) Clinical and neurocognitive outcome in symptomatic isovaleric acidemia. Orphanet J Rare Dis 7:9
Grünert SC, Müllerleile S, De Silva L et al (2013) Propionic acidemia: clinical course and outcome in 55 pediatric and adolescent patients. Orphanet J Rare Dis 8:6
Heinze G, Schemper M (2002) A solution to the problem of separation in logistic regression. Stat Med 21(16):2409–2419
Heinze G, Ploner M, Dunkler D, Southworth H (2013) Logistf: Firth’s bias reduced logistic regression. R package version 1.21. http://CRAN.R-project.org/package=logistf
Heringer J, Boy SPN, Ensenauer R et al (2010) Use of guidelines improves the neurological outcome in glutaric aciduria type I. Ann Neurol 68:743–752
Heringer J, Boy N, Burgard P, Okun JG, Kölker S (2015) Newborn screening for glutaric aciduria type I: benefits and limitations. Int J Neonatal Screen 1:57–68
Herskovitz M, Goldsher D, Sela BA, Mandel H (2013) Subependymal mass lesions and peripheral polyneuropathy in adult-onset glutaric aciduria type I. Neurology 81:849–850
Hörster F, Baumgartner MR, Viardot C et al (2007) Long-term outcome in methylmalonic acidurias is influenced by the underlying defect (mut0, mut−, cblA, cblB). Pediatr Res 62:225–230
Hörster F, Garbade SF, Zwickler T et al (2009) Prediction of outcome in isolated methylmalonic acidurias: combined use of clinical and biochemical parameters. J Inherit Metab Dis 32:630–639
Hothorn T, Hornik K, Zeileis A (2006) Unbiased recursive partitioning: a conditional inference framework. J Comput Graph Stat 15(3):651–674
Hothorn T, Hornik K, Strobl C, Zeileis A (2015) Party: a laboratory for recursive partitioning. R package version 1.0-23. http://CRAN.R-project.org/package=party
Kölker S, Garbade SF, Greenberg CR et al (2006) Natural history, outcome, and therapeutic efficacy in children and adults with glutaryl-CoA dehydrogenase deficiency. Pediatr Res 59:840–847
Kölker S, Garbade SF, Boy N, Maier EM et al (2007) Decline of acute encephalopathic crises in children with glutaryl-CoA dehydrogenase deficiency identified by newborn screening in Germany. Pediatr Res 62:357–362
Kölker S, Christensen E, Leonard JV et al (2011) Diagnosis and management of glutaric aciduria type I—revised recommendations. J Inherit Metab Dis 34:677–694
Kölker S, Boy SP, Heringer J et al (2012) Complemetary dietary treatment using lysine-free, arginine-fortified amino acid supplements in glutaric aciduria type I—a decade of experience. Mol Genet Metab 107:72–80
Kölker S, Garcia Cazorla A, Valayannopoulos V et al (2015a) The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: the initial presentation. J Inherit Metab Dis 38:1041–1057
Kölker S, Valayannopoulos V, Burlina AB et al (2015b) The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: the evolving clinical phenotype. J Inherit Metab Dis 38:1059–1074
Kölker S, Dobbelaere D, Häberle J et al (2015c) Networking across borders for individuals with organic acidurias and urea cycle disorders: the E-IMD consortium. JIMD Rep 22:29–38
Loeber JG, Burgard P, Cornel MC et al (2012) Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 1. From blood spot to screening result. J Inherit Metab Dis 35:603–611
Mardach R, Verity MA, Cederbaum SD (2004) Clinical, pathological, and biochemical studies in a patient with propionic acidemia and fatal cardiomyopathy. Mol Genet Metab 85:286–290
Marquard J, El Scheich T, Klee D, Schmitt M, Meissner T, Mayatepek E, Oh J (2011) Chronic pancreatitis in branched-chain organic acidurias—a case of methylmalonic aciduria and an overview of the literature. Eur J Pediatr 170:241–245
Martinez Alvarez L, Jameson E, Parry NR, Lloyd C, Ashworth JL (2015) Optic neuropathy in methylmalonic and propionic acidemia. Br J Ophthalmol. doi:10.1136/bjophthalmol-2015-306798
Nicolaides P, Leonard J, Surtees R (1998) Neurological outcome of methylmalonic acidaemia. Arch Dis Child 78:508–512
Nizon M, Ottolenghi C, Valayannopoulos V et al (2013) Long-term neurological outcome of a cohort of 80 patients with classical organic acidurias. Orphanet J Rare Dis 8:148
Pena L, Franks J, Chapman KA et al (2012) Natural history of propionic acidemia. Mol Genet Metab 105:5–9
Pfeil J, Listl HGF, Kölker S, Lindner M, Burgard P (2013) Newborn screening by tandem mass spectrometry for glutaric aciduria type 1: a cost-effectiveness analysis. Orphanet J Rare Dis 8:167
Prada CE, Al Jasmi F, Kirk EP, Hopp M, Jones O, Leslie ND, Burrow TA (2011) Cardiac disease in methylmalonic acidemia. J Pediatr 159:862–864
Richter SJ, McCann MH (2007) Multiple comparison of medians using permutation tests. J Mod Appl Stat Methods 6:399–412
Romano S, Valayannopoulos V, Touati G et al (2010) Cardiomyopathies in propionic aciduria are reversible after liver transplantation. J Pediatr 156:128–134
Schreiber J, Chapman KA, Summar ML et al (2012) Neurologic considerations in propionic acidemia. Mol Genet Metab 105:10–15
Smucker MD, Allan J, Carterette B (2007) A comparison of statistical significance tests for information retrieval evaluation. CIKM’07 Proceedings of the sixteenth ACM conference on information and knowledge management. 623–632
Strauss KA, Lazovic J, Wintermark M, Morton DH (2007) Multimodal imaging of striatal degeneration in Amish patients with glutaryl-CoA dehydrogenase deficiency. Brain 130:1905–1920
Strauss KA, Brumbaugh J, Duffy A et al (2011) Safety, efficacy and physiological actions of a lysine-free, arginine-rich formula to treat glutaryl-CoA dehydrogenase deficiency: focus on cerebral amino acid influx. Mol Genet Metab 10:93–106
Sutton VR, Chapman KA, Gropman AL et al (2012) (2012) Chronic management and health supervision of individuals with propionic acidemias. Mol Genet Metab 105:26–33
Viau K, Ernst SL, Vanzo RJ, Botto LD, Pasquali M, Longo N (2012) Glutaric acidemia type 1: outcomes before and after expanded newborn screening. Mol Genet Metab 106:430–438
Wilcken B (2010) Expanded newborn screening: reducing harm, assessing benefit. J Inherit Metab Dis 33(Suppl 2):S205–S210
World Health Organization, Food and Agriculture Organization of the United Nations, United Nations University (2007) Protein and amino acid requirements in human nutrition. Report of a joint WHO/FAO/UNU expert consultation. WHO Technical Report Series, No. 935, WHO Press
Acknowledgments
We are indebted to all patients and their families who have contributed to this study, shared their experience of living with a rare disease, and for their trust. We are grateful for fruitful collaboration with the following clinical partners, patient support groups and industrial partners (in alphabetical order of countries): Lut de Baere, Nathalie Stroobant (Belgische Organisatie voor Kinderen en Volwassenen met een Stofwisselingsziekte VZW [BOKS], Belgium), Nela Carić (Hrvatska udruga za rijetke bolesti, Croatia), Veronika Dvorakova and Tomas Honzik (Charles University and General University of Prague, First Faculty of Medicine, Prague, Czech Republic), Annika and Kenneth Rovsing (PND – Protein Nedbrydnings Defekt Foreningen, Denmark), Samantha Parker (Lysogene, Paris, France), EURORDIS, European Organisation for Rare Disease (France), Erich Bauchart (Hôpital Necker-Enfants Malades, Assistance Publique-Hôpitaux de Paris, Reference Center for Inherited Metabolic Disease, Necker-Enfants Malades University Hospital and IMAGINE Institute, Paris, France), Markus Ott, Beate Szczerbak (Nutricia Metabolics GmbH, Germany), Hubertus von Voss, Raimund Schmid (Kindernetzwerk e.V., Germany), Mandy Kretschmer (Glutarazidurie e.V., Germany), Reinhild Link (Wiesbaden, representing the SSIEM Dieticians Group), Persephone Augoustides-Savvopoulou (University 1st Pediatric Department, Metabolic Laboratory, ‘Hippocration’ General Hospital of Thessaloniki, Greece), Ioannis Anagnostopoulos (KRIKOS ZOIS – Society for patients and friends of patients with inherited metabolic diseases, Greece), Evridiki Drogari (University of Athens, Aghia Sophia Children’s Hospital, Unit of Metabolic Diseases, Athens, Greece), Renza Barbon Galluppi (UNIAMO FIMR, Italy), Susan Udina (COMETA ASMME – Associazione Studio Malattie Metaboliche Ereditarie – ONLUS, Italy), Hanka Meutgeert (Volwassenen en Kinderen met Stoffwisselingsziekten [VKS], Netherlands), Vanessa Ferreira (Associação Portuguesa CDG, Portugal), Miguel Macedo (Apofen, Portugal), Sérgio Braz Antão (Rarrisimas, Portugal), Sergi Faber (Catalana de Trastorns Metabòlics Hereditaris, Spain), Sofia Nordin (Svedish Orphan Biovitrium AB [SOBI], Sweden), Steven Hannigan (CLIMB, Children Living with Inherited Metabolic Diseases, National Information Centre for Metabolic Diseases, and EMDA, the European Metabolic Disorders Alliance), and Robin Lachmann (National Hospital for Neurology and Neurosurgery, Charles Dent Metabolic Unit, London, United Kingdom), Lara Abulhoul, Maureen Cleary and Paula Gissen (Metabolic Unit, Great Ormond Street Hospital, London, United Kingdom).
This publication arises from the project “European registry and network for intoxication type metabolic diseases” (E-IMD; EAHC no 2010 12 01) which has received funding from the European Union, in the framework of the Health Programme. After the end of the EU funding period the E-IMD patient registry has be sustained by funding from the Kindness-for-Kids Foundation (Munich, Germany) and the Dietmar Hopp Foundation (St. Leon-Rot, Germany). M. Baumgartner and J. Häberle (Zurich, Switzerland) are supported by radiz – Rare Disease Initiative Zurich, a clinical research priority program of the University of Zurich. C. Dionisi-Vici (Rome, Italy) is supported by the association “La vita è un dono”. R. Lachmann is supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
Individual contributors
Lise Aksglaede, Paula Avram, Elena Balmaseda-Serrano, Eric Bauchart, Javier Blasco-Alonso, Anaïs Brassier, Anupam Chakrapani, Yin-Hsiu Chien, Maria L. Couce, Corinne de Laet, Pascale de Lonlay, Linda de Meirleir, Carlo Dionisi-Vici, Dries Dobbelaere, Angeles Garcia-Cazorla, Florian Gleich, Wanda Gradowska, Stephanie Grünewald, Gisela Haege, Johannes Häberle, Wuh-Liang Hwu, Harikleia Ioannou, Robin Lachmann, Eveline Langereis, Elisa Leão Teles, Eduardo López-Laso, Shirou Matsumoto, Hélène Ogier de Baulny, Carlos Ortez, Luis Peña-Quintana, Angeles Ruiz-Gomez, Adrijan Sarajlija, Marshall L. Summar, Nicholas Thompson, Roshni Vara, Inmaculada Vives Pinera, Monique Williams, and Matthias Zielonka also contributed to this work.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
All procedures followed were in accordance with the ethical standards of the responsible committee on human studies (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000.
Conflict of interest
None.
Informed consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human studies (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients or their legal guardians prior to inclusion in the study in countries where this was needed by law.
Animal rights
This article does not contain animal subjects.
Additional information
Communicated by: Jerry Vockley
A correction to this article is available online at https://doi.org/10.1007/s10545-017-0116-5.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Suppl. Table 1
(DOC 54 kb)
Suppl. Table 2
(DOC 39 kb)
Suppl. Table 3
(DOC 35 kb)
Suppl. Table 4
(DOC 37 kb)
Suppl. Table 5
(DOC 36 kb)
Suppl. Table 6
(DOC 45 kb)
Suppl. Table 7
(DOC 49 kb)
Suppl. Table 8
(DOC 57 kb)
Suppl. Table 9
(DOC 39 kb)
Suppl. Table 10
(DOC 40 kb)
Suppl. Table 11
(DOC 39 kb)
Suppl. Table 12
(DOC 31 kb)
Suppl. Fig. 1
(DOC 64 kb)
Suppl. Fig. 2
(DOC 44 kb)
Rights and permissions
About this article

Cite this article
Heringer, J., Valayannopoulos, V., Lund, A.M. et al. Impact of age at onset and newborn screening on outcome in organic acidurias. J Inherit Metab Dis 39, 341–353 (2016). https://doi.org/10.1007/s10545-015-9907-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-015-9907-8