
Abstract
Wild pigs (Sus scrofa) are the most widely distributed invasive wild ungulate in the United States, yet the factors that influence wild pig dispersal and colonization at the regional level are poorly understood. Our objective was to use a population genetic approach to describe patterns of dispersal and colonization among populations to gain a greater understanding of the invasion process contributing to the expansion of this species. We used 52 microsatellite loci to produce individual genotypes for 482 swine sampled at 39 locations between 2014 and 2016. Our data revealed the existence of genetically distinct subpopulations (F ST = 0.1170, p < 0.05). We found evidence of both fine-scale subdivision among the sampling locations, as well as evidence of long term genetic isolation. Several locations exhibited significant admixture (interbreeding) suggesting frequent mixing of individuals among locations; up to 14% of animals were immigrants from other populations. This pattern of admixture suggested successive rounds of human-assisted translocation and subsequent expansion across Florida. We also found evidence of genetically distinct populations that were isolated from nearby populations, suggesting recent introduction by humans. In addition, proximity to wild pig holding facilities was associated with higher migration rates and admixture, likely due to the escape or release of animals. Taken together, these results suggest that human-assisted movement plays a major role in the ecology and rapid population growth of wild pigs in Florida.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Biological invasions are one of the most important factors contributing to the loss of biodiversity, degradation of ecosystems, and decline in ecosystem services (Chapin et al. 1997; Sala et al. 2000; Pysek and Richardson 2010). Understanding the pathways of species introductions and range expansions informs wildlife and land management and can help mitigate or prevent further invasions (Hulme et al. 2008). Many different processes contribute to the human-assisted introduction of exotic animals (Hulme et al. 2008; Carpio et al. 2016), which include the unintentional escape of managed animals (e.g. zoo mammals, Cassey and Hogg 2015), the intentional/accidental release of alien animals from managed environments (such as animals from fur farms (e.g. American mink (Neovison vison), Kidd et al. 2009), or unwanted pets (e.g. domestic cats (Felis catus), Dickman 2009), and the intentional release of game species (e.g. roe deer (Capreolus capreolus), Randi 2005).
There has been a long history of introductions of game species for the creation of hunting opportunities (Yiming et al. 2006; Genovesi et al. 2012), but many of these species have proven to be damaging to the function and health of native ecosystems. For example, non-native browsers such as feral goats (Capra hircus), barbary sheep (Ammotragus lervia) and red deer (Cervus elaphus) negatively impact native plant communities, reduce vegetation densities and cause high levels of soil erosion (Wardle et al. 2001; Acevedo et al. 2007). Other exotic game introductions, such as that of nilgai antelope (Boselaphus tragocamelus) in Texas, have facilitated the spread of cattle fever ticks, which transmit bovine babesiosis—one of the most economically costly livestock diseases in the United States (Cárdenas-Canales et al. 2011).
According to the Species Survival Commission of the World Conservation Union (IUCN), wild pigs (Sus scrofa) are among the most ecologically destructive invasive species in the world (Lowe et al. 2000). Multiple factors have contributed to the establishment of wild pig populations including deliberate releases for hunting, the escape of individuals raised as livestock as a consequence of free-range practices, and the deliberate dumping of unwanted pets (e.g. Vietnamese pot-bellied pigs) (Mayer and Brisbin 2008; Caudell et al. 2013; Bevins et al. 2014). Since their first introduction to the continental USA in the sixteenth-century by European explorers (Wood and Barret 1979), the species’ distribution and abundance have expanded dramatically. Although long-established in the USA in regions of California, Texas and the Southeast, recent and rapid range expansion has led to the establishment of wild pig populations in as many as 44 states (Hutton et al. 2006; Barrios-Garcia and Ballari 2012; Bevins et al. 2014). The rapid expansion of wild pigs has been attributed to both intrinsic properties of the species (i.e. ability to adapt to a variety of habitat types, omnivorous foraging behavior, and high reproductive rates) and extrinsic causes (i.e. illegal transportation and release, frequent escapes from farms and hunting preserves, the propensity to thrive in human-altered landscapes, and a lack of native predators) (Seward et al. 2004; Bevins et al. 2014). Regionally, wild pig abundance in Florida is second only to Texas with an estimated 500,000 to one million individuals in the state (Giuliano 2010; FDACS 2016).
The first introduction of domestic swine in Florida is believed to have occurred in the early 1500s when Spanish conquistadors arrived at Charlotte Harbor in Lee County, southwest Florida (Mayer and Brisbin 2009). Through the early 1900s, European colonists raised domestic swine in unfenced, semi-wild conditions, with animals often becoming feral and expanding across the broad central savannah and coastal areas of the state (Mayer and Brisbin 2008). Specifically, it is believed that descendants of free-ranging domestic swine maintained by homesteaders in the Kissimmee River Valley became a substantial component of the wild pig populations established in Florida by the 1980s (Mayer and Brisbin 2008).
Currently, wild pig hunting is permitted in Florida. From the 1940s through the 1970s, domestic pigs were allowed to range freely. Periodic introductions of pure Eurasian wild boar throughout Florida hybridized with domestic and semi-feral swine to establish non-native wild pig populations throughout the state (Mayer and Brisbin 2008; W. Frankenberger pers. comm.).Footnote 1 In addition, legal translocations were conducted to restock state-controlled wildlife management areas. For example, from 1950 through the 1970s, approximately 3000 wild pigs were collected from various state parks and other ecologically sensitive areas and relocated to wildlife management areas in Palm Beach, Glades and Collier/Monroe counties in south Florida to establish or augment locally hunted populations (Belden and Frankenberger 1977; Mayer and Brisbin 2008).
The Florida Department of Agriculture and Consumer Services (FDACS) authorizes registered dealers to capture wild pigs on federal, state, municipal or private lands, and transport them to transitory holding facilities, prior to being sold for meat or released at private game preserves for hunting (Gioeli and Huffman 2012; FDACS 2016). During the course of this study, approximately 400 transitory holding facilities were registered by FDACS in Florida. Despite current state regulations, animals can escape from holding facilities, or alternatively, they can be illegally transported by recreational hunters and landowners over large distances and introduced to hunting areas without documentation of the movement. The willingness of people to translocate wild pigs has facilitated range expansion of this species in Florida and other states in the southern USA (Seward et al. 2004; Mayer and Brisbin 2009; Bevins et al. 2014; W. Frankenberger pers. comm.).Footnote 2 Illegal introductions represent a growing concern because of the impacts that wild pigs have on biodiversity, agriculture production, and animal and human health (Crooks 2002; Hone 2002; Bankovich et al. 2016).
Population genetic analysis can provide information regarding patterns of connectivity and interbreeding among populations and can be useful for differentiating natural patterns of animal dispersal from human-assisted translocations. Microsatellite markers are a widely used molecular tool to infer population connectivity and dispersal among sampling locations, thus allowing a greater understanding of the location-specific ecology of this species (Vernesi et al. 2003; Hampton et al. 2004; Nikolov et al. 2009; Scandura et al. 2011). Previous population genetic studies of wild pigs, largely conducted in Europe and Oceania, have identified individual membership to particular populations and levels of population admixture (i.e. interbreeding among isolated populations which produces offspring with a mixture of alleles from different ancestral populations) (Vernesi et al. 2003; Hampton et al. 2004; Spencer and Hampton 2005; Nikolov et al. 2009; Scandura et al. 2011; Lopez et al. 2014). Although these data will help inform population management and control efforts, little is known about wild pig dispersal and expansion throughout North America.
The goal of this study was to use population genetic techniques to describe movement patterns of wild pigs and to identify the potential factors that may influence their dispersal across Florida. We hypothesized that wild pigs would exhibit genetic population structure consistent with both historic and contemporary patterns of human-assisted introductions. Specifically, in the Kissimmee Valley region, where populations have been long established, we expected to find significant levels of both interbreeding and immigration among wild pig populations, consistent with a long history of natural and human-assisted movement in the valley and surrounding regions. If recent human-assisted introductions from outside the Kissimmee Valley were occurring, we would expect to find pockets of genetically distinct populations with limited genetic exchange with other nearby populations. Finally, because both escapes from holding facilities and intentional release at wildlife management areas have been identified as a source of introductions in the southeastern USA (Seward et al. 2004; Mayer and Brisbin 2009; Bevins et al. 2014; W. Frankenberger pers. comm.),Footnote 3 we hypothesized that populations near animal holding facilities and at wildlife management areas would support higher frequencies of interbred wild pigs and genetic immigrants than other sites around Florida.
Materials and methods
Sample collection of wild pig tissue
From January 2014 to March 2016, we collected tissue samples from 482 wild pigs at 39 sites across the state of Florida, USA (Fig. 1). We sampled animals opportunistically as part of a national wild pig disease monitoring effort led by the United States Department of Agriculture, Animal Plant and Health Inspection, Wildlife Services, National Wildlife Disease Program (NWDP). We acquired genetic samples from wild pigs that were trapped and euthanized during animal control efforts conducted throughout the study period by state or federal agencies. Additionally, we collected samples at check-stations from animals that were legally harvested by hunters on federal and state wildlife management areas, military bases, and private properties. We recorded demographic data for each animal, which included sex, age, and sampling location. Specifically, we used body size, reproductive traits, and tooth eruption patterns (Matschke 1967) to classify animals as adults (≥ 1 yr), subadults (2 mo–1 yr), or juveniles (< 2 mo). From 431 animals, we collected whole blood (0.5 ml) by cardiac puncture or orbital draw and stored the sample immediately in 1 ml mammalian lysis buffer (Qiagen, Valencia, CA, USA). We stored blood samples on ice packs and then refrigerated at 4 °C. From 51 animals, we collected hair, which was stored in paper envelopes in the field. Both whole blood and hair samples were transported to the University of Florida and stored at − 80 °C until DNA could be extracted. This study was approved by University of Florida’s Institutional Animal Care and Use Committee.

Sample size of wild pigs (Sus scrofa) collected per site through the state of Florida (U.S.) (2014–2016)
DNA isolation and microsatellite genotyping
We extracted DNA from blood using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA) and from hair using the QIAamp DNA Micro Kit (Qiagen, Valencia, CA, USA). For both procedures, we followed the manufacturer’s protocol, with slight modifications to increase DNA yields including vigorously mixing blood samples prior to extraction, increasing the amount of starting material (i.e. 200 µl for blood and 1–21 collected hair follicles), using 20 µl 1M DTT to increase hair tissue digestion, and a longer incubation period prior to final DNA elution (i.e. up to 15 min with shaking). We quantified the concentrations of recovered nucleic acids using the Epoch Microplate Spectrophotometer running the Gen5 software, version 2.09 (BioTek Instruments, Inc., Winooski, VT, USA). We stored isolated DNA at − 20 °C.
Sixty-one microsatellite markers were initially selected for multilocus genotyping, 42 of which were previously described (Ellegren et al. 1993; Robic et al. 1994; Alexander et al. 1996; Rohrer et al. 1996) and 19 novel markers that were designed and contributed by us (Online Resource 1). We screened markers and arranged loci into multiplexes using the program Multiplex Manager, version 1.2 (Holleley and Geerts 2009) based on their primer annealing temperatures and the likelihood of primer-product hybridization. Ultimately, 52 markers were either polymorphic or were successfully amplified to produce fragment peaks with a clear topology (see next subsection). We performed multiplex PCRs in 15 µl reaction volumes using the Qiagen Type-it Microsatellite PCR Kit (Qiagen, Valencia, CA, USA) as follows: 1X master mix, 0.2 µM 10X primer mix (see Online Resource 2 for optimized primer concentrations), 0.5X Q-solution additive, 3.5 µl sterile water, and 25 ng template DNA. We used touchdown PCR protocols to reduce the occurrence of non-specific amplification with the following protocol: initial denaturation at 95 °C for 15 min, followed by cycling at 95 °C for 30 s, annealing for 30 s with a 0.5 °C decrease with each subsequent cycle to reach optimum annealing temperature, and elongation at 72 °C for 30 s (see Online Resource 2 for specific starting temperatures) for 20–30 cycles with a final elongation at 72 °C for 40 min. We analyzed PCR products by capillary electrophoresis on an ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) and scored fragments using GeneMarker version 2.6.2 (SoftGenetics, State College, PA, USA) at the University of Florida.
Validation of genotypes and calculation of genotyping error
We attempted to re-amplify loci that were initially unsuccessful; however, if subsequent efforts failed (i.e. second and third attempts), these genotypes were categorized as missing data for the sake of analysis. To assess genotyping error and allelic dropout, 52 blood samples (i.e. approximately 12% of the dataset) were chosen at random and re-genotyped. We then compared the 52 duplicated genotypes to the originals, and any discrepancies were reconciled by conducting a third genotyping run. Six markers (S0215, Susc18, S0005, CGA, SW1680, SW13) exhibited ≥ 5% genotype error and were removed from the final dataset. Additionally, we eliminated two loci that exhibited high amplification failure (> 20%) and one monomorphic locus from the final dataset (SW1816, S0090, Susc11). Ultimately, we considered 52 loci in the final dataset.
Considering that locus amplification and genotyping error rates potentially may be affected by using different tissue types (blood and hair) that yield different quality and quantity of DNA (e.g. noninvasive samples, such as hair, have been shown to have higher allelic dropout because of lower quantity and quality of DNA recovered relative to other tissue types, Bonin et al. 2004), we conducted an independent validation study from parallel kidney and hair samples collected from an additional 34 wild pigs. Specifically, kidney samples were collected from fresh carcasses, placed into a cooler in the field, and then shipped on ice packs overnight to the NWDP for processing. We stored both kidney and hair samples at − 20 °C until DNA could be extracted. For each kidney sample, we extracted DNA independently in triplicate using a Qiagen DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA), following the manufacturer’s recommended protocol. Similarly, we extracted DNA from hair follicles in triplicate using QIAamp DNA Micro Kit (Qiagen, Valencia, CA, USA) with 15 follicles used for each independent extraction. We modified Qiagen’s recommended extraction protocol by disrupting follicle samples immediately prior to incubation by vibrating samples with a TissueLyser LT (Qiagen, Valencia, CA, USA) at 30 Hz for 6 min with sterile stainless steel 5 mm bead as recommended by Smith et al. (2011). We quantified the quality and quantity of DNA extracted from both kidney and hair samples with a Nanodrop (Thermo Fisher Scientific, Waltham, MA, USA) and diluted extractions to 10 ng/µl for PCR amplification. Extraction replicates with an elution concentration < 10 ng/µl were re-extracted. Each replicate of kidney and hair DNA was amplified and genotyped using the same multiplex PCRs and fragment analysis conditions described above. We compared the genotypes derived from triplicate DNA extractions from kidney (n = 3 × 34) and hair (n = 3 × 34) samples to validated multilocus genotypes and quantify genotyping error between putatively high quality (kidney) and putatively low quality (hair) DNA sources. Genotypes were assigned with GeneMapper 4.0 (Applied Biosystems) and we analyzed genotypes with software package ConGenR (Lonsinger and Waits 2015) in R version 3.3.1 (R Core Team 2016) to identify allelic dropout and false alleles among the 6 (3 kidney, 3 hair) replicates.
Estimating genetic diversity
We calculated descriptive statistics of basic measures of genetic diversity to assess sampling bias, population structure, and the robustness of molecular marker data of wild pigs across sampling locations. For all genetic analyses, we only considered genotypic data from sampling sites with ≥ 5 individuals (n = 454 animals). To describe locus polymorphism, we calculated the number of alleles (Na), observed heterozygosity (Ho) and expected heterozygosity (He) using GenAlex version 6.5 (Peakall and Smouse 2012). We calculated the mean allelic richness per sampling location (AR, El Mousadik and Petit 1996), corrected for the smallest sample size, using the R package PopGenReport version 2.2.2 (Gruber and Adamack 2015). We evaluated deviation from Hardy–Weinberg equilibrium (HWE) (i.e. derived from the comparison between observed and expected heterozygosity) at each locus across the entire dataset and per each sampling location using an exact test based on 100,000 Monte Carlo permutations, and linkage disequilibrium (LD) (i.e. significant correlation of alleles at different loci) using the R package pegas version 0.9 (Paradis 2010). We adjusted significance levels for multiple tests of HWE and LD using sequential Bonferroni corrections (Holm 1979; Rice 1989) in R.
Estimating population genetic structure
We characterized wild pig population genetic structure to infer historical and contemporary patterns of animal introduction and dispersal throughout Florida. We calculated F-statistics to examine the hierarchical partitioning of inbreeding within sampling locations (F IS ), relative to the inbreeding that can be explained by drift among different sampling locations (F ST ), and the individual inbreeding relative to the total population (F IT ) (Wright 1951, 1965). The statistical significance of F-statistics was tested using 999 permutations using the G-statistic Monte Carlo test implemented in the R package hierfstat 0.04–26 (Goudet 2005). We calculated pairwise F ST values (Weir and Cockerham 1984) among all sampling locations, and their statistical significance determined by 999 permutations, using GenAlEx version 6.5 (Peakall and Smouse 2012).
Before quantifying migration into and out of populations, we evaluated the level of genetic clustering (i.e., the assortment of genotypes into distinct genetic clusters) using two different Bayesian clustering methods implemented in STRUCTURE version 2.3.4 (Pritchard et al. 2000; Falush et al. 2003) and BAPS version 6.0 (Corander and Marttinen 2006; Corander et al. 2008). Both methods assign individuals to clusters (K) by minimizing deviations from HWE and linkage disequilibrium. STRUCTURE derives a posterior probability for each K examined across multiple Markov Chain Monte Carlo (MCMC) replicates (Pritchard et al. 2000; Falush et al. 2003). BAPS employs a greedy stochastic optimization method to search for the most probably number of clusters (Corander et al. 2008).
To assess genetic clustering of individuals, we tested the likelihood of K = 1–25 clusters using 20 replications at each K using the program STRUCTURE. Because of the long history of human-assisted introductions and high natural dispersal capabilities of wild pigs, we assumed an admixture ancestry model and correlated allele frequencies without including sampling location for each individual. We established 100,000 iterations for the burn-in period (i.e. simulation run previous to data collection to minimize the effect of the starting configuration) and 100,000 iterations post burn-in (i.e. simulations run after the burn-in to obtain parameter estimates). We compared likelihood values across replicates for each value of K and calculated ΔK, a statistic based on the rate of change in log-likelihood of the data (Evanno et al. 2005), with STRUCTURE HARVESTER version 0.6.94 (Earl and vonHoldt 2012) to estimate the optimal value for K. ΔK has been shown to identify only the uppermost hierarchical level of genetic structure (Evanno et al. 2005). Further, the utility of ΔK to accurately identify the genetic structure is limited by unequal sample sizes, as is the case here (Puechmaille 2016). Thus, we also used the suite of metrics developed by Puechmaille (2016) (i.e. corrected PP, MedMeaK, MaxMeaK, MedMedK and MaxMedK) to infer the number of genetic clusters present within our dataset.
To assess the robustness of our genetic clusters, we conducted a Bayesian mixture-clustering analysis among individuals without considering sampling location (i.e. the inclusion of sampling locations did not generate different clustering results in preliminary analyses) using the program BAPS. Initially, we ran the program with 5 replications of K = 1–25 and subsequently, we conducted 20 replications on the best-visited K values with highest likelihood (K = 15–17).
To better visualize our clustering approaches to population assignment, we used a Discriminant Analysis of Principal Components (DAPC, Jombart et al. 2010) using the R package adegenet version 2.0.1 (Jombart and Ahmed 2011). DAPC is a multivariate approach that does not make any assumption about HWE or linkage equilibrium, maximizing the among-population variation and minimizing the variation within predefined groups (Jombart et al. 2010). The optimal value of K was determined based on Bayesian Information Criterion (BIC) scores.
We tested the relationship between genetic differentiation (F ST /1 − F ST ) and geographic (Euclidean) distance (km) to assess patterns of isolation-by-distance. This hypothesis posits that the regular increase in genetic differentiation among individuals (or populations) is positively correlated with geographic distance due to geographically limited but continuous dispersal. When isolation by distance is not observed, it is inferred that factors other than proximity, such as human-assisted movement, shape the population movement patterns. Using data from individuals from all sampling locations, we ran a Mantel test with 10,000 permutations implemented in the R package ade4 version 1.7-4 (Dray and Dufour 2007). Geographical distances among sampling locations were calculated from either the geometric centroid of hunting check-stations or the mean center point of trap clusters (depending on collection method).
Estimating migration rates and admixed individuals
We assigned individuals as recent immigrants into a population or as nonimmigrants. These individual assessments were derived from population migration rates within each sampling location using BAYESASS version 3.0 (Wilson and Rannala 2003). We ran 100,000,000 MCMC iterations following a 10,000,000 burn-in period, and we used a sampling interval of 500 steps. We tested multiple delta values for the mixing parameters of migration rates, allele frequencies and inbreeding values. Delta values set to 1 resulted in optimal acceptance rates for changes to each mixing parameter (between 20 and 60%). We conducted multiple runs initialized with different random seeds and compared the posterior mean parameter estimates for convergence. We calculated 95% credible intervals (CI) for pairwise migration rate estimates between sampling locations, considering credible intervals that did not include zero to be statistically significant. Finally, we compared the significant rates of recent (first and second-generation) descendants of migrants with their corresponding genetic cluster assignment to evaluate congruence among all the conducted analyses.
To determine whether interbreeding among individuals from isolated clusters was occurring, we estimated individual genetic admixture using BAPS. This measure assessed whether an individual had the signature of one or more distinct genetic clusters. Once the most likely K value was determined, admixture inference was conducted using 100 simulations from posterior allele frequencies. We compared the mean posterior proportion of each individual’s ancestry (admixture coefficient) relating to each estimated K (clusters with ≥ 5 individuals). The admixture coefficient was bound between 0 and 1; animals with a coefficient closer to 1 had a less admixed ancestry than coefficient estimates closer to 0. Statistical significance was set at α = 0.05 to determine whether individuals had evidence of admixture. The p value reflected the proportion of simulated individuals (n = 200) from the cluster to which one specific individual was originally assigned that had admixture coefficient ≤ to that specific individual.
Predictors of wild pig admixture and migration rates
We tested whether human-related land use practices by hunters and trappers were related to the probability that an individual was (1) a genetic mixture of two genetically distinct populations (admixture) or was a recent immigrant (as inferred from the population migration rate). Admixed offspring or recent immigrants would be present if individuals from one genetically distinct population migrated (either naturally or with human assistance) to another area and mated with individuals from a different and genetically distinct population.
Land use factors were categorical (i.e. public hunting = 1 and no public hunting = 0) and continuous (i.e. geographic distance to the nearest wild pig holding facility) variables. Individual traits were age (i.e. juvenile, sub-adult, or adult) and sex. We used generalized linear regression models within an Akaike information criterion (AIC) framework for model comparison (Burnham and Anderson 2002) to test for relationships between predictor (i.e. human-related land use and individual age and sex) and response (i.e. probabilities of admixture and migration) variables. We used the R package MuMIn version 1.15.6 (Barton 2015) to fit the global multinomial model and all additive subsets, and to calculate model-averaged regression coefficients, 95% confidence intervals and cumulative AIC weight of evidence of each predictor variable (Burnham and Anderson 2002). Prior to their inclusion of predictor variables in the models, we tested all predictor variables for collinearity using Pearson correlation coefficients. We conducted statistical tests in R using α = 0.05 for determination of statistical significance.
Results
Genetic diversity and tissue validation
The 52 microsatellite loci in the final dataset produced a genotyping error rate (both allelic dropout and false alleles) of 0.7% (21 genotyping discrepancies/2704 scored genotypes) across the 52 replicate genotypes from blood samples. From the comparison of genotypes from paired triplicate hair follicle and kidney samples, we obtained a genotyping error of 1.7% (196 genotyping discrepancies/11,628 scored loci) indicating that we were able to generate robust genotypes from hair samples. Thus, for subsequent analyses, we treated genotypes generated from hair follicles as equivalent to genotypes generated from blood.
Allelic diversity across loci ranged from 6 (locus SW174) to 42 (locus SW856) alleles per locus (Online Resource 2). Within sampling locations, we detected an average of 5.1 alleles per locus with a mean allelic richness of 3.35 (when resampling individuals within site to an n = 5) (Table 1). H o and H e values across loci ranged from 0.163 (locus Susc34) to 0.866 (locus SW1067), and from 0.352 (locus S0227) to 0.942 (locus SW856), respectively. Although we found significant differences between H o and H e , the difference was small (Online Resource 2). Mean H o and H e were 0.626 and 0.616, respectively, across all the locations (Table 1). Of the 52 loci, we detected significant deviations from Hardy–Weinberg equilibrium (HWE) at 46 loci (Bonferroni adjusted p < 0.05) across the entire dataset. Forty-three of 46 markers with HWE deviations exhibited a deficit of heterozygotes (H o < H e ) (Online Resource 2). Evidence of linkage disequilibrium (LD) between genotyped loci was demonstrated for 63% (830/1326) of pairwise loci comparisons (Bonferroni adjusted p < 0.05). The relatively high HWE deviations and LD likely due to the genetic population structure we observed across sampling locations (see next subsection). We detected significant deviations from HWE at 13 out of the 1508 tests conducted (Bonferroni adjusted p < 0.05) across all sampling locations. A maximum of three deviations from HWE were detected per location, and one marker (locus Susc2) exhibited HWE deviation in six out of 29 sampling locations (Table 1).
Genetic population structure
The overall F-statistics resulted in significant values for F IS = 0.0281, F IT = 0.1419, and F ST = 0.1170 (G-statistic = 38,470, p = 0.001). The small but significant F IS value indicated low levels of inbreeding within sampling locations, which was likely driven by related individuals (within family units called sounders) being sampled within a site. This pattern also likely influenced the finding of a significant LD and heterozygote deficit.
All pairwise F ST values estimated between sampling locations were significantly different from zero (p < 0.01), which indicated genetic differentiation among sampling locations. F ST values ranged from 0.020 (between locations 14 and 22) to 0.256 (between locations 3 and 10). Fourteen of 29 sampling sites showed moderate level of genetic differentiation (all F ST values > 0.05) compared to the rest of sampling sites (Online Resource 3). A Mantel test did not reveal a significant correlation between genetic and geographic distances across the sampling locations (r = 0.081, p > 0.05) (Online Resource 4), suggesting that the patterns of genetic differentiation could not be explained by Euclidian distance.
STRUCTURE analyses revealed a peak in the mean posterior probabilities L(K) at K = 21 accompanied by the lowest variance (L(K) = − 70,901.035 ± 204.443) among replicates (Online Resource 5). We detected the highest ΔK peak at K = 2 (ΔK = 9.9906), and a second highest ΔK peak at K = 21 (ΔK = 2.1179) (Online Resource 5). Evaluation of the STRUCTURE results with the statistics introduced by Puechmaille (2016) provided support for a range of K values varying by metric (corrected PP = 20; MedMeaK = 21, MaxMeaK = 23, MedMedK = 22 and MaxMedK = 23). We chose to interpret K = 21 given that was included within the distribution of Puechmaille’s statistics and coincided with the L(K) peak with the lowest variance according to STRUCTURE. Thus, the mean membership coefficient (Q) of each sampling location to the inferred clusters was divided into 21 distinct groups, considering locations with Q ≥ 0.5 in any inferred cluster. Mean membership coefficient per location ranged from 0.546 to 0.976 for individuals across the 21 inferred clusters (Online Resource 6). One distinct cluster included all the animals from location 3 in northwest Florida. We found other clusters where the majority of individuals comprising each cluster were from one sampling location, such as location 1 in the northwest; locations 2 and 4 in the northcentral; locations 6, 8, 9, 10, 13, 19 and 21 in the northeast; locations 15, 18, 20, 24, 25, 26 and 29 in the southwest; and location 28 in the south. Two other clusters were composed of individuals from two sampling locations in each cluster, such as locations 5 (northeast) and 17 (southwest), and locations 12 (northeast) and 16 (southwest), respectively (Fig. 3a). The rest of the six locations (7, 11, 14, 22, 23 and 27) had Q < 0.5 in any inferred cluster and were not assigned to a specific cluster (Online Resource 6, Fig. 2a).

Geographic location and number of genetic clusters (K) inferred by three statistical methods across the 29 sampling locations of wild pigs in Florida. a STRUCTURE (corrected by Puechmaille’s statistics): sampling sites colored according to the predominant assignment of individuals to one of 21 genetic clusters (left) and Bayesian clustering output (right) (sites not assigned to a specific cluster are colored in white), b BAPS: sampling sites colored according to the predominant assignment of individuals to one of 16 genetic clusters (left) and mixture clustering output (right), and c DAPC: sampling sites colored according to the predominant assignment of individuals to one of 5 genetic clusters (above) and projection of clusters in discriminant space using the first two principal components (proportion of variance conserved by PCA principal components = 0.932) (below)
Mixture-clustering analyses in BAPS resulted in a probability of > 0.999 (log (ml) of optimal partition: − 80159.3593) of there being K = 16 genetic clusters of wild pigs in the study area (Fig. 2b). Other cluster partitions, such as K = 15 and K = 14, had lower log (ml) values (− 80188.3407 and − 80224.8726, respectively). BAPS analyses were able to detect a similar fine-scale population structuring as STRUCTURE. BAPS identified 13 clusters where the majority of individuals comprising each cluster were from one sampling location (i.e. locations 1, 2, 3, 4, 6, 9, 10, 13, 18, 20, 21, 28 and 29). One major cluster was comprised of animals from the rest of the 16 locations (Online Resource 7). Two clusters only included two individuals in each cluster (from locations 26 and 27, respectively), and they were not included in the admixture analysis.
DAPC analyses suggested an ‘optimal’ value of K = 5 (i.e. lower BIC value at K = 5); but the relatively flat pattern of the elbow in the curve (representing the relationship between BIC and number of clusters) suggested that values of K ranging from 3 to 10 may also represent ‘optimal’ number of clusters summarizing the observed genetic structure of wild pigs. Considering a K = 5 as the most probable number of clusters, animals sampled at locations 3 (K5), 9 and 10 (K2), and 13 (K3) seemed to be genetically distinct from individuals sampled at all the rest of 25 locations (divided in both K1 and K4) (Online Resource 8, Fig. 2c).
Deviations from genetic equilibrium were likely a product of biological processes and not null alleles. We reran STRUCTURE without the four loci that had the highest deviations from HWE (loci Susc2, Susc15, Susc34, and Susc20), and found no effect on the clustering results. Thus, we left the loci in all of our analyses.
Migration and ancestry analysis
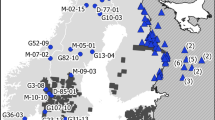
Analysis of gene flow patterns revealed low and statistically insignificant migration rates for the majority of sampling locations (i.e. mean migration rates for which the 95% CI included the zero). However, we found statistically significant migration rates between one particular ‘core’ site (location 22 in the southwest) and 16 other sampling sites throughout Florida (ranging from 3 to 14% immigrants between sites, Fig. 3). For other locations that had significant migration rates with the core location, 84.2% (16/19) of these animals exhibited a probability > 0.9 to be either first or second-generation migrants which suggested that these animals were recent migrants or the descendant of recent migrants. Except for two locations, all the sites with significant migration into or out of location 22 were assigned to the major genetic cluster inferred by the BAPS analysis. Six out of 16 locations with significant migration into or out of location 22 corresponded to the locations that were not assigned to any specific genetic cluster by STRUCTURE, after calculation of Puechmaille’s statistics.

Significant migration rates between one particular ‘core’ site (location 22, red circle) in the Kissimmee Valley and 16 other surrounding locations (green circles). Entire lines denote 3–6.7% immigrants between sites, and dashed lines denote 7.4–14% immigrants between sites
Mean posterior proportion of each individual’s ancestry showed that 6.2% (28/450) of wild pigs had significant evidence (p < 0.05) of genetic admixture (i.e. mixture of alleles from different ancestral populations due to interbreeding events) across 14 out of 16 inferred clusters, and 75% (21/28) of admixed individuals were assigned to the major cluster. Individual wild pigs from other genetic clusters were not significantly admixed, and thus, the majority of their genome was related to one particular ancestral population.
Predictors of wild pig admixture and migration
A total of 390 transitory holding facilities were identified and located throughout the state of Florida. Proximity to the nearest wild pig holding facility (range: 2–40 km) was the only variable that significantly predicted both the probability of admixture and individual migration patterns among all top models. The best-ranked AIC model (Log-lik = − 148.26, AIC = 302.51) predicting wild pig admixture only included distance to nearest holding facility as a predictor variable. This model presented the highest AIC weight of evidence (w r = 0.32) and the cumulative AIC weight of evidence of the predictor variable was 0.72 across the four best-ranked candidate models (Online Resource 9). Probability of wild pig admixture was higher in wild pigs collected in sites near holding facilities (β1 = 0.0048, 95% CI 0.0011, 0.0085, p = 0.0105), i.e. the closer the proximity to a holding facility the lower the ancestry coefficient and the higher the individual admixture.
The best-ranked AIC model (Log-lik = − 269, AIC = 546) predicting wild pig migration included distance to nearest holding facility and sex as predictor variables. This model presented the highest AIC weight of evidence (w r = 0.19) and the cumulative AIC weights of evidence for both predictor variables were 1 (distance to nearest holding facility) and 0.56 (sex), respectively, across the eight best-ranked candidate models (Online Resource 9). Probability of an individual being a first/second-generation migrant significantly increased with the proximity of the sampling site to animal holding facilities (β1 = − 0.0106, 95% CI − 0.0157, − 0.0059, p < 0.001), but sex was not significantly related to individual migration (β1 = − 0.0684, 95% CI − 0.1521, 0.0174, p = 0.113).
Although candidate models including the rest of predictor variables exhibited ΔAIC < 2 and similar weight of evidence compared to the best-ranked AIC models, neither public hunting nor individual covariates of age or sex were significantly related to admixture or migration patterns across all the candidate generalized linear regression models (i.e. all β1 coefficients with p > 0.05). No correlation was detected between the predictor variables included in the global multinomial model (all Pearson correlation coefficients with p > 0.05).
Discussion
The genetic patterns of wild pigs observed in Florida support the hypothesis that ongoing human-assisted movement is a source of their introduction and dispersal throughout the state. We found evidence of multiple unique genetic groupings, and patterns of both admixture and isolation that are not easily explained by natural dispersal. The lack of isolation by distance signal suggests that patterns of dispersal are driven by processes other than geographic proximity as would be expected under a stepping-stone model of gene flow. We suggest that human-assisted movement at least partially explains the observed pattern, aligning with a previous population genetic study of wild pigs in California (Tabak et al. 2017).
We demonstrated that locations proximal to wild pig holding facilities were associated with a higher probability of (1) individuals with a mix of genetic signatures from two or more genetically distinct populations, and (2) first or second-generation immigrant individuals. Human-assisted movement also explains the high migration rates in populations near the holding facilities. Our results suggest that holding facilities may act as foci for genetic exchange within landscapes through multiple potential routes, such as escapes from the facility, escapes during animal transport, escapes during transfer from dealers to holding facility, and/or deliberate releases. These arguments support previous research that speculates that the influence of animal escapes from farms and hunting preserves (Bratton 1975), and illegal transport and release (Waithman et al. 1999; Zivin et al. 2000) is responsible for increasing the range expansion and population densities of wild pigs across other states in U.S.
Three genetic clusters associated with unique locations were consistently inferred by each clustering method. Recent wild pig introductions from multiple genetic sources may explain the existence of these three distinct genetic clusters that were genetically distinct from wild pigs found elsewhere in Florida. One cluster was composed of animals sampled on an island located in Franklin County (northwest Florida) where purebred domestic Brown Russian and Poland China swine were introduced in the early 1940 s to restock hunted wild pigs on the island. Since the last introduction, the insular population was assumed to be disconnected from other wild pigs inhabiting the mainland (Mayer and Brisbin 2008), and these data suggest that, indeed, no migration to the island from the mainland has occurred. The other two clusters were composed of animals sampled at locations from Lake and Orange counties, respectively (northeast Florida). No official records exist about wild pig translocations into these particular sites, but the genetic uniqueness compared to wild pigs from other surrounding locations suggests that animals at these sites were recently introduced. This introduction would likely be associated with unreported/illegal transport and release to increase local hunting opportunities (Vernesi et al. 2003; Spencer and Hampton 2005; Scandura et al. 2011; Lopez et al. 2014).
Human-assisted movement is a likely explanation for the signal of admixed, recent immigrants detected between a particular ‘core’ site in southwest Florida and the other 16 sites that were mainly in one cluster, yet distributed across the state. Several anecdotal reports suggest that trappers have successively introduced up to several thousand wild pigs per year (from 2000 through 2008) into a private hunting club on the northern border of the ‘core’ site (both sites were located on the border of Polk and Highlands Counties). These animals could have been trapped at multiple unidentified preserves and parks across northeast and southwest Florida (W. Frankenberger pers. comm.Footnote 4), creating stocks from different genetic sources in the hunting club. Intensive and prolonged hunting pressures may expand the movement ranges of wild pigs from the hunting club to the ‘core’ site (Choquenot et al. 1996; Mayer and Brisbin 2009), likely resulting in the admixture and production of F1/F2 individuals from the mating between animals from the ‘core’ site and other source populations (up to 84% of admixed individuals were first/second immigrants from another population). Ultimately the emerging picture of wild pigs in the Kissimmee Valley region of Florida is a long and continuous history of movement, both natural and human-assisted within the valley.
The small but significant inbreeding (as measured by F IS ) we detected across all populations can be explained by an interaction between wild pigs breeding strategy/social structure, and our collection scheme. Aggregation of related individuals in family groups, high levels of female philopatry, and a few polygynous males siring the next generations, contribute to increase the genetic similarities among individuals of the same group (Gabor et al. 1999; Hampton et al. 2004; Kaminski et al. 2005; Poteaux et al. 2009). Considering our collection scheme of wild pigs, where multiple individuals were often harvested or trapped simultaneously in sampling locations, we likely genotyped related individuals belonging to the same family group, increasing the estimation of inbreeding with subpopulation (F IS ). The large number of loci with small deviations in HWE is likely due to the large number of admixed and migrant individuals in our samples. When individuals are the product of parents from genetically distinct populations this produces linkage disequilibrium, which by definition produces deviations in HWE.
As a whole, our study contributes novel insights regarding the role of human-assisted movement in the maintenance and spread of wild pigs in Florida and the influence of holding facilities as foci of translocation activities. We identified areas where long-term and ongoing wild pig introductions have taken place, reflected in high interbreeding due to wild pig dispersion between different locations through the Kissimmee Valley region and surrounding regions. We have also identified isolated genetic groupings with limited genetic exchange with other nearby populations, which is suggestive of recent translocations. Finally, we have shown that transition holding facilities for wild pigs are not secure and likely result in escapes or intentional releases into surrounding areas. These human activities have shaped the demographic structure of wild pigs at the regional level. Our findings inform both legislative and regulatory management focused on this invasive wild ungulate in Florida and other southeastern states in the U.S. by highlighting the role of transportation and escapes from holding facilities in maintaining and expanding invasive wild pigs in Florida.
Notes
December 2016, Gainesville, Florida (U.S.).
December 2016, Gainesville, Florida (U.S.).
December 2016, Gainesville, Florida (U.S.).
December 2016, Gainesville, Florida (U.S.).
References
Acevedo P, Cassinello J, Hortal J, Gortázar C (2007) Invasive exotic aoudad (Ammotragus lervia) as a major threat to native Iberian ibex (Capra pyrenaica): a habitat suitability model approach. Divers Distrib 13:587–597
Alexander LJ, Troyer DL, Rohrer GA, Smith TPL, Schook LB, Beattie CW (1996) Physical assignments of 68 porcine cosmid and lambda clones containing polymorphic microsatellites. Mamm Genome 7:368–372
Bankovich B, Boughton E, Boughton R, Avery ML, Wisely SM (2016) Plant community shifts caused by feral swine rooting devalue South Florida rangelands. Agric Ecosyst Environ 220:45–54
Barrios-Garcia MN, Ballari SA (2012) Impact of wild boar (Sus scrofa) in its introduced and native range: a review. Biol Invasions 14:2283–2300
Barton K (2015) R package ‘MuMIn’: multi-model inference (version 1.15.6). http://CRAN.R-project.org/package=MuMIn. Accessed 21 Apr 2017
Belden RC, Frankenberger WG (1977) Management of feral hogs in Florida—past, present, and future. In: Wood GW (ed) Research and management of wild hog populations. Clemson University, Georgetown, pp 5–10
Bevins SN, Pedersen K, Lutman MW, Gidlewski T, Deliberto TJ (2014) Consequences associated with the recent range expansion of nonnative feral swine. Bioscience 64:291–299
Bonin A, Bellemain E, Bronken Eidesen P, Pompanon F, Brochmann C, Taberlet P (2004) How to track and assess genotyping errors in population genetics studies. Mol Ecol 13:3261–3273
Bratton SP (1975) The effect of the European wild boar, Sus scrofa, on gray beech forest in the Great Smoky Mountains. Ecology 56:1356–1366
Burnham KP, Anderson DR (2002) Model selection and multimodel inference: a practical information-theoretic approach. Springer, New York
Cárdenas-Canales EM, Ortega-Santos JA, Campbell TA, García-Vázquez Z, Cantú-Covarrubias A, Figueroa-Millán JV, DeYoung RW, Hewitt DG, Bryant FC (2011) Nilgai antelope in northern Mexico as a possible carrier for cattle fever ticks and Babesia bovis and Babesia bigemina. J Wildl Dis 47:777–779
Carpio AJ, Guerrero-Casado J, Barasona JA, Tortosa FS, Vicente J, Hillstrom L, Delibes-Mateos M (2016) Hunting as a source of alien species: a European review. Biol Invasions 19:1–15
Cassey P, Hogg CJ (2015) Escaping captivity: the biological invasion risk from vertebrate species in zoos. Biol Conserv 181:18–26
Caudell JN, McCann BE, Newman RA, Simmons RB, Backs SE, Schmit BS, Sweitzer RA (2013) Identification of putative origins of introduced pigs in Indiana using microsatellite markers and oral history. Proc Wildl Damage Manag Conf 15:39–41
Chapin FS, Walker BH, Hobbs RJ, Hooper DU, Lawton JH, Sala OE, Tilman D (1997) Biotic control over the functioning of ecosystems. Science 277:500–504
Choquenot D, McIlroy J, Korn T (1996) Managing vertebrate pests: feral pigs. Australian Government Publishing Service, Bureau of Resource Sciences, Canberra
Corander J, Marttinen P (2006) Bayesian identification of admixture events using multilocus molecular markers. Mol Ecol 15:2833–2843
Corander J, Marttinen P, Sirén J, Tang J (2008) Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinform 9:539
Crooks JA (2002) Characterizing ecosystem-level consequences of biological invasions: the role of ecosystem engineers. Oikos 97:153
Dickman CR (2009) House cats as predators in the Australian environment: impacts and management. Hum Wildl Interact 3:41–48
Dray S, Dufour AB (2007) The ade4 package: implementing the duality diagram for ecologists. J Stat Softw 22:1–20
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor Appl Genet 92:832–839
Ellegren H, Johansson M, Chowdhary BP, Marklund S, Ruyter D, Marklund L, Bräuner-Nielsen P, Edfors-Lilja I, Gustavsson I, Juneja RK, Andersson L (1993) Assignment of 20 microsatellite markers to the porcine linkage map. Genomics 16:431–439
Evanno G, Regnault S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 51:672–681
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
FDACS (Florida Department of Agricultural and Consumer Services) (2016) Intrastate movement of feral swine. www.freshfromflorida.com/Divisions-Offices/Animal-Industry/Consumer-Resources/Consumer-Protection/Animal-Movement/Intrastate-Movement-of-Feral-Swine. Accessed 15 Dec 2016
Gabor TM, Hellgren EC, Van Den Bussche RA, Silvy NJ (1999) Demography, sociospatial behaviour and genetics of feral pigs in a semi-arid environment. J Zool 247:311–322
Genovesi P, Carnevali L, Alonzi A, Scalera R (2012) Alien mammals in Europe: updated numbers and trends, and assessment of the effects on biodiversity. Integr Zool 7:247–253
Gioeli KT, Huffman J (2012) Land managers’ feral hog management practices inventory in Florida. Proc Fla State Hort Soc 125:2012
Giuliano W (2010) Wild hogs in Florida: ecology and management. IFAS# WEC277 University of Florida Institute of Food and Agricultural Sciences
Goudet J (2005) Hierfstat, a package for R to compute and test variance components and F-statistics. Mol Ecol Notes 5:184–186
Gruber B, Adamack AT (2015) Landgenreport: a new R function to simplify landscape genetic analysis using resistance surface layers. Mol Ecol Resour 15:1172–1178
Hampton JO, Spencer PBS, Alpers D, Twigg L, Woolnough A, Doust J, Higgs T, Pluske J (2004) Applying molecular ecology to wildlife management: population structure and dynamics of feral pigs in south-western Australia. J Appl Ecol 41:735–743
Holleley CE, Geerts PG (2009) Multiplex manager 1.0: a cross-platform computer program that plans and optimizes multiplex PCR. Biotechniques 46:511–517
Holm S (1979) A simple sequentially rejective multiple test procedure. Scand J Stat 6:65–70
Hone J (2002) Feral pigs in Namadgi National Park, Australia: dynamics, impacts and management. Biol Conserv 105:231–242
Hulme PE, Bacher S, Kenis M, Klotz S, Kuhn I, Minchin D, Nentwig W, Olenin S, Panov V, Pergl J, Pysek P, Roques A, Sol D, Solarz W, Vila M (2008) Grasping at the routes of biological invasions: a framework for integrating pathways into policy. J Appl Ecol 45:403–414
Hutton T, DeLiberto T, Owen S, Morrison B (2006) Disease risks associated with increasing feral swine numbers and distribution in the United States. Michigan Bovine Tuberculosis Bibliography and Database
Jombart T, Ahmed I (2011) adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:1–15
Kaminski G, Brandt S, Baubet E, Baudoin C (2005) Life-history patterns in female wild boars (Sus scrofa L., 1758): mother–daughter postweaning associations. Can J Zool 83:474–480
Kidd A, Bowman J, Lesbarrères D, Schulte-Hostedde A (2009) Hybridization between escaped domestic and wild American mink (Neovison vison). Mol Ecol 18:1175–1186
Lonsinger RC, Waits LP (2015) ConGenR: rapid determination of consensus genotypes and estimates of genotyping errors from replicated genetic samples. Conserv Genet Resour 7:841–843
Lopez J, Hurwood D, Dryden B, Fuller S (2014) Feral pig populations are structured at fine spatial scales in tropical Queensland, Australia. PLoS ONE 9:e91657
Lowe S, Browne M, Boudjelas S, Poorter M (2000) 100 of the world’s worst invasive alien species: a selection from the global invasive species database. The Invasive Species Specialist Group, Species Survival Commission, World Conservation Union IUCN, p 12
Matschke GH (1967) Aging European wild hogs by dentition. J Wildl Manag 31:109–113
Mayer JJ, Brisbin IL Jr. (2009) Wild pigs: biology, damage, control techniques and management. SRNL-RP-2009-00869. Savannah River National Laboratory: Aiken, SC
Mayer JJ, Brisbin IL Jr (2008) Wild pigs in the United States: their history, comparative morphology, and current status. University of Georgia Press, Athens
Nikolov IS, Gum B, Markov G, Kuehn R (2009) Population genetic structure of wild boar Sus scrofa in Bulgaria as revealed by microsatellite analysis. Acta Theriol 54:193–205
Paradis E (2010) pegas: an R package for population genetics with an integrated–modular approach. Bioinformatics 26:419–420
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539
Poteaux C, Baubet E, Kaminski G, Brandt S, Dobson FS, Baudoin C (2009) Socio-genetic structure and mating system of a wild boar population. J Zool 278:116–125
Pritchard J, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Puechmaille SJ (2016) The program STRUCTURE does not reliably recover the correct population structure when sampling is uneven: subsampling and new estimators alleviate the problem. Mol Ecol Resour 16:608–627
Pysek P, Richardson DM (2010) Invasive species, environmental change and management, and health. Annu Rev Environ Resour 35:25–55
R Core Team (2016) R: a language and environment for statistical computing. R foundation for statistical computing, Vienna, Austria. ISBN 3-900051-07-0. http://www.R-project.org/
Randi E (2005) Management of wild ungulate populations in Italy: captive-breeding, hybridization and genetic consequences of translocations. Vet Res Commun 29:71–75
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Robic A, Dalens M, Woloszyn N, Milan D, Riquet J, Gellin J (1994) Isolation of 28 new porcine microsatellites revealing polymorphism. Mamm Genome 5:580–583
Rohrer GA, Alexander LJ, Hu Z, Smith TPL, Keele JW, Beattie CW (1996) A comprehensive map of the porcine genome. Genome Res 6:371–391
Sala OE, Chapin FS, Armesto JJ, Berlow E, Bloomfield J, Dirzo R, Huber-Sanwald E, Huenneke LF, Jackson RB, Kinzig A, Leemans R, Lodge DM, Mooney HA, Oesterheld M, Poff NL, Sykes MT, Walker BH, Walker M, Wall DH (2000) Global biodiversity scenarios for the year 2100. Science 287:1770–1774
Scandura M, Iacolina L, Cossu A, Apollonio M (2011) Effects of human perturbation on the genetic make-up of an island population: the case of the Sardinian wild boar. Heredity 106:1012–1020
Seward NW, VerCauteren KC, Witmer GW, Engeman RM (2004) Feral swine impacts on agriculture and the environment. Sheep Goat Res J 19:34–40
Smith B, Li N, Andersen AS, Slotved HC, Krogfelt KA (2011) Optimising bacterial DNA extraction from faecal samples: comparison of three methods. Open Microbiol J 5:14–17
Spencer PBS, Hampton JO (2005) Illegal translocation and genetic structure of feral pigs in Western Australia. J Wildl Manag 69:377–384
Tabak MA, Piaggio AJ, Miller RS, Sweitzer RA, Ernest HB (2017) Linking anthropogenic factors with the movement of an invasive species. Ecosphere 8:e01844
Vernesi C, Crestanello B, Pecchioli E, Tartari D, Caramelli D, Hauffe H, Bertorelle G (2003) The genetic impact of demographic decline and reintroduction in the wild boar (Sus scrofa): a microsatellite analysis. Mol Ecol 12:585–595
Waithman JD, Sweitzer RA, Van Vuren D, Drew JD, Brinkhaus AJ, Gardner IA, Boyce WM (1999) Range expansion, population sizes, and management of wild pigs in California. J Wildl Manag 63:298–308
Wardle DA, Barker GM, Yeates GW, Bonner KI, Ghani A (2001) Introduced browsing mammals in New Zealand forests: aboveground and belowground consequences. Ecol Monogr 71:587–614
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Wood G, Barrett R (1979) Status of wild pigs in the United States. Wildl Soc B 7:237–246
Wright S (1951) The genetical structure of populations. Ann Eugenic 15:323–354
Wright S (1965) The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 19:395–420
Yiming L, Zhengjun W, Duncan RP (2006) Why islands are easier to invade: human influences on bullfrog invasion in the Zhoushan archipelago and neighbouring mainland China. Oecologia 148:129–136
Zivin J, Hueth BM, Zilberman D (2000) Managing a multiple-use resource: the case of feral pig management in California rangeland. J Environ Econ Manag 39:189–204
Acknowledgements
We thank USDA/APHIS/WS field personnel and several hunting check station operators for graciously collecting samples on our behalf. We thank JC Griffin, R. Boughton and M. Legare for repeatedly assisting us with our sampling efforts in the field; M. Lopez, M. Anderson, P. Royston, B. Pace-Aldana, D. Watkins, and B. Camposano for quickly getting us the necessary sampling permits. We thank M. Tabak, H. Ernest and R. Beasley for providing essential technical support and molecular data to work with wild pig microsatellite loci. This study was partially funded by U.S. Department of Energy under Award Number DE--FC09--07SR22506 to the University of Georgia Research Foundation, and by funding to SMW from USDA NIFA McIntire-Stennis Cooperative Forestry Research Program, Award No. FLA-WEC-005166. FAH was supported by the Comisión Nacional de Ciencia y Tecnología de Chile (CONICYT-Chile).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hernández, F.A., Parker, B.M., Pylant, C.L. et al. Invasion ecology of wild pigs (Sus scrofa) in Florida, USA: the role of humans in the expansion and colonization of an invasive wild ungulate. Biol Invasions 20, 1865–1880 (2018). https://doi.org/10.1007/s10530-018-1667-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-018-1667-6