
Abstract
Purpose of work
A simple and rapid DNA extraction protocol capable of obtaining high-quality and quantity DNA from a large number of individuals is essential for assaying population and phylogenetic studies of plant pathogens. Most DNA extraction protocols used with oomycetes are relatively lengthy and cumbersome for high throughput analysis. Commercial kits are widely used, but low quantities of DNA are usually obtained, and with large scale analysis multiple isolations are required.
A protocol for DNA isolation from Phytophthora and Pythium suitable for the evaluation of a large set of molecular markers was modified from one previously developed for soybean seed. There was a one to three fold increase in the amount of DNA that was extracted using the modified protocol compared to a commercial kit. The DNA obtained using the modified protocol was suitable for the amplification of microsatellite markers as well as the ITS region. This protocol is inexpensive, easy, quick, and efficient in terms of the volume of reagents and the number of steps involved in the procedure. The method may be applicable to other oomycetes and effectively implemented in other laboratories.
Similar content being viewed by others


Introduction
Plant pathologists have studied the genetic structure of oomycete populations intensively to understand the evolutionary potential of these pathogens and to optimize management strategies (McDonald 1997; McDonald and Linde 2002). Phytophthora and Pythium are two of the major oomycete genera responsible of many destructive plant diseases around the world and efforts have been focused on understanding their biology, diversity, and evolutionary development. Genetic population studies require the analysis of large numbers of individuals for comparisons within and between populations with numerous molecular markers. Nevertheless, most extraction protocols for large quantities of high quality DNA essential for genetic studies of species of these genera are relatively lengthy, many require more than 1 day to be carried out, thus they are cumbersome for high throughput analysis; and others need specialized equipment to be completed (Lamour and Finley 2006; Villa et al. 2006; Winton and Hansen 2001). Commercial kits are widely used, they are less time-consuming and the DNA obtained is of high-quality; however, they are expensive, require the execution of many steps in a short period of time, and low quantities of DNA are regularly obtained.
Kamiya and Kiguchi (2003) developed a protocol for DNA extraction from soybean to expedite genotype analyses. Their protocol allows for processing of a large number of samples quickly. The combination of three detergents: SDS, Tween 20, and nonidet P-40 (NP-40) along with proteinase K in the digestion buffer, and the incubation at 55°C were crucial for the inactivation of nucleases, lysis of the cells, and release of DNA from the nuclear components and degradation of proteins (Merante et al. 1998). The objective of this study was to evaluate the Kamiya and Kiguchi (2003) protocol for the extraction of high quantity and quality DNA from Phytophthora and Pythium species that could also be implemented in large scale molecular studies. The DNA obtained from 14 different isolates was evaluated by amplifying markers commonly used in phylogenetic and population studies: the ITS region (Cooke et al. 2000) and two polymorphic microsatellites, PS36 (Dorrance and Grünwald 2009) and P74-75 (Schena et al. 2008), respectively. In addition, a side-by-side comparison was made between Kamiya and Kiguchi (2003) protocol and the commercial kit, DNeasy Plant Mini Kit (Qiagen, USA), the most commonly used in oomycete studies (Belbahri et al. 2006, 2008; Bowers et al. 2007; Ioos et al. 2006; Nechwatal et al. 2008).
Materials and methods
Oomycete isolates and culture conditions
Mycelia of 14 isolates of Phytophthora sojae, Phytophthora sansomeana, and Pythium heterothallicum (Table 1), from the soybean pathology collection at OARDC, were grown on half-strength lima bean media plates (LB50) at 27°C for 1 week. Ten 0.5 mm2 pieces of media were cut, transferred to 30 ml clarified V8 broth, and incubated at 27°C. After 3–5 days the mycelia was decanted, rinsed twice with sterilized distilled water, and dried on sterilized paper towel for 5 min. Dry mycelia was transferred to a mortar, frozen with liquid N2, and crushed with a pestle to fine powder. Fifty to 100 mg fine tissue was placed in a 2 ml microcentrifuge tube, and the tubes were then stored at −20°C until all the samples were processed.
DNA extraction protocols
The protocol of Kamiya and Kiguchi (2003) was slightly modified; the modifications were the substitution of nonidet P-40 (NP-40), which is no longer produced, with the detergent Triton X-100, and the increase in the volume of the reagents from 200 μl by four to 800 μl in order to obtain more DNA from each sample. DNA was extracted from 14 samples as follows, 800 μl of modified digestion buffer (10 mM Tris/HCl pH 8.0, 50 mM EDTA, 0.5% SDS, 0.5% Triton X-100, 0.5% Tween 20) and 2 μl of 20 mg proteinase K/ml (Amresco, USA) were added to a 2 ml microcentrifuge tube containing the frozen ground mycelia. The tube was vortexed for 30 s and incubated at 55°C for 30 min, with inversions every 10 min. After the sample was cooled to 27°C, 800 μl chloroform/isoamyl alcohol (24:1, v/v) were added, the tube was vortexed until the two phases formed an emulsion and centrifuged for 10 min at ~10,000×g. The supernatant was transferred to a 2 ml tube, and the chloroform/isoamyl alcohol step was repeated. To precipitate the DNA, the supernatant was transferred to another 2 ml tube containing 1 ml isopropanol. The tube was inverted 5 times and held at −20°C for 15 min. A DNA pellet was formed by centrifugation for 10 min at 10,000×g. The supernatant was carefully discarded, and the pellet was washed with 1 ml 70% (v/v) ethanol and then 1 ml absolute ethanol. The tube was then placed upside down over a paper towel for 15 min or until the pellet was completely dried. The pellet was resuspended in 100 μl warm (55°C) TE buffer (10 mM Tris/HCl, pH 8.0, 1 mM EDTA). After the pellet was dissolved and the DNA solution cooled to 27°C, 2 μl 20 mg Ribonuclease A/ml (Sigma, USA) was added and the tube was then incubated at 37°C for 30 min. DNA was also extracted simultaneously from the same ground tissue samples using the commercial DNeasy Plant Mini Kit (Qiagen, USA) following manufacturer’s instructions, and the DNA was eluted in 50 μl of TE buffer.
Comparison of DNA extraction methods
The quality and quantity of the DNA obtained from both protocols was evaluated by measuring the concentration (ng/μl), and A260/A280 and A260/A230 ratios in a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, USA). Two micrograms of each DNA sample was electrophoresed on a 2% (w/v) agarose gel with Rapid Run Agarose buffer (USB, USA) for 1 h at 100 V. After electrophoresis, the gel was stained in a 0.5% GelRed (Biotum, USA) for 10 min. DNA was visualized and photographed with a UV light transilluminator.
Amplification of DNA markers
DNA quality of the samples obtained with Kamiya and Kiguchi (2003) modified protocol was assessed by PCR amplification of the ITS region and PS36 and P74-75 SSR markers (Cooke et al. 2000; Dorrance and Grünwald 2009; Schena et al. 2008). The ITS region was amplified in a 25 μl reaction using 20 pmol of ITS4 (5′-CCTCCGCTTATTGATATGC-3′) and ITS6 (5′-GAAGGTGAAGTCGTAACAAGG-3′) primers, 20 ng DNA, 1× Taq Flexi Buffer (Promega, USA), 2.5 mM MgCl2, 100 mM BSA, and 2 U of Go Taq Flexi DNA polymerase (Promega). Amplifications were carried out in a DNA Engine Tetrad 2 Peltier Thermal Cycler (BioRad, USA). The PCR conditions were as follows: 95°C for 2 min, 30 cycles of 95°C for 20 s, 55°C for 30 s, and 72°C for 1 min, and a final cycle at 72°C for 10 min. The PCR products for the ITS were visualized as mentioned above. SSR marker PS36 was amplified in a 15 μl reaction volume containing 50 ng of template DNA, 0.1 mM of each dNTP, 1 mM MgCl2, 0.5 μM each of primers PS36F (5′-CAAAAATCATCAGCACCTTCG-3′) and PS36R (5′-TAGCCAAAAGAGCGACAACC-3′), 2 U of Go Taq Flexi DNA polymerase, and 1× Green Go Taq Flexi Buffer. The PCR conditions were: 94°C for 5 min, followed by 8 cycles at 92°C for 45, 45 s at 60, 58, 56, 55, 54, 53, 52, and 51°C, then 70°C for 45 s. Followed by 29 cycles of 92°C for 45 s, 48°C for 45 s, 70°C for 45 s and a final extension of 3 min at 70°C. SSR marker P74-75 was also amplified in a 15 μl reaction volume with 10 μM of primers S74F (5′-CGGTGGTACTTGTCGTCCTC-3′) and S75R (5′-TSTCCGGCTACATCATCATC-3′), 20 ng DNA, 1× Green Go Taq Flexi Buffer, 100 μM dNTPs, 1 mM MgCl2, and 2 U of Go Taq Flexi DNA polymerase. The PCR conditions were: 95°C for 2 min, followed by 40 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C 1 min, and 72°C for 10 min. PCR products of PS36 and P74-75 markers were electrophoresed on a 4% (w/v) agarose 3:1 High Resolution Blend (Amresco, USA) gel with Rapid Run at 300 V for 1.2 h, and stained with GelRed as described above.
The ITS PCR products were processed using ExoSAP-IT (USB, USA) following manufacturer instructions, and directly cycle-sequenced with the same primers ITS6/ITS4 in an ABI Prism 3100xl Genetic Analyzer at the Molecular Center and Imaging Center (OARDC, OSU). The sequences were visualized and edited using BioEdit ver. 7.0.9.0 (Hall 1999), and compared to the NCBI database using BLAST (Zhang et al. 2000) to confirm the identity of the species.
Results and discussion
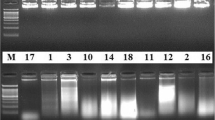
DNA, using the modified protocol of Kamiya and Kiguchi (2003), was successfully obtained from isolates of both Phytophthora and Pythium. One of the most critical steps in this procedure was the age of the culture. Winton and Hansen (2001) grew cultures at 27°C for 1–3 weeks; however, in the three species analyzed in this study the mycelia from the 10 pieces covered the area of the Petri plate after 3–5 days of growth. Mechanical grinding with liquid N2 was required to process the tissue that was used for the DNA extraction with both Kamiya and Kiguchi (2003) modified protocol and DNeasy Plant Mini Kit. The 28 DNA samples extracted with both protocols were visualized as solid bands of high molecular weight in a 2% (w/v) agarose gel (Fig. 1a, b). A light smear indicating DNA degradation was observed in some samples, specifically from the samples of P. sojae obtained with the DNeasy Plant Mini Kit (Fig. 1b, lanes 2–6). This smear may be a consequence of the high sensitivity of the GelRed dye.

Comparison of DNA extraction methods with 2 μg of genomic DNA and PCR products of Phytophthora and Pythium. a Kamiya and Kiguchi modified protocol; b DNeasy Plant Mini Kit (Qiagen); c 900 bp bands of ITS region; d polymorphic bands of microsatellite P74–75; e polymorphic bands of microsatellite PS36. Lanes 2–6, P. sojae, isolates: OH 1999 1.S.1.1, OH 2000 Wood 25, OH 2000 Wood 31, OH SS05 MB 4-4-1. Lanes 7–10, P. sansomeana, isolates: OH Mad 245, OH SS05 3B-2, OH Brown 2312. Lanes 11–15, P. heterothallicum, isolates: OH Blan B101-32, OH Blan B101-41, OH Blan B101-31, OH Blan B408-14, OH Blan B101-22. Lane 1: DNA ladders, a, b 0.2 μg of 1 Kb DNA ladder (Promega, USA); c, d, 0.15 μg; e 0.2 μg of 100 bp DNA ladder (Biolabs, USA)
The DNA samples were quantified spectrophotometrically, and the concentrations ranged from 0.5 to 2.9 μg/μl with Kamiya and Kiguchi (2003) modified method, with a mean of 1.9 μg/μl; and 0.3–0.8 μg/μl with the DNeasy Plant Mini Kit, with a mean of 0.6 μg/μl (Table 1). In both methods DNA was isolated from 50 to 100 mg of frozen mycelia, and the total amount of DNA was greater from the samples of Kamiya and Kiguchi modified protocol (46–296 μg) than those of the DNeasy Plant Mini Kit (15–43 μg). One of the samples obtained with Kamiya and Kiguchi modified protocol was 14 times greater than the same sample obtained with the DNeasy Plant Mini Kit (Table 1).
When the quality of the DNA was evaluated, the A260/A280 ratio was greater than 1.8 for all the samples, indicating that the DNA was almost free of proteins (Glasel 1995; Maniatis et al. 1982). With regard to the A260/A230 ratio, the values of almost all the samples obtained with Kamiya and Kiguchi modified protocol were approx. 2, indicating that the samples do not have impurities of carbohydrates, peptides, phenols, salts, and aromatic compounds (Hansen et al. 2007). In contrast, the A260/A230 ratio from the samples obtained with the DNeasy Plant Mini Kit were lower than 2, reflecting the presence of some contaminants that were not removed during the DNA isolation.
Three DNA markers were successfully amplified from the DNA isolated with Kamiya and Kiguchi modified protocol. PS36 and P74-75 SSR markers generated PCR products of 150–220 bp (Fig. 1d, e). PS36 was used previously in a P. sojae genetic population study (Dorrance and Grünwald 2009), and P74-75 amplified interspecific polymorphic bands of P. sojae, P. rubi, and P. europea (Schena et al. 2008). In addition, the same markers also amplified segments in P. sansomeana and P. heterothallicum.
A band of approx. 900 bp corresponding to the ITS region was obtained with the amplification of the primers ITS4/ITS6 (Fig. 1c). The sequences obtained from direct sequencing of the PCR products were of high quality, the sequence traces presented peaks evenly-spaced and easy to score. Taking into account that the ITS region is one of the most used genes for identification and phylogenetic analysis of oomycetes species (Cooke et al. 2000), all of the isolates studied were identified with a Blast search. A threshold of 100% homology between the sequences from the isolates in this study and sequences from strains which were previously deposited in Genbank was obtained and used to identify the individual species of each of the 14 isolates.
The combination of three detergents and proteinase K in the Kamiya and Kiguchi (2003) buffer was better at separating DNA from almost all the polysaccharides and protein complexes. Temperatures of 56–65°C were important to completely digest all the proteins. Residual proteins and lipids could readily be removed by extraction with chloroform-isoamyl alcohol (Merante et al. 1998). After these few steps, high quality DNA was obtained with a single precipitation with isopropanol.
The purpose of this project was to evaluate a simple, rapid, and cost-effective extraction method capable of obtaining high-quantity and quality DNA from large numbers of oomycete samples. The quality of the DNA obtained with Kamiya and Kiguchi modified protocol was suitable to amplify and sequence markers for both oomycete phylogenetics and population studies. The costs associated with the modified Kamiya and Kiguchi protocol was $0.32 per sample and these reagents are common to labs that routinely conduct molecular studies; while the costs of DNA isolation with the DNeasy Plant Mini Kit were $3.64 per sample. Thus, this protocol is inexpensive, simple, quick and efficient in terms of the volume of reagents, materials and number of steps involved in the procedure. The protocol will be suitable when large quantities of high quality DNA from Phytophthora and Pythium species are required.
References
Belbahri L, McLeod A, Paul B, Calmin G, Moralejo E, Spies CFJ, Botha WJ, Clemente A, Descals E, Sánchez-Hernández E, Lefort F (2006) Intraspecific and within-isolate sequence variation in the ITS rRNA gene region of Pythium mercuriale sp. nov. (Pythiaceae). FEMS Microbiol Lett 284:17–27
Belbahri L, Calmin G, Mauch F, Andersson JO (2008) Evolution of the cutinase gene family: evidence for lateral gene transfer of a candidate Phytophthora virulence factor. Gene 408:1–8
Bowers JH, Martin FN, Tooley PW, Luz EDMN (2007) Genetic and morphological diversity of temperate and tropical isolates of Phytophthora capsici. Phytopathology 97:492–503
Cooke DEL, Drenth A, Duncan JM, Wagels G, Brasier CM (2000) A molecular phylogeny of Phytophthora and related oomycetes. Fungal Genet Biol 30:17–32
Dorrance AE, Grünwald NJ (2009) Phytophthora sojae: diversity among and within populations. In: Lamour K, Kamoun S (eds) Oomycete genetics and genomics: diversity, interactions and research tools. Wiley-Blackwell, Hoboken, pp 197–212
Glasel JA (1995) Validity of nucleic acid purities monitored by A260/A280 absorbance ratios. Biotechniques 18:62–63
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acid Symp Ser 41:95–98
Hansen TVO, Simonsen MK, Nielsen FC, Hundrup YA (2007) Collection of blood, saliva, and buccal cell samples in a pilot study on the Danish nurse cohort: comparison of the response rate and quality of genomic DNA. Cancer Epidemiol Biomark Prev 16:2072–2076
Ioos R, Andrieux A, Marçais B, Frey P (2006) Genetic characterization of the natural hybrid species Phytophthora alni as inferred from nuclear and mitochondrial DNA analyses. Fungal Genet Biol 43:511–529
Kamiya K, Kiguchi T (2003) Rapid DNA extraction method from soybean seeds. Breed Sci 53:277–279
Lamour K, Finley L (2006) A strategy for recovering high quality genomic DNA from a large number of Phytophthora isolates. Mycologia 98:514–517
Maniatis T, Fritsch RF, Sambrook J (1982) Molecular cloning a laboratory manual. Cold Spring Harbor Laboratory, New York
McDonald BA (1997) The population genetics of fungi: tools and techniques. Phytopathology 87:448–453
McDonald BA, Linde C (2002) Pathogen population genetics, evolutionary potential, and durable resistance. Ann Rev Phytopathol 40:349–379
Merante F, Raha S, Ling M (1998) Isolation of total cellular DNA from tissues and cultured cells. In: Rapley R, Walker J (eds) Molecular biomethods handbook. Human Press Inc., Totawa, pp 9–16
Nechwatal J, Wielgoss A, Mendgen K (2008) Diversity, host, and habitat specificity of oomycete communities in declining reed stands (Phragmites australis) of a large freshwater lake. Mycol Res 112:689–696
Schena L, Cardle L, Cooke DEL (2008). Use of genome sequence data in the design and testing of SSR markers for Phytophthora species. BMC Genomics. doi:10.1186/1471-2164-9-620
Villa NO, Kageyama K, Asano T, Suga H (2006) Phylogenetic relationships of Pythium and Phytophthora species based on ITS rDNA, cytochrome oxidase II and β-tubulin gene sequences. Mycologia 98:410–422
Winton LM, Hansen EM (2001) Molecular diagnosis of Phytophthora lateralis in trees, water, and foliage baits using multiplex polymerase chain reaction. For Pathol 31:275–283
Zhang Z, Schwartz S, Wagner L, Miller W (2000) A greedy algorithm for aligning DNA sequences. J Comput Biol 7:203–214
Acknowledgments
Funding for this study was received in part from the Iowa Soybean Association and Ohio Soybean Council. Salaries and research support were provided by State and Federal funds appropriated to the Ohio Agricultural Research and Development Center, The Ohio State University. Sequencing was done at the Molecular Cellular Imaging Center at the OARDC. We wish to give special thank you to Damitha Wickramasinghe for editing, testing, and calculating the costs of the protocol.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Zelaya-Molina, L.X., Ortega, M.A. & Dorrance, A.E. Easy and efficient protocol for oomycete DNA extraction suitable for population genetic analysis. Biotechnol Lett 33, 715–720 (2011). https://doi.org/10.1007/s10529-010-0478-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-010-0478-3