
Abstract
Insufficient data on the origins of the first introduced propagule and the initial stages of invasion complicate the reconstruction of a species’ invasion history. Phylogeography of the native area profoundly shapes the genomic patterns of the propagules on which subsequent demographic processes of the invasion are based. Thus, a better understanding of this aspect helps to disentangle native and invasive histories. Here, we used genomic data together with clustering methods, explicit admixture tests combined with ABC models and Machine Learning algorithms, to compare patterns of genetic structure and gene flow of native and introduced populations, and infer the most likely invasion pathways of the highly invasive freshwater fish Pseudorasbora parva. This species is the vector of a novel lethal fungal-like pathogen (Sphaerothecum destruens) that is responsible for the decline of several fish species in Europe. We found that the current genetic structuring in the native range of P. parva has been shaped by waves of gene flow from populations in southern and northern China. Furthermore, our results strongly suggest that the genetic diversity of invasive populations results from recurrent global invasion pathways of admixed native populations. Our study also illustrates how the combination of admixture tests, ABC and Machine Learning can be used to detect high-resolution demographic signatures and reconstruct an integrative biological invasion history.


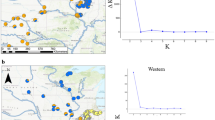
source populations predicted for the invasive range. a (Native range, Asia) Mean admixture proportions estimated with STRUCTURE in the native range (Asia), for the chosen K = 6. Native sampled sites are pooled into putative demes for subsequent analyses. Pie chart colors correspond to the genetic clusters inferred by STRUCTURE, as in Fig. 1. (Invasive range, Europe & Middle-East) Assignment predictions of invasive individuals to native demes with AssignPOP’s SVM algorithm with a relative posterior probability > 2 (n = 292 for training, n = 100 for predictions). Pie chart colors correspond to the putative demes defined in the native range to which invasive individuals were assigned. b Posterior assignment probabilities to putative native demes estimated with AssignPop. c The demo-genetic scenario that was inferred with Approximate Bayesian Computation demonstrating three independent introductions from three independent admixed source populations. Branch lengths are not scaled. Bottleneck events are represented in thin red lines in branches. Colored branches correspond to invasive demes history

Similar content being viewed by others

References
Andreou D, Gozlan RE (2016) Associated disease risk from the introduced generalist pathogen Sphaerothecum destruens: management and policy implications. Parasitology 143:1204–1210. https://doi.org/10.1017/S003118201600072X
Aparicio E, Peris B, Torrijos L, Prenda J, Nieva A, Perea S (2012) Expansion of the invasive Pseudorasbora parva (Cyprinidae) in the Iberian Peninsula: first record in the Guadiana River basin. Cybium 36:585–586
Arnold A (1985) Pseudorasbora parva (Schlegel 1842) nun auch in der DDR. Z Binnenfisch DDR 32:182–183
Baltazar Soares M, Blanchet S, Cote J, Tarkan AS, Záhorská E, Gozlan RE, Eizaguirre C (2020) Genomic footprints of a biological invasion: Introduction from Asia and dispersal in Europe of the topmouth gudgeon (Pseudorasbora parva). Mol Ecol 29:71–85
Barker BS, Cocio JE, Anderson SR, Braasch JE, Cang FA, Gillette HD et al (2019) Potential limits to the benefits of admixture during biological invasion. Mol Ecol 28:100–113. https://doi.org/10.1111/mec.14958
Barnes GL (2003) Origins of the Japanese Islands: The New “Big Picture”. Nichibunken Jpn Rev 3–50
Beaumont MA, Zhang W, Balding DJ (2002) Approximate bayesian computation in population genetics. Genetics 162:2025–2035
Becker RA, Wilks AR, Brownrigg R, Minka TP, Deckmyn A (2018) Maps: draw geographical maps. R package version 3.3. 0. https://CRAN.R-project.org/package=maps
Beichman AC, Huerta-Sanchez E, Lohmueller KE (2018) Using genomic data to infer historic population dynamics of nonmodel organisms. Annu Rev Ecol Evol Syst 49:433–456. https://doi.org/10.1146/annurev-ecolsys-110617-062431
Blum MG, François O (2010) Non-linear regression models for approximate bayesian computation. Stat Comput 20:63–73. https://doi.org/10.1007/s11222-009-9116-0
Bossdorf O, Auge H, Lafuma L, Rogers WE, Siemann E, Prati D (2005) Phenotypic and genetic differentiation between native and introduced plant populations. Oecologia 144:1–11. https://doi.org/10.1007/s00442-005-0070-z
Brandley MC, Guiher TJ, Pyron RA, Winne CT, Burbrink FT (2010) Does dispersal across an aquatic geographic barrier obscure phylogeographic structure in the diamond-backed watersnake (Nerodia rhombifer)? Mol Phylogenetics Evol 57:552–560. https://doi.org/10.1016/j.ympev.2010.07.015
Britton JR, Gozlan RE (2013) Geo-politics and freshwater fish introductions: how the cold war shaped Europe’s fish allodiversity. Glob Environ Change 23:1566–1574. https://doi.org/10.1016/j.gloenvcha.2013.09.017
Browning SR, Browning BL (2007) Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 81:1084–1097. https://doi.org/10.1086/521987
Browning BL, Zhou Y, Browning SR (2018) A one-penny imputed genome from next-generation reference panels. Am J Hum Genet 103:338–348. https://doi.org/10.1016/j.ajhg.2018.07.015
Cabrera AA, Palsbøll PJ (2017) Inferring past demographic changes from contemporary genetic data: a simulation based evaluation of the ABC methods implemented in DIYABC. Mol Ecol Resour 17:e94–e110. https://doi.org/10.1111/1755-0998.12696
Caiola N, De Sostoa A (2002) First record of the Asiatic cyprinid Pseudorasbora parva in the Iberian Peninsula. J Fish Biol 4:1058–1060. https://doi.org/10.1006/jfbi.2002.2103
Carosi A, Ghetti L, Lorenzoni M (2016) Status of Pseudorasbora parva in the Tiber river basin (Umbria, central Italy) 20 years after its introduction. Knowl Manag Aquat Ecosyst 417:22. https://doi.org/10.1051/kmae/2016009
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140. https://doi.org/10.1111/mec.12354
Chan KO, Alexander AM, Grismer LL, Su Y-C, Grismer JL, Quah ESH, Brown RM (2017) Species delimitation with gene flow: a methodological comparison and population genomics approach to elucidate cryptic species boundaries in Malaysian Torrent Frogs. Mol Ecol 26:5435–5450
Chapin Iii FS, Zavaleta ES, Eviner VT, Naylor RL, Vitousek PM, Reynolds HL et al (2000) Consequences of changing biodiversity. Nature 405:234–242. https://doi.org/10.1038/35012241
Chen KY, Marschall EA, Sovic MG, Fries AC, Gibbs HL, Ludsin SA (2018) Assign POP: an r package for population assignment using genetic, non-genetic, or integrated data in a machine-learning framework. Methods Ecol Evol 9:439–446. https://doi.org/10.1111/2041-210X.12897
Chiang TY, Lin HD, Zhao J, Kuo PH, Lee TW, Hsu KC (2013) Diverse processes shape deep phylogeographical divergence in Cobitis sinensis (Teleostei: Cobitidae) in East Asia. J Zool Syst Evol Res 51:316–326. https://doi.org/10.1111/jzs.12030
Clavero M, García-Berthou E (2005) Invasive species are a leading cause of animal extinctions. Trends Ecol Evol 20:110. https://doi.org/10.1016/j.tree.2005.01.003
Combe M, Gozlan RE (2018) The rise of the rosette agent in Europe: an epidemiological enigma. Transbound Emerg Dis 65:1474–1481. https://doi.org/10.1111/tbed.13001
Combe M, Cherif E, Charrier A, Barbey B, Chague M, Carrel G et al (2022) Towards unravelling the Rosette agent enigma: spread and emergence of the co-invasive host-pathogen complex. Pseudorasbora Parva-Sphaerothecum Destruens Sci Total Environ 806:150427. https://doi.org/10.1016/j.scitotenv.2021.150427
Cornuet JM, Pudlo P, Veyssier J, Dehne-Garcia A, Gautier M, Leblois R et al (2014) DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism. DNA Seq Microsatellite Data Bioinf 30:1187–1189. https://doi.org/10.1093/bioinformatics/btt763
Côte J, Roussel JM, Le Cam S, Evanno G (2014) Outbreeding depression in Atlantic salmon revealed by hypoxic stress during embryonic development. Evol Biol 41:561–571. https://doi.org/10.1007/s11692-014-9289-0
Crispo E, Moore JS, Lee-Yaw JA, Gray SM, Haller BC (2011) Broken barriers: human-induced changes to gene flow and introgression in animals: an examination of the ways in which humans increase genetic exchange among populations and species and the consequences for biodiversity. BioEssays 33:508–518. https://doi.org/10.1002/bies.201000154
Cristescu ME (2015) Genetic reconstructions of invasion history. Mol Ecol 24:2212–2225. https://doi.org/10.1111/mec.13117
Crowl TA, Crist TO, Parmenter RR, Belovsky G, Lugo AE (2008) The spread of invasive species and infectious disease as drivers of ecosystem change. Front Ecol Environ 6:238–246. https://doi.org/10.1890/070151
Csilléry K, François O, Blum MG (2012) abc: an R package for approximate Bayesian computation (ABC). Methods Ecol Evol 3:475–479. https://doi.org/10.1111/j.2041-210X.2011.00179.x
Didham RK, Tylianakis JM, Hutchison MA, Ewers RM, Gemmell NJ (2005) Are invasive species the drivers of ecological change? Trends Ecol Evol 20:470–474. https://doi.org/10.1016/j.tree.2005.07.006
Dlugosch KM, Parker IM (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17:431–449
Dong Y, Zhang G, Neubauer F, Liu X, Genser J, Hauzenberger C (2011) Tectonic evolution of the Qinling orogen, China: review and synthesis. J Asian Earth Sci 41:213–237. https://doi.org/10.1016/j.jseaes.2011.03.002
Durand EY, Patterson N, Reich D, Slatkin M (2011) Testing for ancient admixture between closely related populations. Mol Biol Evol 28:2239–2252. https://doi.org/10.1093/molbev/msr048
Earl DA, vonHoldt BM (2012) Structure harvester: a website and program for visualizing structure output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Ekmekçi FG, Kirankaya ŞG (2006) Distribution of an invasive fish species, Pseudorasbora parva (Temminck & Schlegel, 1846) in Turkey. Turk J Zool 30:329–334
Estoup A, Guillemaud T (2010) Reconstructing routes of invasion using genetic data: Why, how and so what? Mol Ecol 19:4113–4130. https://doi.org/10.1111/j.1365-294X.2010.04773.x
Estoup A, Lombaert E, Marin JM, Guillemaud T, Pudlo P, Robert CP, Cornuet JM (2012) Estimation of demo-genetic model probabilities with approximate Bayesian computation using linear discriminant analysis on summary statistics. Mol Ecol Resour 12:846–855. https://doi.org/10.1111/j.1755-0998.2012.03153.x
Estoup A, Ravigné V, Hufbauer R, Vitalis R, Gautier M, Facon B (2016) Is there a genetic paradox of biological invasion? Annu Rev Ecol Evol Syst 47:51–72. https://doi.org/10.1146/annurev-ecolsys-121415-032116
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Heckel G (2006) Computer programs for population genetics data analysis: a survival guide. Nat Rev Genet 7:745–758
Facon B, Genton BJ, Shykoff J, Jarne P, Estoup A, David P (2006) A general eco-evolutionary framework for understanding bioinvasions. Trends Ecol Evol 21:130–135. https://doi.org/10.1016/j.tree.2005.10.012
Fengshu Z (1990) Preliminary study of geological geomorphological conditions of ling canal. Carsologica Sin 1
Fitzpatrick BM, Fordyce JA, Niemiller ML, Reynolds RG (2012) What can DNA tell us about biological invasions? Biol Invasions 14:245–253
Fonseca DM, Widdel AK, Hutchinson M, Spichiger SE, Kramer LD (2010) Fine-scale spatial and temporal population genetics of Aedes japonicus, a new US mosquito, reveal multiple introductions. Mol Ecol 19:1559–1572. https://doi.org/10.1111/j.1365-294X.2010.04576.x
Gallardo B, Clavero M, Sánchez MI, Vilà M (2016) Global ecological impacts of invasive species in aquatic ecosystems. Glob Chang Biol 22:151–163. https://doi.org/10.1111/gcb.13004
Ganjali Z, Esmaeili HR, Zarei F, Sayyadzadeh G, Eagderi S, Gozlan RE (2020) West Asian colonisation of topmouth gudgeon, Pseudorasbora parva (Teleostei: Gobionidae): genetic admixture at the crossroad of Europe and east Asia. Freshw Biol 66:699–715. https://doi.org/10.1111/fwb.13671
Genton BJ, Shykoff JA, Giraud T (2005) High genetic diversity in French invasive populations of common ragweed, Ambrosia artemisiifolia, as a result of multiple sources of introduction. Mol Ecol 14:4275–4285. https://doi.org/10.1111/j.1365-294X.2005.02750.x
Gillis NK, Walters LJ, Fernandes FC, Hoffman EA (2009) Higher genetic diversity in introduced than in native populations of the mussel Mytella charruana: evidence of population admixture at introduction sites. Divers Distrib 15:784–795. https://doi.org/10.1111/j.1472-4642.2009.00591.x
Gong M, Tu F (1991) Fishery in contemporary China. Contemporary China Press, Beijing
Goudet J (2005) Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol Ecol Notes 5:184–186. https://doi.org/10.1111/j.1471-8278.2004.00828.x
Goudet J, Jombart T (2015) Hierfstat: estimation and tests of hierarchical F-statistics. R package version 0.04–22. 10
Gozlan RE (2008) Introduction of non-native freshwater fish: is it all bad? Fish Fish 9:106–115. https://doi.org/10.1111/j.1467-2979.2007.00267.x
Gozlan RE, Pinder AC, Shelley J (2002) Occurrence of the Asiatic cyprinid Pseudorasbora parva in England. J Fish Biol 61:298–300. https://doi.org/10.1006/jfbi.2002.2042
Gozlan RE, Andreou D, Asaeda T, Beyer K, Bouhadad R, Burnard D et al (2010) Pan-continental invasion of Pseudorasbora parva: towards a better understanding of freshwater fish invasions. Fish Fish 11:315–340. https://doi.org/10.1111/j.1467-2979.2010.00361.x
Gozlan RE, Záhorská E, Cherif E, Asaeda T, Britton JR, Chang CH et al (2020) Native drivers of fish life history traits are lost during the invasion process. Ecol Evol 10:8623–8633. https://doi.org/10.1002/ece3.6521
Gozlan RE (2012) Pseudorasbora parva temminck and schlegel (topmouth gudgeon). Handb glob freshw invasive species. Earthscan, Abingdon
Graebner RC, Callaway RM, Montesinos D (2012) Invasive species grows faster, competes better, and shows greater evolution toward increased seed size and growth than exotic non-invasive congeners. Plant Ecol 213:545–553. https://doi.org/10.1007/s11258-012-0020-x
Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M et al (2010) A draft sequence of the Neandertal genome. Science 328:710–722. https://doi.org/10.1126/science.1188021
Guillemaud T, Ciosi M, Lombaert E, Estoup A (2011) Biological invasions in agricultural settings: insights from evolutionary biology and population genetics. C R Biol 334:237–246. https://doi.org/10.1016/j.crvi.2010.12.008
Guivier E, Gilles A, Pech N, Duflot N, Tissot L, Chappaz R (2019) Canals as ecological corridors and hybridization zones for two cyprinid species. Hydrobiologia 830(1):1–16. https://doi.org/10.1007/s10750-018-3843-1
Hardouin EA, Andreou D, Zhao Y, Chevret P, Fletcher DH, Britton JR, Gozlan RE (2018) Reconciling the biogeography of an invader through recent and historic genetic patterns: the case of topmouth gudgeon Pseudorasbora parva. Biol Invasions 20(8):2157–2171. https://doi.org/10.1007/s10530-018-1693-4
Hasselman DJ, Argo EE, McBride MC, Bentzen P, Schultz TF, Perez-Umphrey AA, Palkovacs EP (2014) Human disturbance causes the formation of a hybrid swarm between two naturally sympatric fish species. Mol Ecol 23:1137–1152. https://doi.org/10.1111/mec.12674
Hegediš A, Lenhardt M, Mićković B, Cvijanović G, Jarić I, Gačić Z (2007) Amur sleeper (Perccottus glenii Dubowski, 1877) spreading in the Danube River basin. J Appl Ichthyol 23:705–706. https://doi.org/10.1111/j.1439-0426.2007.00867.x
Holsbeek G, Mergeay J, Hotz H, Plötner J, Volckaert FAM, De Meester L (2008) A cryptic invasion within an invasion and widespread introgression in the European water frog complex: consequences of uncontrolled commercial trade and weak international legislation. Mol Ecol 17:5023–5035. https://doi.org/10.1111/j.1365-294X.2008.03984.x
Huang H, Knowles LL (2016) Unforeseen consequences of excluding missing data from next-generation sequences: simulation study of RAD sequences. Syst Biol 65:357–365. https://doi.org/10.1093/sysbio/syu046
Huff DD, Miller LM, Chizinski CJ, Vondracek B (2011) Mixed-source reintroductions lead to outbreeding depression in second-generation descendents of a native North American fish. Mol Ecol 20:4246–4258. https://doi.org/10.1111/j.1365-294X.2011.05271.x
Hufford KM, Krauss SL, Veneklaas EJ (2012) Inbreeding and outbreeding depression in Stylidium hispidum: implications for mixing seed sources for ecological restoration. Ecol Evol 2:2262–2273. https://doi.org/10.1002/ece3.302
Hulme PE (2009) Trade, transport and trouble: managing invasive species pathways in an era of globalization. J Appl Ecol 46:10–18. https://doi.org/10.1111/j.1365-2664.2008.01600.x
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806. https://doi.org/10.1093/bioinformatics/btm233
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jombart T, Ahmed I (2011) Adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071. https://doi.org/10.1093/bioinformatics/btr521
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. https://doi.org/10.7717/peerj.281
Kamvar ZN, Brooks JC, Grünwald NJ (2015) Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front Genet 6:208. https://doi.org/10.3389/fgene.2015.00208
Karsten M, Jansen van Vuuren B, Addison P, Terblanche JS (2015) Deconstructing intercontinental invasion pathway hypotheses of the Mediterranean fruit fly (Ceratitis capitata) using a Bayesian inference approach: are port interceptions and quarantine protocols successfully preventing new invasions? Divers Distrib 21:813–825. https://doi.org/10.1111/ddi.12333
Kolbe JJ, Glor RE, Schettino LR, Lara AC, Larson A, Losos JB (2004) Genetic variation increases during biological invasion by a Cuban lizard. Nature 431:177–181. https://doi.org/10.1038/nature02807
Konishi M, Hosoya K, Takata K (2003) Natural hybridization between endangered and introduced species of Pseudorasbora, with their genetic relationships and characteristics inferred from allozyme analyses. J Fish Biol 63:213–231. https://doi.org/10.1046/j.1095-8649.2003.00146.x
Konishi M, Sakano H, Iguchi KI (2009) Identifying conservation priority ponds of an endangered minnow, Pseudorasbora pumila, in the area invaded by Pseudorasbora parva. Ichthyol Res 56:346. https://doi.org/10.1007/s10228-009-0106-1
Lelek A, Köhler C (1989) Zustandsanalyse der fischartengemeinschaften im Rhein (1987–1988). Fischökologie 1:47–64
Leuven RS, van der Velde G, Baijens I, Snijders J, van der Zwart C, Lenders HR, de Bij Vaate A (2009) The river Rhine: a global highway for dispersal of aquatic invasive species. Biol Invasions 11:1989. https://doi.org/10.1007/s10530-009-9491-7
Li S (1981) Studies on zoogeographical divisions for fresh water fishes of China. Science Press
Li YL, Liu JX (2018) StructureSelector: a web-based software to select and visualize the optimal number of clusters using multiple methods. Mol Ecol Resour 18:176–177. https://doi.org/10.1111/1755-0998.12719
Lombaert E, Guillemaud T, Lundgren J, Koch R, Facon B, Grez A et al (2014) Complementarity of statistical treatments to reconstruct worldwide routes of invasion: the case of the Asian ladybird Harmonia axyridis. Mol Ecol 23:5979–5997. https://doi.org/10.1111/mec.12989
Mergeay J, Verschuren D, De Meester L (2005) Cryptic invasion and dispersal of an American Daphnia in East Africa. Limnol Oceanogr 50:1278–1283. https://doi.org/10.4319/lo.2005.50.4.1278
Milanesi M, Capomaccio S, Vajana E, Bomba L, Garcia JF, Ajmone-Marsan P, Colli L (2017) BITE: an R package for biodiversity analyses. BioRxiv. https://doi.org/10.1101/181610
Mooney HA, Cleland EE (2001) The evolutionary impact of invasive species. Proc Natl Acad Sci U S A 98:5446–5451. https://doi.org/10.1073/pnas.091093398
Muirhead JR, Gray DK, Kelly DW, Ellis SM, Heath DD, Macisaac HJ (2008) Identifying the source of species invasions: sampling intensity vs. genetic diversity. Mol Ecol 17:1020–1035. https://doi.org/10.1111/j.1365-294X.2008.03669.x
Nichols JT (1928) Chinese fresh-water fishes in the American Museum of Natural History's collections: a provisional check-list of the fresh-water fishes of China. Bull AMNH; v. 58, article 1
Nolte AW, Freyhof J, Stemshorn KC, Tautz D (2005) An invasive lineage of sculpins, Cottus sp. (Pisces, Teleostei) in the Rhine with new habitat adaptations has originated from hybridization between old phylogeographic groups. Proc R Soc Lond B Biol Sci 272:2379–2387. https://doi.org/10.1098/rspb.2005.3231
Nolte AW, Gompert Z, Buerkle CA (2009) Variable patterns of introgression in two sculpin hybrid zones suggest that genomic isolation differs among populations. Mol Ecol 18:2615–2627. https://doi.org/10.1111/j.1365-294X.2009.04208.x
Oikonomou A, Leprieur F, Leonardos ID (2014) Biogeography of freshwater fishes of the Balkan Peninsula. Hydrobiologia 738:205–220. https://doi.org/10.1007/s10750-014-1930-5
Olden JD, Poff NL, Douglas MR, Douglas ME, Fausch KD (2004) Ecological and evolutionary consequences of biotic homogenization. Trends Ecol Evol 19:18–24. https://doi.org/10.1016/j.tree.2003.09.010
Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y et al (2012) Ancient admixture in human history. Genetics 192:1065–1093. https://doi.org/10.1534/genetics.112.145037
Peischl S, Excoffier L (2015) Expansion load: recessive mutations and the role of standing genetic variation. Mol Ecol 24:2084–2094. https://doi.org/10.1111/mec.13154
Perdereau E, Dedeine F, Christidès JP, Dupont S, Bagnères AG (2011) Competition between invasive and indigenous species: an insular case study of subterranean termites. Biol Invasions 13:1457–1470. https://doi.org/10.1007/s10530-010-9906-5
Phillips BL, Brown GP, Webb JK, Shine R (2006) Invasion and the evolution of speed in toads. Nature 439:803–803. https://doi.org/10.1038/439803a
Pickrell JK, Pritchard JK (2012) Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet 8:e1002967. https://doi.org/10.1371/journal.pgen.1002967
Pickrell JK, Patterson N, Barbieri C, Berthold F, Gerlach L, Güldemann T et al (2012) The genetic prehistory of southern Africa. Nat Commun 3(1):1–6. https://doi.org/10.1038/ncomms2140
Plummer M, Best N, Cowles K, Vines K (2006) CODA: convergence diagnosis and output analysis for MCMC. R News 6:7–11
Price PW, Westoby M, Rice B, Atsatt PR, Fritz RS, Thompson JN, Mobley K (1986) Parasite mediation in ecological interactions. Annu Rev Ecol Syst 17:487–505
Price SJ, Garner TWJ, Cunningham AA, Langton TES, Nichols RA (2016) Reconstructing the emergence of a lethal infectious disease of wildlife supports a key role for spread through translocations by humans. Proc R Soc B 283:20160952. https://doi.org/10.1098/rspb.2016.0952
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/sysbio/sys038
Pudlo P, Marin JM, Estoup A, Cornuet JM, Gautier M, Robert CP (2016) Reliable ABC model choice via random forests. Bioinformatics 32:859–866. https://doi.org/10.1093/bioinformatics/btv684
Puechmaille SJ (2016) The program structure does not reliably recover the correct population structure when sampling is uneven: subsampling and new estimators alleviate the problem. Mol Ecol Resour 16:608–627. https://doi.org/10.1111/1755-0998.12512
Raynal L, Marin JM, Pudlo P, Ribatet M, Robert CP, Estoup A (2019) ABC random forests for Bayesian parameter inference. Bioinformatics 35:1720–1728. https://doi.org/10.1093/bioinformatics/bty867
Rius M, Darling JA (2014) How important is intraspecific genetic admixture to the success of colonising populations? Trends Ecol Evol 29:233–242. https://doi.org/10.1016/j.tree.2014.02.003
Rius M, Turon X (2020) Phylogeography and the description of geographic patterns in invasion genomics. Front Ecol Evol 8:595711. https://doi.org/10.3389/fevo.2020.595711
Roman J, Darling JA (2007) Paradox lost: genetic diversity and the success of aquatic invasions. Trends Ecol Evol 22:454–464. https://doi.org/10.1016/j.tree.2007.07.002
Salo P, Korpimäki E, Banks PB, Nordström M, Dickman CR (2007) Alien predators are more dangerous than native predators to prey populations. Proc R Soc Lond B Biol Sci 274:1237–1243. https://doi.org/10.1098/rspb.2006.0444
Sanz N, Araguas RM, Vidal O, Diez-del-Molino D, Fernández-Cebrián R, García-Marín JL (2013) Genetic characterization of the invasive mosquitofish (Gambusia spp.) introduced to Europe: population structure and colonization routes. Biol Invasions 15:2333–2346. https://doi.org/10.1007/s10530-013-0456-5
Sax DF, Stachowicz JJ, Brown JH, Bruno JF, Dawson MN, Gaines SD et al (2007) Ecological and evolutionary insights from species invasions. Trends Ecol Evol 22:465–471. https://doi.org/10.1016/j.tree.2007.06.009
Shafer AB, Gattepaille LM, Stewart RE, Wolf JB (2015) Demographic inferences using short-read genomic data in an approximate Bayesian computation framework: in silico evaluation of power, biases and proof of concept in Atlantic walrus. Mol Ecol 24:328–345. https://doi.org/10.1111/mec.13034
Simberloff D (2013) Biological invasions: much progress plus several controversies. Contrib Sci. https://doi.org/10.2436/20.7010.01.158
Simon A, Britton R, Gozlan R, Van Oosterhout C, Volckaert FA, Hänfling B (2011) Invasive cyprinid fish in Europe originate from the single introduction of an admixed source population followed by a complex pattern of spread. PLoS ONE 6:e18560. https://doi.org/10.1371/journal.pone.0018560
Simon A, Gozlan RE, Britton JR, Van Oosterhout C, Hänfling B (2015) Human induced stepping-stone colonisation of an admixed founder population: the spread of topmouth gudgeon (Pseudorasbora parva) in Europe. Aquat Sci 77:17–25. https://doi.org/10.1007/s00027-014-0374-3
Sinama M, Gilles A, Costedoat C, Corse E, Olivier JM, Chappaz R, Pech N (2013) Non-homogeneous combination of two porous genomes induces complex body shape trajectories in cyprinid hybrids. Front Zool 10:1–16. https://doi.org/10.1186/1742-9994-10-22
Skoglund P, Mallick S, Bortolini MC, Chennagiri N, Hünemeier T, Petzl-Erler ML et al (2015) Genetic evidence for two founding populations of the Americas. Nature 525:104–108. https://doi.org/10.1038/nature14895
South A (2017) Rnaturalearth: world map data from natural earth. R package version 0.1.0. https://CRAN.R-project.org/package=rnaturalearth
Stemshorn KC, Reed FA, Nolte AW, Tautz D (2011) Rapid formation of distinct hybrid lineages after secondary contact of two fish species (Cottus sp.). Mol Ecol 20:1475–1491. https://doi.org/10.1111/j.1365-294X.2010.04997.x
Stiers I, Crohain N, Josens G, Triest L (2011) Impact of three aquatic invasive species on native plants and macroinvertebrates in temperate ponds. Biol Invasions 13:2715–2726. https://doi.org/10.1007/s10530-011-9942-9
Tange O (2011) Gnu parallel-the command-line power tool. USENIX Mag 36:42–47
Team RC (2019) R: a language and environment for statistical computing (version 3.1.2) Vienna, Austria. R Foundation for Statistical Computing; 2014
Vallejo-Marín M, Friedman J, Twyford AD, Lepais O, Ickert-Bond SM, Streisfeld M et al (2021) Population genomic and historical analysis suggests a global invasion by bridgehead processes in Mimulus guttatus. Commun Biol 4:1–12. https://doi.org/10.1038/s42003-021-01795-x
van Boheemen LA, Lombaert E, Nurkowski KA, Gauffre B, Rieseberg LH, Hodgins KA (2017) Multiple introductions, admixture and bridgehead invasion characterize the introduction history of Ambrosia artemisiifolia in Europe and Australia. Mol Ecol 26:5421–5434. https://doi.org/10.1111/mec.14293
Vandelannoote A, Yseboodt R (1998) Atlas van de Vlaamse beek-en riviervissen. Water-Energik-vLario
Verhoeven KJ, Macel M, Wolfe LM, Biere A (2011) Population admixture, biological invasions and the balance between local adaptation and inbreeding depression. Proc R Soc Lond B Biol Sci 278:2–8. https://doi.org/10.1098/rspb.2010.1272
Vilcinskas A (2015) Pathogens as biological weapons of invasive species. PLoS Pathog 11:e1004714. https://doi.org/10.1371/journal.ppat.1004714
Villéger S, Blanchet S, Beauchard O, Oberdorff T, Brosse S (2011) Homogenization patterns of the world’s freshwater fish faunas. Proc Natl Acad Sci U S A 108:18003–18008. https://doi.org/10.1073/pnas.1107614108
Watanabe K, Iguchi KI, Hosoya K, Nishida M (2000) Phylogenetic relationships of the Japanese minnows, Pseudorasbora (Cyprinidae), as inferred from mitochondrial 16S rRNA gene sequences. Ichthyol Res 47:43–50. https://doi.org/10.1007/BF02674312
Weber E (1984) Die ausbreitung der pseudokeilfleckbarben im donauraum. Österreichs Fischerei 37:63–65
Weiss S, Persat H, Eppe R, Schlötterer C, Uiblein F (2002) Complex patterns of colonization and refugia revealed for European grayling Thymallus thymallus, based on complete sequencing of the mitochondrial DNA control region. Mol Ecol 11:1393–1407. https://doi.org/10.1046/j.1365-294x.2002.01544.x
Wolter C, Röhr F (2010) Distribution history of non-native freshwater fish species in Germany: how invasive are they? J Appl Ichthyol 26:19–27. https://doi.org/10.1111/j.1439-0426.2010.01505.x
Yang JQ, Hsu KC, Liu ZZ, Su LW, Kuo PH, Tang WQ et al (2016) The population history of Garra orientalis (Teleostei: Cyprinidae) using mitochondrial DNA and microsatellite data with approximate Bayesian computation. BMC Evol Biol 16:73. https://doi.org/10.1186/s12862-016-0645-9
Yuan JH, Cheng FY, Zhou SL (2012) Genetic structure of the tree peony (Paeonia rockii) and the Qinling mountains as a geographic barrier driving the fragmentation of a large population. PLoS ONE 7:e34955. https://doi.org/10.1371/journal.pone.0034955
Záhorská E, Kováč V (2009) Reproductive parameters of invasive topmouth gudgeon Pseudorasbora parva (Temminck and Schlegel, 1846) from Slovakia. J Appl Ichthyol 25:466–469. https://doi.org/10.1111/j.1439-0426.2009.01190.x
Zhang C, Zhao Y (2016) Species diversity and distribution of inland fishes in China. Science Press, Beijing
Zhao Y, Gozlan RE, Zhang C (2015) Current state of freshwater fisheries in China. Wiley-Blackwell, Oxford
Acknowledgements
We would like to thank all co-authors of Gozlan et al. 2020 for their help in collecting P. parva samples across the native and invasive range as well as B. Haenfling. We would also like to thank Eric Lombaert for kindly agreeing to discuss the best practices of Approximate Bayesian Computation methods. This study has been conducted in accordance with the relevant animal or human ethics approvals.
Funding
This study is part of the project GENESIS ANR-AF 13-ADAP-0005-001 supported by the Agence Nationale de la Recherche. S.B. was co-funded by the project PROBIS (Biodiversa) and E.C. was funded by an IRD postdoctoral fellowship. R.E.G., A.G., J.F.M. and S.B. received funding for this research from ANR Bioadapt.
Author information
Authors and Affiliations
Contributions
T.B., E.C. performed research, analysed the data. T.B., E.C. and R.E.G. wrote the manuscript; R.E.G., R.J.S.McC., J.F.M., A.G. and S.B. designed research and contributed with data; Y.Z., R.E.G., M.C. contributed with data; R.J.S.McC. assisted with analyses. All authors commented on the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Data availability
All sequencing data (GBS reads) are publicly-available under the BioProject ID PRJNA799666. The original genetic data yielded by genotyping (in vcf format), additional metadata (e.g. geographical coordinates) and all scripts necessary to reproduce the results presented in the paper are available in a publicly-available Github repository: https://github.com/ThomasBrazier/PseudorasboraGENESIS. Data are available from the authors upon reasonable request.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Brazier, T., Cherif, E., Martin, JF. et al. The influence of native populations’ genetic history on the reconstruction of invasion routes: the case of a highly invasive aquatic species. Biol Invasions 24, 2399–2420 (2022). https://doi.org/10.1007/s10530-022-02787-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-022-02787-6