
Abstract
Background
The clinical presentation of patients with organic acidurias (OAD) and urea cycle disorders (UCD) is variable; symptoms are often non-specific.
Aims/methods
To improve the knowledge about OAD and UCD the E-IMD consortium established a web-based patient registry.
Results
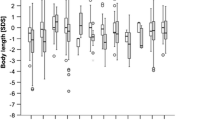
We registered 795 patients with OAD (n = 452) and UCD (n = 343), with ornithine transcarbamylase (OTC) deficiency (n = 196), glutaric aciduria type 1 (GA1; n = 150) and methylmalonic aciduria (MMA; n = 149) being the most frequent diseases. Overall, 548 patients (69 %) were symptomatic. The majority of them (n = 463) presented with acute metabolic crisis during (n = 220) or after the newborn period (n = 243) frequently demonstrating impaired consciousness, vomiting and/or muscular hypotonia. Neonatal onset of symptoms was most frequent in argininosuccinic synthetase and lyase deficiency and carbamylphosphate 1 synthetase deficiency, unexpectedly low in male OTC deficiency, and least frequently in GA1 and female OTC deficiency. For patients with MMA, propionic aciduria (PA) and OTC deficiency (male and female), hyperammonemia was more severe in metabolic crises during than after the newborn period, whereas metabolic acidosis tended to be more severe in MMA and PA patients with late onset of symptoms. Symptomatic patients without metabolic crises (n = 94) often presented with a movement disorder, mental retardation, epilepsy and psychiatric disorders (the latter in UCD only).
Conclusions
The initial presentation varies widely in OAD and UCD patients. This is a challenge for rapid diagnosis and early start of treatment. Patients with a sepsis-like neonatal crisis and those with late-onset of symptoms are both at risk of delayed or missed diagnosis.




Similar content being viewed by others


Abbreviations
- ARG1:
-
Arginase 1
- ASL:
-
Argininosuccinate lyase
- ASS:
-
Argininosuccinate synthetase
- CPS1:
-
Carbamylphosphate synthetase 1
- E-HOD:
-
European network and registry for homocystinurias and methylation defects
- E-IMD:
-
European registry and network for intoxication type metabolic diseases
- GA1:
-
Glutaric aciduria type 1
- HHH:
-
Hyperornithinemia-hyperammonemia-homocitrullinuria
- IVA:
-
Isovaleric aciduria
- MMA:
-
Methylmalonic aciduria
- NAGS:
-
N-acetylglutamate synthase
- OAD:
-
Organic aciduria
- OTC:
-
Ornithine transcarbamylase
- PA:
-
Propionic aciduria
- Q:
-
Quartile
- QoL:
-
Quality of life
- UCD:
-
Urea cycle disorder
References
Urea Cycle Disorders Consortium of the Rare Diseases Clinical Research Network, Ah Mew N, Krivitzky L, McCarter R, Batshaw M, Tuchman M (2013) Clinical outcomes of neonatal onset proximal versus distal urea cycle disorders do not differ. J Pediatr 162:324–329
Members of the Urea Cycle Disorders Consortium, Batshaw ML, Tuchman M, Summar M, Seminara J (2014) A longitudinal study of urea cycle disorders. Mol Genet Metab 113:127–130
Baumgartner MR, Hörster F, Dionisi-Vici C et al (2014) Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis 9:130
Boy N, Haege G, Heringer J et al (2013) Low lysine diet in glutaric aciduria type I — effect on anthropometrical and biochemical follow-up parameters. J Inherit Metab Dis 36:525–533
Burgard P, Rupp K, Lindner M et al (2012) Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 2. From screening laboratory results to treatment, follow-up and quality assurance. J Inherit Metab Dis 35:613–625
Chandler RJ, Zerfas PM, Shanske S, Sloan J, Hoffmann V, DiMauro S, Venditti CP (2009) Mitochondrial dysfunction in mut methylmalonic acidemia. FASEB J 23:1252–1261
Chapman KA, Gropman A, MacLeod E et al (2012) Acute management of propionic acidemia. Mol Genet Metab 105:16–25
Cole TJ (1990) The LMS method for constructing normalized growth standards. Eur J Clin Nutr 44:45–60
Cole TJ, Freeman JV, Preece MA (1998) British 1990 growth reference centiles for weight, height, body mass index and head circumference fitted by maximum penalized likelihood. Stat Med 17:407–429
Cole TJ, Williams AF, Wright CM (2011) Revised birth centiles for weight, length and head circumference in the UK-WHO growth charts. Ann Hum Biol 38:7–11
DeBrosse, Okajima K, Zhang S et al (2012) Spectrum of neurological and survival outcomes in pryruvate dehydrogenase complex (PDC) deficiency: lack of correlation with genotype. Mol Genet Metab 107:394–402
de Keyzer Y, Valayannopoulos V, Benoist JF et al (2009) Multiple OXPHOS deficiency in the liver, kidney, heart, and skeletal muscle of patients with methylmalonic and propionic aciduria. Pediatr Res 66:91–95
Dionisi-Vici C, Deodato F, Röschinger W, Rhead W, Wilcken B (2006) 'Classical' organic acidurias, propionic aciduria, methylmalonic aciduria and isovaleric aciduria: long-term outcome and effects of expanded newborn screening using tandem mass spectrometry. J Inherit Metab Dis 29:383–389
Engelhardt B, Liebner S (2014) Novel insights into the development and maintenance of the blood–brain barriers. Cell Tissue Res 355:687–699
Enns GM, Berry SA, Berry BT, Rhead WJ, Brusilow SW, Hamosh A (2007) Survival after treatment with phenylacetate and benzoate for urea cycle disorders. N Engl J Med 356:2282–2292
Gabbe SG, Landon MB, Warren-Boulton E, Fradkin J (2012) Promoting health after gestational diabetes: a national diabetes education program call to action. Obstet Gynecol 119:171–176
Gallagher RC, Lam C, Wong D, Cederbaum S, Sokol RJ (2014) Significant hepatic involvement in patients with ornithine transcarbamylase deficiency. J Pediatr 164:720–725
Grünert SC, Wendel U, Lindner M et al (2012) Clinical and neurocognitive outcome in symptomatic isovaleric acidemia. Orphanet J Rare Dis 7:9
Grünert SC, Müllerleile S, De Silva L et al (2013) Propionic acidemia: clinical course and outcome in 55 pediatric and adolescent patients. Orphanet J Rare Dis 8:6
Gutiérrez Junquera C, Balmaseda E et al (2009) Acute liver of pregnancy and neonatal long-chain 3-hydroxyacyl-coenzyme A dehydrogenase (LCHAD) deficiency. Eur J Pediatr 168:103–106
Häberle J, Burlina A, Chakrapani A et al (2012) Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis 7:32
Harting I, Neumaier-Probst E, Seitz A et al (2009) Dynamic changes of striatal and extrastriatal abnormalities in glutaric aciduria type I. Brain 132:1764–1782
Heringer J, Boy SP, Ensenauer R et al (2010) Use of guidelines improves the neurological outcome in glutaric aciduria type I. Ann Neurol 68:743–752
Hoffmann GF, Athanassopoulos S, Burlina AB et al (1996) Clinical course, early diagnosis, treatment, and prevention of disease in glutaryl-CoA dehydrogenase deficiency. Neuropediatrics 27:115–123
Hörster F, Garbade SF, Zwickler T et al (2009) Prediction of outcome in isolated methylmalonic aciduria: combined use of clinical and biochemical parameters. J Inherit Metab Dis 32:630–639
Kido J, Nakamura K, Mitsubuchi H et al (2012) Long-term outcome and intervention of urea cycle disorders in Japan. J Inherit Metab Dis 35:777–785
Kölker S, Köhr G, Ahlemeyer B et al (2002) Ca2+ and Na + dependence of 3-hydroxyglutarate-induced excitotoxicity in primary neuronal cultures from chick embryo telencephalons. Pediatr Res 52:199–206
Kölker S, Garbade SF, Greenberg CR et al (2006) Natural history, outcome, and therapeutic efficacy in children and adults with glutaryl-CoA dehydrogenase deficiency. Pediatr Res 59:840–847
Kölker S, Garbade SF, Boy N et al (2007) Decline of acute encephalopathic crises in children with glutaryl-CoA dehydrogenase deficiency identified by newborn screening in Germany. Pediatr Res 62:357–362
Kölker S, Christensen E, Leonard JV et al (2011) Diagnosis and management of glutaric aciduria type I — revised recommendations. J Inherit Metab Dis 34:677–694
Kölker S, Burgard P, Sauer SW, Okun JG (2013) Current concepts in organic acidurias: understanding intra- and extracerebral disease manifestation. J Inherit Metab Dis 36:635–644
Kölker S, Dobbelaere D, Häberle J et al (2015) Networking across borders for individuals with organic acidurias and urea cycle disorders: the E-IMD consortium. J Inherit Metab Dis in press
Lamp J, Keyser B, Koeller DM, Ullrich K, Braulke T, Mühlhausen C (2011) Glutaric aciduria type 1 metabolites impair the succinate transport from astrocytic to neuronal cells. J Biol Chem 286:17777–17784
Leonard JV, Vijayaraghavan S, Walter JH (2003) The impact of screening for propionic and methylmalonic acidaemia. Eur J Pediatr 162(Suppl 1):S21–S24
Liu J, Gallagher AE, Carta CM, Torres ME, Moran R, Wilcox S (2014) Racial differences in gestational weight gain and pregnancy-related hypertension. Ann Epidemiol 24:441–447
Loeber JG, Burgard P, Cornel MC et al (2012) Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 1. From blood spot to screening result. J Inherit Metab Dis 35:603–611
McClelland VM, Bakalinova DB, Hendriksz C, Singh RP (2009) Glutaric aciduria type 1 presenting with epilepsy. Dev Med Child Neurol 51:235–239
Mustafa A, Clarke JT (2006) Ornithine transcarbamylase deficiency presenting with acute liver failure. J Inherit Metab Dis 29:586
Nassogne MC, Héron B, Touati G, Rabier D, Saudubray JM (2005) Urea cycle defects: management and outcome. J Inherit Metab Dis 28:407–414
Nettesheim S, Häberle J, Karall D et al (2013) Neu diagnostizierte Harnstoffzyklusdefekte bei Patienten unter 16 Jahren. Monatsschr Kinderheilkd 161(Suppl 2):166, abstract
Okun JG, Hörster F, Farkas LM et al (2002) Neurodegeneration in methylmalonic aciduria involves inhibition of complex II and tricarboxylic acid cycle, and synergistically acting excitotoxicity. J Biol Chem 277:14674–14680
Olsen RK, Andresen BS, Christensen E, Bross P, Skovby F, Gregersen N (2003) Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl-CoA dehydrogenation deficiency. Hum Mutat 22:12–23
Pena L, Franks J, Chapman KA et al (2012) Natural history of propionic acidemia. Mol Genet Metab 105:5–9
R Core Team (2014) R: A language and environment for statistical computing. R foundation for statistical computing. Vienna, Austria, http://CRAN.R-project.org/
Richter SJ, McCann MH (2007) Multiple Comparison of Medians Using Permutation Tests. J Mod Appl Stat Methods 6:399–412
Rüegger CM, Lindner M, Ballhausen et al (2014) Cross-sectional observational study of 208 patients with non-classical urea cycle disorders. J Inherit Metab Dis 37:21–30
Sachs L, Hedderich J (2012) Angewandte Statistik. Springer, Heidelberg
Sauer SW, Okun JG, Schwab MA et al (2005) Bioenergetics in glutaryl-coenzyme A dehydrogenase deficiency: a role for glutaryl-coenzyme A. J Biol Chem 280:21830–21836
Sauer SW, Okun JG, Fricker G et al (2006) Intracerebral accumulation of glutaric and 3-hydroxyglutaric acids secondary to limited flux across the blood–brain barrier constitute a biochemical risk factor for neurodegeneration in glutaryl-CoA dehydrogenase deficiency. J Neurochem 97:899–910
Sauer SW, Opp S, Mahringer A et al (2010) Glutaric aciduria type I and methylmalonic aciduria: simulation of cerebral import and export of accumulating neurotoxic dicarboxylic acids in in vitro models of the blood–brain barrier and the choroid plexus. Biochim Biophys Acta 1802:552–560
Schwab MA, Sauer SW, Okun JG et al (2006) Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogenous mitochondrial toxins. Biochem J 398:107–112
Smucker MD, Allan J, Carterette B (2007) A comparison of statistical significance tests for information retrieval evaluation. CIKM’07 Proceedings of the sixteenth ACM conference on information and knowledge management: 623–632
Strauss AW, Bennett MJ, Rinaldo P et al (1999) Inherited long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency and a fetal-maternal interaction cause maternal liver disease and other pregnancy complications. Semin Perinatol 23:100–112
Strauss KA, Lazovic J, Wintermark M, Morton DH (2007) Multimodal imaging of striatal degeneration in Amish patients with glutaryl-CoA dehydrogenase deficiency. Brain 130:1905–1920
Strauss KA, Donnelly P, Wintermark M (2010) Cerebral hemodynamics in patients with glutaryl-coenzyme A dehydrogenase deficiency. Brain 133:76–92
Summar ML, Dobbelaere D, Brusilow S, Lee B (2008) Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicenter study of acute hyperammaemic episodes. Acta Paediatr 97:1420–1425
Summar ML, Koelker S, Freedenberg D et al (2013) The incidence of urea cycle disorders. Mol Genet Metab 110:179–180
Sutton VR, Chapman KA, Gropman AL et al (2012) Chronic management and health supervision of individuals with propionic acidemias. Mol Genet Metab 105:26–33
Tavares MV, Santos MJ, Domingues AP et al (2013) Antenatal manifestation of mitochondrial disorders. J Inherit Metab Dis 36:805–811
Teufel U, Weitz J, Flechtenmacher C et al (2011) High urgency liver transplantation in ornithine transcarbamylase deficiency presenting with acute liver failure. Pediatr Transpl 15:E110–E115
Van der Meer SB, Poggi F, Spada M et al (1994) Clinical outcome of long-term management of patients with vitamin B12-unresponsive methylmalonic acidemia. J Pediatr 125:903–908
Vockley J, Ensenauer R (2006) Isovaleric acidemia: new aspects of genetic and phenotypic heterogeneity. Am J Med Genet C: Semin Med Genet 142:95–103
Whitfield, Hurst D, Bennett MJ, Sherwood WG, Hogg R, Gonsoulin W (1996) Fetal polycystic kidney disease associated with glutaric acidurias type II: an inborn error of energy metabolism. Am J Perinatol 13:131–134
Zwickler T, Haege G, Riderer A, Hörster F, Hoffmann GF, Burgard P, Kölker S (2012) Metabolic decompensation in methylmalonic aciduria: which biochemical parameters are discriminative? J Inherit Metab Dis 35:797–806
Zwickler T, Riderer A, Haege G, Hoffmann GF, Kölker S, Burgard P (2014) Usefulness of biochemical parameters in decision-making on the start of emergency treatment in patients with propionic acidemia. J Inherit Metab Dis 37:31–37
Acknowledgments
We are indebted to all patients and their families who have been willing to contribute to this study, to share their experience on living with a rare disease, and for their trust, and we thank all colleagues very much for their contribution to the project. We are grateful for fruitful collaboration with the following clinical partners, patient support groups and industrial partners (in alphabetical order of countries): Lut de Baere, Nathalie Stroobant (Belgische Organisatie voor Kinderen en Volwassenen met een Stofwisselingsziekte VZW [BOKS], Belgium), Nela Carić (Hrvatska udruga za rijetke bolesti, Croatia), Tomas Honzik (Charles University and General University of Prague, First Faculty of Medicine, Prague, Czech Republic), Annika and Kennet Rovsing (PND - Protein Nedbrydnings Defekt Foreningen, Denmark), Samantha Parker (Orphan Europe SARL, France), EURORDIS, European Organisation for Rare Disease (France), Eric Bauchart (Hôpital Necker-Enfants Malades, Assistance Publique-Hôpitaux de Paris, Reference Center for Inherited Metabolic Disease, Necker-Enfants Malades University Hospital and IMAGINE Institute, Paris, France), Markus Ott, Beate Szczerbak (Nutricia Metabolics GmbH, Germany), Hubertus von Voss, Raimund Schmid (Kindernetzwerk e.V., Germany), Mandy Kretschmer (Glutarazidurie e.V., Germany), Reinhild Link (Wiesbaden, representing the SSIEM Dieticians Group), Harikleia Ioannou (University A’Pediatric Department, Metabolic Laboratory, ‘Hippocration’ General Hospital of Thessaloniki), Zarifis Dimitroulis (KRIKOS ZOIS – Society for patients and friends of patients with inherited metabolic diseases), Evridiki Drogari (University of Athens, Aghia Sophia Children's Hospital, Unit of Metabolic Diseases, Athens), Renza Barbon Galluppi (UNIAMO FIMR, Italy), Susan Udina (COMETA ASMME – Associazione Studio Malattie Metaboliche Ereditarie – ONLUS, Italy), Hanka Meutgeert (Volwassenen en Kinderen met Stoffwisselingsziekten [VKS], Netherlands), Vanessa Ferreira (Associação Portuguesa CDG, Portugal), Miguel Macedo (Apofen, Portugal), Sérgio Braz Antão (Rarrisimas, Portugal), Sergi Faber (Catalana de Trastorns Metabòlics Hereditaris, Spain), Sofia Nordin (Svedish Orphan Biovitrium AB [SOBI], Sweden), and Steven Hannigan (CLIMB, Children Living with Inherited Metabolic Diseases, National Information Centre for Metabolic Diseases, and EMDA, the European Metabolic Disorders Alliance).
This publication arises from the project “European registry and network for intoxication type metabolic diseases” (E-IMD; EAHC no 2010 12 01) which has received funding from the European Union, in the framework of the Health Programme. After the end of the EU funding period the E-IMD patient registry will be sustained by funding from the Kindness-for-Kids Foundation (Munich, Germany).
M. Baumgartner, J. Häberle and A. Laemmle (Zurich, Switzerland) are supported by radiz – Rare Disease Initiative Zurich, a clinical research priority programme of the University of Zurich.
Drs Murphy and Lachmann were supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
Compliance with ethics guidelines
ᅟ
Conflict of interest
none.
Human and animal rights and informed consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human studies (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients or their legal guardians prior to being included in the study in countries where this was needed by law.
This article does not contain animal subjects.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: K. Michael Gibson
For a complete list of affiliations of all authors see end of this article.
Rights and permissions
About this article

Cite this article
Kölker, S., Cazorla, A.G., Valayannopoulos, V. et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: the initial presentation. J Inherit Metab Dis 38, 1041–1057 (2015). https://doi.org/10.1007/s10545-015-9839-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-015-9839-3