
Abstract
Lipids are essential for physiological processes such as maintaining membrane integrity, providing a source of energy and acting as signalling molecules to control processes including cell proliferation, metabolism, inflammation and apoptosis. Disruption of lipid homeostasis can promote pathological changes that contribute towards biological ageing and age-related diseases. Several age-related diseases have been associated with altered lipid metabolism and an elevation in highly damaging lipid peroxidation products; the latter has been ascribed, at least in part, to mitochondrial dysfunction and elevated ROS formation. In addition, senescent cells, which are known to contribute significantly to age-related pathologies, are also associated with impaired mitochondrial function and changes in lipid metabolism. Therapeutic targeting of dysfunctional mitochondrial and pathological lipid metabolism is an emerging strategy for alleviating their negative impact during ageing and the progression to age-related diseases. Such therapies could include the use of drugs that prevent mitochondrial uncoupling, inhibit inflammatory lipid synthesis, modulate lipid transport or storage, reduce mitochondrial oxidative stress and eliminate senescent cells from tissues. In this review, we provide an overview of lipid structure and function, with emphasis on mitochondrial lipids and their potential for therapeutic targeting during ageing and age-related disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ageing can be considered as a progressive failure in the maintenance of tissue homeostasis leading to impaired function, health decline and the emergence of age-related disease. Ageing likely involves the dysregulation of several interconnected biological processes including redox state, metabolism, cell signalling, repair, replicative and adaptive capacity (Calabrese et al. 2010). Tightly regulated metabolism, the synthesis and degradation of macromolecules, provides a fundamental quality control check and energy resource. Recent evidence is emerging to indicate that in addition to the longstanding link between impaired lipid metabolism and atherosclerosis, lipid metabolism is also related to ageing. However, while altered lipid metabolism has been associated with and related to age-related diseases (Tindale et al. 2017; Dmitriev and Titov 2010; van Diepen et al. 2013), it is often difficult to discern whether such changes are a cause or a consequence of ageing.
Age-associated impairment in mitochondrial function, including dysregulation of lipid metabolism and increased reactive oxygen species (ROS) production, may be one mechanism contributing to changes in cellular energetics. The majority of mitochondrial lipids are synthesised in the endoplasmic reticulum (ER) and transported to the mitochondria, but some lipids (cardiolipin, phosphatidylethanolamine) are known to be synthesised within the inner membrane of the mitochondria and are critical for maintaining the unique architecture of the mitochondrial membranes, the cristae (Schlame and Greenberg 2017). An increase in ROS production may further exacerbate mitochondrial dysfunction, partly via oxidative damage to lipids (peroxidation), oxidation of respiratory chain proteins affecting metabolism and protein import, and in inducing DNA damage. The age-related accumulation of mitochondria DNA (mtDNA) mutations and respiratory chain dysfunction may lead to a decline in mitochondrial function associated with increased mitochondrial ROS production and a decline in ROS-scavenging enzymes (Simoncini et al. 2015). Mechanistic links between lipid metabolism and inflammation have also been established, where lipids can directly activate inflammatory pathways and vice versa (Pararasa et al. 2016; van Diepen et al. 2013). In addition, mitochondrial dysfunction has been linked to a shift in cellular metabolism towards glycolysis, highlighting the tight relationship between metabolism, energetics and ageing (Selfridge et al. 2013).
If indeed mitochondrial dysfunction contributes to significant lipid peroxidation, impaired lipid biosynthesis and ageing itself, then therapeutic stabilisation and restoration of mitochondrial function via targeted treatment with antioxidants (Zhang et al. 2016; Cai et al. 2015) may prove beneficial for alleviating age-related decline.
Lipid metabolism and lipid composition changes with ageing
Lipids are essential and multifunctional; they are structural components of cell membranes, provide energy reserves, mediate signal transduction and indirectly, can regulate gene expression (Mecocci et al. 1996; Elustondo et al. 2017). Interactions with specific receptors or enzymes mediate the biological effects of many lipid classes including ceramides, eicosanoids, phosphoinositides, cholesterol and sphingolipids. In turn, lipids play important roles in the regulation of cell proliferation, apoptosis, inflammation, migration and adhesion (Ng et al. 2017; Helkin et al. 2016; Mollinedo and Gajate 2015; Greig et al. 2012). Lipids, especially phospholipids (PL) have important mitochondrial functions due to their prominent roles in cell membrane formation, cell signalling pathways and energy storage. Free fatty acids (FFAs) are the major lipid constituents of membrane phospholipids and during disease conditions phospholipases are activated inducing PL hydrolysis and the release of FFAs. Phosphatidylcholine (PC) is the most abundant phospholipid present in the outer layer of the cellular membrane and its concentration typically increases with age (Hong et al. 2016). In Alzheimer’s disease (AD), Hong et al. recorded a decrease in PCs and increase in lysophatidylcholine (LPC) formed by hyperactivated phospholipase 2 (Hong et al. 2016). PL enable important neuronal functions including neurotransmission, stabilisation of ion channels and the localisation of β-amyloid plaques to PL cores (Hong et al. 2016).
Many age-related diseases including cardiovascular diseases, diabetes, kidney disease, rheumatoid arthritis and neurodegenerative diseases are associated with altered lipid metabolism and consequently changes in lipid profiles (Greig et al. 2012; AbouAssi et al. 2017; Zhang and Forman 2016; Helkin et al. 2016; Pararasa et al. 2015). Whiley et al. identified that some long chain PCs (16:0/20:5; 16:0/22:6 and 18:0/22:6) were at significantly lower concentrations in AD patients, particularly 16:0/20:5 (Whiley et al. 2014); noticeable reduction of sulfatides has also been found in AD (Hong et al. 2016). Gaudin et al. also found that PC with very long chain fatty acids (32:0 and 34:0) increased in AD (Gaudin et al. 2012). Conversely, sphingomyelin is reported to be at lower concentrations in AD patients compared to controls (Hong et al. 2016; Whiley et al. 2014).
Dysregulation in lipid desaturation may disrupt normal cell processes, for example, the decline in omega 3 fatty acids that is observed in ageing is considered proinflammatory. This is likely to arise from lower expression of the delta 6 desaturase enzyme. An inter-relationship between mitochondrial gene expression and lipid profiles was demonstrated in an elegant GWAS study; ten genetic variants-mtSNPs were associated with HDL cholesterol and triglyceride levels, highlighting the importance of the mitochondrial genome to the regulation of lipid levels (Flaquer Rospleszcz et al. 2015) in cardiovascular disease.
One of the most visible signs of ageing is an alteration in visceral and subcutaneous fat deposition; there is less subcutaneous fat deposited under the skin which not only impairs thermostasis but also reduces the potential for glucose and fatty acid disposal and insulin resistance (Torres-Villalobos et al. 2016), probably due to changes in hormone secretion (Pararasa et al. 2015). Fat accumulates at ectopic sites with age, such as the bone marrow, muscle and the liver. This may be cytotoxic to emergent immune cells, satellite cells and increase non-alcoholic fatty liver disease (Heilbronn et al. 2004) and can exacerbate insulin resistance (Gao et al. 2009). The most important fatty acid in lipotoxicity is palmitate that is converted to ceramides intracellularly (Gao et al. 2012). Palmitate, probably via ceramide, is an uncoupler of mitochondria, promoting release of ROS in a range of cell types (Phillips et al. 2002).
Deposition of visceral and subcutaneous fat is dependent on the size of the adipocyte pool and efficiency of converting fatty acids into triglyceride. The peroxisome proliferator activated receptor γ (PPARγ) transcription factor family regulates the expression of genes that catalyse the conversion of fatty acids into triglyceride for storage in a tissue specific manner (Nakamura et al. 2014). Metabolic switching between energy sources can also be controlled by PPARγ in immune cells (Griffiths et al. 2017) highlighting a tight relationship between metabolism and immune function (Torrao et al. 2014). Intriguingly, PPARγ expression by Treg cells is necessary to restore insulin sensitivity in obese mice (Cipolletta et al. 2012). In common with adipocytes, PPARγ is phosphorylated at Ser273 by Cdk5 in visceral adipose tissue Treg cells (Cipolletta et al. 2015).
The importance of fatty acid storage for longevity was elegantly demonstrated in a PPARγ deficient mouse that showed reduced lifespan; moreover, an in silico study suggested that the PPARγ signalling pathway was part of a longevity network (Argmann et al. 2009). With age, there is a decline in pre-adipocyte proliferation and differentiation (Sepe et al. 2011). This is likely to contribute to the increased systemic load of free fatty acids that we and others have described during healthy ageing (Pararasa et al. 2016).
In our previous study of fatty acids in plasma, we showed that the longer chain fatty acids (C14–C22) were increased in the healthy over 65 years of age group. This effect may be related to the observed increase in methylation of the Elongation of Very Long Chain Fatty Acids 2 (ELOVL2) promoter that has been reported in monocytes and T cells with age (Reynolds et al. 2014). Decreased expression of ELOVL2, which catalyses the extension of C20 and C22 poly unsaturated fatty acid (PUFA) (arachidonic acid and eicosopentanoic acid) to form very long chain unsaturated fatty acids, offers an explanation for the accumulation of ELOVL2 substrates that we observed. Both n−6 and n−3 PUFAs are a component of membrane phospholipid thus changes in their frequency will influence both the structure and function of the membrane (Ma et al. 2004). Finally, ELOVL2 also plays an important role in triglyceride storage and lower expression promotes an increase in fatty acid release by adipocytes (Kobayashi et al. 2007).
Consistent with other changes seen in fatty acid metabolism, plasma cholesterol levels tend to increase with age. However, this effect is not uniform across lipid classes; plasma LDL increases whereas HDL concentrations tend to be lower (Davidson et al. 2002; Walter 2009). The net effect is that cholesterol transport into cells is increased but reverse cholesterol transport out of cells to HDL decreases. Cholesterol together with ceramides can confer changes in membrane fluidity in the phospholipid bilayer and affect the formation of lipid raft microdomains; accumulating data suggest that the function of lipid microdomains is altered with ageing and can impair immune signalling (Fulop et al. 2012).
Several studies have highlighted differences in the lipid profile of male and females relating to age. Higher total cholesterol (TC), low-density lipoprotein cholesterol (LDL), and non-high-density lipoprotein cholesterol (N-HDL) in the non-elderly compared with the elderly for both males and females (Wei et al. 2014). Compared with females, males have higher HDL while females have high LDL with no difference in HDL among various age groups for females but there was significance for males (Goh et al. 2007; Wei et al. 2014). Triacylglycerol (TG) was reported to be significantly different in males of different age groups but no difference was observed in TG for females. TG levels were higher in 40–49, 50–59, and 60–69 year age groups in males than those in 70–79, 80–89, and ≥90 year age groups (Wei et al. 2014).
Goh et al. (2007) evaluated the impact of age and gender on lipid and lipoprotein profiles in a cohort of healthy Chinese-Singaporean and found no significant difference in the lipid and lipoprotein levels in men of different age groups but observed significantly higher level of some lipids in older women (>50 years old) than in younger women (30–46 years old). They also reported TG to be significantly correlated with lipids and lipoproteins differently in men and women (Goh et al. 2007).
Reynolds et al. (2010) evaluated the relationship between serum lipids and cognitive decline in men and women from the ages of 50–96 years. Among their findings, TG predicted memory ability in women across all ages with low TG predicting general cognitive ability while in men apolipoprotein B and TC levels correlated with better cognitive function. The gender effect seen in the lipid profile has been associated with the hormone androgen (Reynolds et al. 2010).
Metabolism is required to provide ATP for cell survival. The most efficient mode of ATP production is via oxidative phosphorylation in the mitochondrion and is supplied from the degradation of carbohydrates, proteins and lipids. Normally, oxidative phosphorylation is a tightly coupled process of two electron transfer in the presence of oxygen, yielding ATP and water. However, in conditions of nutrient excess or when components of the electron transport chain are modified, single electrons may leak (Jackson et al. 2002). The first product of single electron leakage is the reactive oxygen species (ROS), superoxide anion. Superoxide itself has relatively low reactivity and probably does not contribute to any major oxidative damage. However, superoxide spontaneously dismutates to hydrogen peroxide which is membrane permeable and is a powerful oxidant generating hydroxyl radicals via the Fenton reaction in the presence of catalytic iron. During oxidative stress, the imbalance in oxidants and antioxidant levels results in oxidative damage to macromolecules (Simoncini et al. 2015; Gandhi and Abramov 2012). The uncontrolled oxidation of lipids mediated by reactive oxygen species (ROS) is known as peroxidation. Peroxidation causes fundamental changes in molecular properties. For example phospholipids are essential for membrane architecture, but peroxidation causes changes in fluidity which can influence receptor clustering and yield bioactive and inflammatory oxidised phospholipids. Peroxidation is considered in the next section.
Hydrogen peroxide can mediate oxidation of thiolate anions found in cysteine residues that have a pKa ~5 and this is considered to be an important reaction for inactivation of tyrosine phosphatases and for propagating inflammatory signalling (Ratnayake et al. 2013). The intermediate sulphenic acid can be reduced by glutathione (GSH), catalysed by glutathione reductase, although this reaction is limited during ageing as GSH concentrations tend to be lower. Superoxide may also combine with nitric oxide to form peroxynitrite, another lipophilic species that can cross biological membranes and exert oxidising and nitrating reactions. Oxidation of cysteine residues in the iron-sulphur clusters of the mitochondria can further exacerbate ROS leakage (Sheeran and Pepe 2016).
During ageing, the integrity of the mitochondrial respiratory chain is compromised. ROS-induced mitochondria DNA mutation/damage, lipid and protein oxidation, affect physiological functions (Cui et al. 2012). In this state, cellular antioxidant defences are insufficient to keep the ROS levels below a toxic level (Simoncini et al. 2015). Age related accumulation of ROS has been suggested to play a part in the damage to major cell components in age-related diseases (Simoncini et al. 2015).
ROS induced lipid peroxidation
Lipid peroxidation is a biologically important process that results in intermediate unstable oxidised lipid species and stable end products, which act as bioactive lipid mediators. Being more polar than their parent lipids, lipid peroxides (LPOs) can perturb membrane bilayers, affect membrane structure and interfere with intracellular functions. Phospholipid peroxidation changes membrane biophysical properties, decreasing fluidity, inactivating membrane-bound proteins and ultimately leads to destruction of the membrane (Wong-ekkabut et al. 2007).
Products of peroxidation, lipid-oxides and –peroxides, are reported to be involved in many cellular processes including cellular metabolism, signalling, and cell survival (Aufschnaiter et al. 2017). Lipid molecules, especially PUFA and cholesterol, undergo oxidation at varied rates initiated by ROS [e.g. hydroxyl radicals (OH·), superoxide (O2·–), peroxyl radical (ROO–), nitric oxide (NO·), peroxynitrite (ONOO·–) or nitrogen dioxide (NO2·)], and in PUFA this initiates a self-propagating chain reaction (Halliwell 2001). The propagation stage of lipid peroxidation involves electron transfer and oxidation of other PUFA in close proximity. Propagation and termination have been reviewed extensively elsewhere (Yin et al. 2011; Girotti 1998). Non-enzymatically generated lipid peroxides often have a very short half-life and therefore pose technical challenges for precise measurement and biological study. Mass spectrometry is seen as the method of choice for absolute determination of lipid peroxidation.
In our recent work, we explained some challenges with oxidative lipidomic techniques and reported the detection of lipid oxidation by analysing the novel oxidised phospholipid biomarker 1-palmitoyl-2-(5’-oxo-valeroyl)-sn-glycero-3-phosphocholine (POVPC), a truncated oxidation product of 1-palmitoyl-2-arachidonoyl-sn-phosphatidylcholine (PAPC) using electrospray ionization tandem mass spectrometry (MS) with multiple reaction monitoring (MRM) (Ademowo et al. 2017). Several authors have used antioxidant activities and oxidative stress derived biomarkers to detect and analyse lipid peroxidation (Cai et al. 2017; Umeno et al. 2017; Ademowo et al. 2017). Gaschler and Stockwell reviewed in details several other methods for the detection and analysis of lipid peroxidation in biological samples [see (Gaschler and Stockwell 2017)].
Oxidised phospholipids (oxPLs) are generated from (poly)unsaturated diacyl- and alk(en)ylacyl glycerophospholipids. OxPLs exert a wide variety of biological effects and play a role in the development of several chronic diseases (Philippova et al. 2014). In pathological conditions associated with activation of oxidative stress, unsaturated phosphatidylcholines, the most abundant phospholipids undergo oxidation leading to generation of fragmented phospholipids such as 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine (lysoPC), or 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (PAPC) full length oxygenation products (oxPAPC) (Heffern et al. 2013). Other oxidised derivative of PAPC are 1–palmitoyl-2–(5–oxovaleroyl)- sn -glycero-3–phosphocholine (POVPC), 5–hydroxy-8–oxo-6–octenoyl-phosphocholine (HOOA-PC), 1–palmitoyl-2–(5,6–epoxyisoprostane E2)- sn -glycero-3–phosphocholine (PEIPC), and 1–palmitoyl-2–glutaroyl- sn -glycero-3–phosphocholine (PGPC) (Philippova et al. 2014). Eicosanoids including prostaglandins, thromboxanes and leukotrienes are also products of arachidonic acid oxidation and they play major regulatory roles in immune and inflammatory functions (Rubio-Perez and Morillas-Ruiz 2012).
Although oxPLs are recognized to be involved in disease pathogenesis, others reported that the converse is true under certain biological conditions (Mauerhofer et al. 2016). Mauerhofer et al. have demonstrated that OxPLs can also induce protective effects such as inhibition of inflammatory signaling pathways through Nrf2-dependent and -independent mechanisms, upregulation of antioxidant genes, antagonism of Toll-like receptors. The immuno-modulating and immuno-suppressive action of OxPLs may influence adaptive immunity and autoimmune disease through activation of PPARs which are known for their anti-inflammatory action. OxPLs may also inhibit inflammation induced via TLR4 (Bochkov et al. 2010). Hence, phospholipid oxidation may provide a negative feedback to prevent damage to host tissues from uncontrolled inflammation and oxidative stress (Mauerhofer et al. 2016).
Whilst cholesterol-derived hydroperoxides are more stable and have been studied both in model systems and cells (van Lier et al. 2015), the analysis of the predominant lipid peroxide species in fluids and tissue in health and how they change in disease requires methodological precision and sensitivity (Reis 2017). Due to the instability of many other intermediate products, PUFA-derived stable aldehydes such as 4-hydroxy-2-nonenal (HNE), malondialdehyde (MDA) and acrolein are the most extensive species that are studied (Esterbauer et al. 1991; Ayala et al. 2014; Zhang and Forman 2016). HNE and MDA show high chemical reactivity and interact with proteins, resulting in the formation of covalent adducts that can be measured by ELISA and often modulating protein activity (Liu et al. 2014b) including in proteins in the membrane (Pizzimenti et al. 2013). The majority of studies have focussed on the aforementioned three stable aldehydes, however, advances in analytical capability using advanced MS methods are revealing many other interesting and less well-studied carbonyl compounds (Sousa et al. 2017). Products of lipid peroxidation are often reactive themselves and are classified as either hydroxyl acids or reactive aldehydes (Gaschler and Stockwell 2017). Due to the ability of lipid peroxidation products such as aldehydes to covalently modify proteins, Shibati et al. used mass spectrometry methods to detect product ions generated from positively ionised adducts (Shibata et al. 2017). In future, in depth studies using state of the art MS methods will help us to understand more about targets and potentially mechanisms of lipid peroxidation involving protein adducts in ageing.
Zheng et al. (2005) measured HNE-adduct accumulation in ageing in fruit flies using an ELISA method and found that HNE adduct concentrations are significantly increased in the second half of the adult life by two fold (Zheng et al. 2005). Another study using ageing Wistar rats at 7, 15, 22 and 30 weeks found significantly increased levels of HNE-protein adducts with ageing (Asselin et al. 2006).
Isoprostanes (IsoPs) are prostaglandin F2-like compounds and neuroprostanes (NPs) that are formed by the non-enzymatic, free radical-mediated oxidation of arachidonate and docosahexaenoate, respectively. IsoPs have been measured in biological fluids such as urine, plasma, bile, cerebrospinal fluids, and in many cell and tissue types (Dias et al. 2015; Cracowski 2004; Ademowo et al. 2017). Choksi et al. (2007) showed lower serum and liver IsoP levels at all ages in Dwarf mice that show inherently low levels of ROS. The authors suggested that lower levels of endogenous ROS production may be a factor in their resistance to oxidative stress and longevity (Choksi et al. 2007).
García-Flores et al. (2017) analysed F2-dihomo-isoprostanes, F3-neuroprostanes, and F4-neuroprostanes- in urine samples from 158 healthy volunteers ranging from 4 to 88 years old. They found a significant, positive correlation between age and total F2-dihomo-IsoP concentration (García-Flores et al. 2017). To further investigate any relationship between lipid peroxidation and ageing, a cohort study was undertaken in a geriatric population; circulating MDA was related to frailty and not to age or sex (Inglés et al. 2014).
An increase of LPOs is commonly found in many age-related diseases. Skewing of the redox balance towards oxidation is observed in biological ageing and in many inflammatory diseases that increase with age. Table 1 provides examples of organs and diseases that are associated with increased LPOs.
To minimise lipid peroxide and reactive aldehyde accumulation to subtoxic levels, cells have a vast network of antioxidant and detoxification systems. Removal of aldehydes is achieved by glutathione (GSH) that reacts with aldehydes to form GSH adducts either spontaneously or catalysed by glutathione S-transferases (GSTs), by aldo–keto reductases (AKRs) to alcohols or oxidation to acids by aldehyde dehydrogenases. The redoxins (thioredoxin, peroxiredoxin, glutaredoxin) and dismutase enzymes also play a role in the detoxification process and have been reviewed elsewhere (Zimniak 2008; Krag 2011). Increased LPO products observed in biological ageing are likely to be a result of increased production and decreased removal caused by age-associated dysfunction of detoxification.
Mitochondrial lipids in biological ageing and age-related diseases
Mitochondria are cytoplasmic organelles with a characteristic double-membrane structure and have been described as the “powerhouses” of the cell, generating the energy needed to function and survive (Reddy and Beal 2005; Yin et al. 2015; Simoncini et al. 2015). Mitochondria perform electron transport producing adenosine triphosphate (ATP) (Swerdlow and Khan 2004) and regulate the initiation of apoptosis by releasing proteins that activate the caspase family proteases. Disruption of the electron transport chain (ETC) affects the reduction–oxidation (redox) potential of the cell. This impacts on signalling pathways through modulation of SIRT (Griffiths et al. 2017) and by oxidising reduced NADH to NAD+ needed in glycolysis (Selfridge et al. 2013). Mitochondria are the greatest cellular source of ROS as the electron transport consumes about 85% of the oxygen that the cell uses (Reddy and Beal 2005). An impaired ETC reduces ATP energy but increases ROS production. During biological ageing, aberrations in the control of cell death, impaired metabolism and perturbed redox homeostasis have been attributed to mitochondrial dysfunction (Yin et al. 2015) and in the pathogenesis of inflammation (Wan et al. 2014) and age related diseases including neurodegenerative diseases (Di Domenico et al. 2016), diabetes (Vadvalkar et al. 2017) and cardiovascular diseases (Li et al. 2017).
As a source of ROS production, mitochondria are particularly susceptible to locally mediated oxidative damage to lipids, DNA and proteins; the latter are normally limited by effective oxidative DNA damage repair enzymes and the Lon protease respectively. Secondary, oxidative DNA damage from lipid peroxidation products may contribute to the increasing numbers of mitochondrial mutations seen in diseases and inactive respiratory complexes, so promoting further ROS leakage in a vicious cycle of oxidative damage (Yao and Brinton 2011; Tanaka et al. 1996).
The structural and functional arrangement of mitochondrial protein complexes within the membrane changes with age. These structural changes affect the ion mobility disrupting the carrier proteins that shuttle ions, ATP, ADP and small metabolites between the cytoplasm and the matrix; these proteins include the 33 kDa ATP/ADP carrier, the soluble electron carrier protein, cytochrome c and enzymes such as the ATP synthase, succinate dehydrogenase and other oxido-reductases (Pebay-Peyroula et al. 2003; Kühlbrandt 2015). However, whether or not the morphological changes seen in mitochondria during ageing are direct or indirect consequences of oxidative damage to proteins and lipids is not known. The biogenesis of mitochondrial proteins during ageing is not well explored and due to the dynamic and fragile nature of mitochondria protein complexes, they are not easily analysed by established methods (Kühlbrandt 2015). Hence the effects of age-related changes to mitochondrial lipid and protein complexes are emerging areas of research.
Cardiolipin (CL), a form of glycerophospholipid found in the inner membrane of the mitochondrial lipid bilayer, has an important structural role acting as an anchor for respiratory super-complexes and for mitochondrial DNA during replication. CL is essential for mitochondrial health (Shen et al. 2015) and CL oxidation has been identified as a primary event in the release of cytochrome c and to increase permeability of the mitochondrial membrane to apoptosis factors such as BAX and BAD. The release of cytochrome c was suppressed when the production of ox-CL in mitochondria was inhibited by the overexpression of mitochondrial phospholipid hydroperoxide glutathione peroxidase (Nomura et al. 2000).
As an anionic phospholipid, cardiolipin is very susceptible to oxidation; this affects respiration, the assembly and stability of the mitochondrial protein import machinery, may cause abnormal mitochondrial morphology and even promote cell death (Horvath and Daum 2013). Recently, a pentapaptide-SS31 has been discovered to bind and protect cardiolipin from oxidation. It has been shown to improve mitochondrial function in ageing muscle, maintain the structure of cristae and protect neurons from hypoxia reperfusion injury (Birk et al. 2014).
Mitochondrial lipids in neurodegeneration
Although the central nervous system (CNS) function is dependent on efficient mitochondrial activity due to its energy requirement, it is still unclear whether or not mitochondrial impairment and oxidative stress are actually involved at the onset or progression of neurodegeneration and some other diseases (Simoncini et al. 2015). Mitochondria are rapidly transported to areas of high energy demand such as the neuronal projections (Selfridge et al. 2013). The mitochondrial transport is important for the development, function and stability of synapses and dendritic spines (Selfridge et al. 2013).
Mitochondrial dysfunction has been reported to be one of the earliest and prominent features in neurodegenerative disorders as ROS are produced locally and target the mitochondria causing the oxidation (Simoncini et al. 2015). Emerging research has strongly implicated mitochondrial dysfunction as causal in the brain energy deficit and failure to adapt to stressors observed in neurodegenerative diseases such as AD; mitochondrial abnormalities are part of AD phenotype (Selfridge et al. 2013). The decline in glucose metabolism and mitochondrial function with increased oxidative stress often appears decades before onset of clinical/pathological symptoms of the disease (Yao and Brinton 2011), hence, ROS have been suggested to be the cause rather than the consequence of neurodegeneration (Moreira et al. 2009). During AD progression, lower glucose utilization and exhaustion of the ketone reservoir increases mitochondrial dysfunction and the fatty acid oxidation pathway which results in white matter degeneration and eventually neuronal death (Yao and Brinton 2011).
Fundamental changes in the composition and distribution of lipids within the brain are believed to contribute to the cognitive decline associated with ageing. The mechanisms by which these changes in lipid composition affect cellular function and ultimately cognition are not well understood (Kennedy et al. 2016; Hong et al. 2016; Whiley et al. 2014). However, Kennedy et al. identified the mitochondria as an important downstream target of PC(O-16:0/2:0), a neurotoxic lipid species found to be increased in AD and that PC(O-16:0/2:0) promotes a global increase in ceramide accumulation in human neurons, associated with mitochondrial-derived ROS and toxicity. These data suggest that PC(O-16:0/2:0)-dependent mitochondrial dysfunction may be an underlying factor for increased ROS production associated with AD (Kennedy et al. 2016). The mitochondrion-located antioxidant enzyme copper/zinc superoxide dismutase is significantly decreased in the frontal and temporal cortex of AD brain, rendering the AD mitochondrion at increased risk for oxidation and offering a possible explanation as to why brain cells (astrocytes, neurones and glial cells) are more susceptible to mitochondrial dysfunction in AD (Reddy and Beal 2005; Moreira et al. 2009).
Mitochondrial lipids in diabetes
Type 2 diabetes mellitus (T2DM) is the most common human endocrine disease and is characterized by peripheral insulin resistance and pancreatic islet β-cell failure resulting fat deposition in adipose tissue. Pancreatic islet β-cell mitochondria promote lipid oxidation, increase lipogenesis and activate several stress-response pathways in diabetes (Chattopadhyay et al. 2015).
Kahle et al. (2015) took a targeted metabolomics approach to identify high fat (HF) diet-induced modifications in membrane lipid and mitochondrial-membrane protein signatures in mouse liver cells. After 7, 14, and 21 days exposure to an HF diet rich in polyunsaturated fatty acids, hepatosteatosis was induced and major lipid constituent signatures in liver were modified e.g. increased total unsaturated, long-chain fatty acid-containing acyl-carnitine or membrane-associated diacylglycerol moieties and decreased total short-chain acyl-carnitines, glycerophosphocholines, lysophosphatidylcholines, or sphingolipids. The HF diet progressively decreased the abundance of protein-components of all mitochondrial respiratory chain complexes, inner and outer mitochondrial membrane substrate transporters impacting on energy metabolism in liver cells (Kahle et al. 2015).
Using shotgun lipidomics data with multidimensional mass spectrometry, Han et al. (2005) identified a significant decrease in cardiolipin levels and alterations of cardiolipin acyl chain remodelling in diabetic murine myocardium, linking altered substrate utilization and metabolic flux with the diabetic heart (Han et al. 2005). Transgenic expression of cardiolipin synthase in a streptozotocin-induced diabetic mouse model, accelerated cardiolipin remodelling, improved mitochondrial function and attenuated mitochondrial dysfunction during diabetes (Kiebish et al. 2012). This study highlights an avenue to target mitochondrial bioenergetics via cardiolipin synthase.
Mitochondrial lipids in cardiovascular diseases
Mitochondria play an important role in cardiomyocytes by sustaining ATP production to satisfy the high demand for energy and the normal function of the heart. Even subtle alterations in mitochondrial function or membrane potential e.g. through peroxidation, can cause a significant change in cardiomyocyte energy production and deleterious consequences in cardiovascular health (Pytel et al. 2016). Increased mitochondrial ROS and reactive nitrogen species production during the early stages of hypoxia are associated with cardiomyocyte toxicity (Kolamunne et al. 2013). Lower levels of CL were observed in the rat ischemia–reperfusion model and a decrease in mitochondrial complex III activity was also observed (Paradies et al. 2004). A proteomic study by (Pavón et al. 2011) revealed that proteins of the electron transport chain constituted the greatest percentage of altered lipid-adducted proteins in rat cardiac mitochondria treated with a spectrum of lipid peroxidation products including HNE and 4-hydroxyhexanal (HHE). While a high level (≥20 µM) of HNE was cardio-toxic, levels around 5 µM of HNE induced endogenous anti-oxidant systems through Nrf2 activation (Dasuri et al. 2009).
As mitochondria have been referred to as the most important determinant of life span (Sun et al. 2014), there is a need to understand mitochondrial bioenergetics in prodromal and extant age-related diseases; to explore signature markers of disease that identify onset and progression; and to investigate different interventions that could prevent or delay the progression of such diseases.
Mitochondrial ROS production during cellular senescence
As we have discussed previously, impaired mitochondrial function and elevated ROS production associate with altered lipid metabolism and increased lipid oxidation that is observed in several age-related diseases. Senescent cells are one potential source of mitochondrial dysfunction during ageing.
Cellular senescence can be considered as a programmed change in cell state, associated with irreversible proliferative arrest and often accompanied by conversion to a pro-inflammatory, immunogenic phenotype (Burton and Krizhanovsky 2014). In contrast, cell ageing occurs as a result of accumulating stochastic damage over time, leading to gradual impairment in cell function. Senescent cells are not aged/old cells. Although, cell senescence can be induced at random, independent of age, the accompanying changes associated with cell senescence are highly regulated.
Cells become senescent in response to various stress-inducing stimuli such as ROS, telomere dysfunction and oncogene activation, which consequently initiates a persistent DNA damage response (DDR) (Di Micco et al. 2006; Rodier et al. 2009; Rossiello et al. 2014). In recent years, cell senescence has emerged as a major mechanism promoting biological ageing and age-related diseases (Childs et al. 2015; Muñoz-Espín and Serrano 2014; van Deursen 2014) and as such, a discussion on the potential connection between cellular senescence, mitochondrial dysfunction and lipid metabolism is warranted.
The senescent state has been shown to be associated with a shift from mitochondrial respiration towards glycolysis (Aird and Zhang 2014; James et al. 2015) and whilst mitochondrial dysfunction may contribute to this metabolic shift, induction of glycolysis itself could possibly contribute to mitochondrial dysfunction (Senyilmaz and Teleman 2015). An elevation in ROS production in response to impaired mitochondrial function could theoretically activate a DDR, triggering cellular senescence and activating their pro-ageing responses.
To determine whether mitochondrial dysfunction can promote cellular senescence, Wiley et al. depleted mtDNA in IMR90 fibroblasts, treated cells with electron transport chain inhibitors (rotenone or antimycin A) or depleted cells of the mitochondrial chaperone HSPA9 (Wiley et al. 2016). All treatments were shown to induce cell senescence as evident by reduced BrdU incorporation and increased senescence-associated beta galactosidase staining. To evaluate the potential role of mitochondria in orchestrating the pro-ageing features of the senescent phenotype, Correia‐Melo et al. demonstrated that senescent cells lacking mitochondria reduced many aspects of the senescent phenotype including the pro-inflammatory secretome, whilst maintaining ATP production via enhanced glycolysis (Correia-Melo et al. 2016). Findings by Passos et al. suggest that ROS are initially important for the maintenance of cell senescence through sustaining a persistent DNA damage response (DDR) (Passos et al. 2010). This may suggest that ROS production is initially beneficial during the transient appearance of cell senescence, but if senescent cells persist in tissues, likely owing to impaired clearance by the immune system (Burton and Faragher 2015), persistent senescent cells later become harmful.
In principle, these data suggest that disruption to mitochondrial function could promote cellular senescence, in addition to regulating the pro-ageing features of senescent cells. However, these studies do not provide evidence that such mechanistic processes occur during natural ageing in the absence of experimental intervention. Nevertheless, because senescent cells are associated with many age-related diseases, whether mitochondrial dysfunction is a cause or a consequence of cell senescence may not be a primary point of issue. What is of concern is the long-term biological impact of altered processes such as lipid metabolism and lipid peroxidation when senescent cells persist.
Lipids and lipid peroxidation during cellular senescence
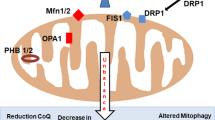
Mitochondrial dysfunction, increased lipid peroxidation and altered catabolism will affect the cellular lipidome during cell senescence. Alterations in lipid metabolism and the generation of oxidised lipids may be beneficial for the senescent program during the early stages of senescence induction, possibly through modulating inflammatory and immune responses (Lawrence et al. 2002; van Diepen et al. 2013; Yaqoob 2003). However, if senescent cells persist in tissues, changes in lipid composition can result in cell dysfunction, altered rates of fatty acid oxidation that can induce inflammation and increased lipid peroxidation that can promote damage to neighbouring cells (Fig. 1). These factors may contribute to ageing and age-related diseases. Research focused on altered lipid metabolism during cellular senescence, particularly regarding mitochondrial lipids, is in its infancy. However, in recent years, several studies have made progress in evaluating the senescent lipidome of fibroblasts.

Schematic demonstrating the possible link between cellular senescence, altered lipid metabolism and organismal ageing
One group investigated the alterations in a number of metabolites associated with the extracellular metabolome of fibroblasts induced to senesce via proliferative exhaustion or via γ-irradiation (James et al. 2015). They reported that a number of fatty acids and their precursors such as eicosapentaenoate, malonate, 7-alpha-hydroxy-3-oxo-4-cholestenoate and 1-stearoylglycerophosphoinositol were elevated during fibroblast senescence when compared with proliferating and quiescent cells, whereas linoleate, dihomo-linoleate, 10-heptadecenoate were depleted. Also amongst the secretory lipidome from senescent fibroblasts was an accumulation of monohydroxy fatty acids (2-hydroxypalmitate, 2-hydroxystearate, 3-hydroxydecanoate, 3-hydroxyoctanoate) and a phospholipid catabolite (glycerophosphorylcholine). It was suggested that whilst some of these changes may be due to oxidative stress, other observed increases may be a response to increased biomass commonly observed amongst senescent cells.
Maeda et al. (2009) investigated the regulation of fatty acid synthesis and ∆9-desaturation during cell senescence in human fibroblasts (Maeda et al. 2009). They found that the levels of fatty acid synthase and stearoyl-CoA desaturase-1 were decreased in senescent fibroblasts compared to proliferating fibroblasts, consequently leading to a decrease in monounsaturated fatty acids. In addition, reduced de novo synthesis of phospholipids with an associated increase in the formation of cholesterol in senescent cells was also observed and exogenous fatty acids were shown to be preferentially incorporated into the triacylglycerol pool of senescent cells.
In another study, the metabolic alterations associated with oncogene-induced senescence (OIS), using Ras-induced senescent human fibroblasts as a model were investigated (Quijano et al. 2012). Through the profiling of ~300 different intracellular metabolites, these authors showed that cells that have undergone OIS develop a metabolic signature which is distinct from cells which have undergone replicative senescence in response to extended in vitro cell culture. In the latter, a switch towards glycolysis has been observed that precedes the onset of senescence (Bittles and Harper 1984). In OIS, an increase in certain intracellular long chain fatty acids, including eicosanoate, dihomo-linoleate, mead acid and docosadienoate were observed. This altered metabolome was shown to associate with a decline in lipid synthesis and increases in fatty acid oxidation. Interestingly, the pro-inflammatory activity of the senescent secretome was reduced by inhibition of carnitine palmitoyltransferase 1, the rate limiting step in mitochondrial fatty acid oxidation, suggesting that alterations in lipid metabolism during OIS may play a role in regulating the pro-inflammatory senescent secretome. Although the mechanism underlying the increase in fatty acid levels during OIS were not fully explored, it may be due to promyelocytic leukemia (PML) activation of the fatty acid oxidation pathway through PPAR signalling (Aird and Zhang 2014). The differences between replicative senescence and OIS are intriguing; they may relate to the physiological need in preventing cancer to switch away from glycolysis as a rapid source of energy that is harnessed by cancer cells to enable them to proliferate rapidly versus the increasing insulin resistance that is seen in ageing and which associates with impaired oxidative metabolism (Burkart et al. 2016). However, while this and other studies have indicated an increase in glucose uptake during OIS, a number of other studies have observed either no change or a significant decrease in glucose uptake. This may relate to the timing of senescence induction, the cell type or the oncogene responsible.
A further study compared global lipid profiles and associated mRNA levels of proliferating and replicative senescent BJ fibroblasts; 19 specific polyunsaturated triacylglycerol species were identified as undergoing significant changes in lipid composition during cell senescence (Lizardo et al. 2017). In addition, significant changes in the expression of genes involved in specific lipid-related pathways, including glycerolipid metabolism, glycerophospholipid metabolism, unsaturated fatty acid synthesis and sphingolipid metabolism were observed during cell senescence. Based on these lipidomic and transcriptomic analysis, the authors postulated that activation of CD36-mediated fatty acid uptake and alteration to glycerolipid biosynthesis may contribute to the accumulation of triacylglycerols during cell senescence. It was suggested that these changes may be a mechanism to prevent lipotoxicity during elevated oxidative stress conditions during cell senescence.
In addition to an altered lipidome during cellular senescence, elevated ROS, likely from uncoupled mitochondria, can promote lipid peroxidation which potentiates cellular damage at distant sites. For example, stable aldehydes can diffuse from their site of generation and form adducts at distant locations, thereby propagating the responses and injury initiated by ROS (Ramana et al. 2013), including the induction of cell senescence in neighbouring cells. Flor and Kron observed an accumulation of lipid-derived aldehydes such as 4-hydroxy-2-nonenal (4-HNE) during accelerated senescence (Flor and Kron 2016). Whereas, the treatment of cells with either 4-HNE or low (5 Gy) γ-irradiation only generated low levels of cell senescence, combining both 4-HNE and 5 Gy γ-irradiation significantly elevated the senescence response. Furthermore, the use of the aldehyde-sequestering drug hydralazine blocked cell senescence induction by 25 Gy and etoposide treatment, demonstrating the potential importance of lipid peroxidation during therapy-induced senescence (Flor et al. 2016). Despite the highly damaging and pro-ageing potential of senescence-derived lipid peroxidation, little research has been conducted in this area and this requires further study.
Research on cell senescence has primarily been undertaken on fibroblasts and more research is required to explore whether the same phenomena are observed in cell-types linked to age-related disease such as in senescent adipocytes, pancreatic beta cells, renal proximal tubular epithelial cells and vascular endothelial cells. Whilst different types of senescent cells may share similarities in lipid metabolism, there may also be differences that are cell type-dependent or due to the mechanism of senescence induction and these require further study to better assess the role of altered lipid metabolism during ageing and disease. Finally, an important question to contemplate is whether the alterations in ROS, lipid metabolism and mitochondrial lipids observed during ageing and diseases are due solely to the presence of senescent cells or whether lipidomic changes can occur in absence of senescent cell accumulation.
Therapeutic opportunities
Agents that are capable of improving mitochondrial bioenergetics, enhancing glucose metabolism and reducing oxidative stress can promote healthy span (Zhang et al. 2016). Exercise has been found to stimulate mitochondrial biogenesis and activate the phase II antioxidant defence system and improved mitochondrial function is implicated in preventing age-related diseases (Sun et al. 2014). A class of mitochondria-located natural antioxidants are the carotenoids. AD patients exhibit significantly lower serum concentrations of carotenoids such as lutein, lycopene and zeaxanthin associated with increased lipid peroxidation products (Nolan et al. 2014). The enzymes responsible for metabolizing carotenoids are partitioned in the mitochondrial membranes and chronic lycopene administration significantly restores the mitochondrial respiratory enzyme activities in β-A1-42 treated rats (Horvath and Daum 2013). Prakash and Kumar studied the neuroprotective effect of lycopene against the β-amyloid induced cognitive impairment and mitochondria oxidative damage in rats. β-A1-42 treated animals showed poor memory retention and marked oxidative stress was indicated by significant increase in oxidative mitochondrial damage, IL-6, TNF-α, and caspase-3 activity. However, the administration of lycopene improved memory retention, attenuated mitochondrial-oxidative damage, reduced neuro-inflammation and restored brain-derived neurotrophic factor levels in β-A1-42 treated rats. The study also indicated that lycopene protected against β-A1-42 induced cognitive dysfunction and modulated amyloidogenesis (Prakash and Kumar 2014). Interventions using targeted carotenoids has the potential to rescue brain mitochondrial lipids, metabolism, redox state and energy stability, thereby increasing lifespan and delaying the appearance of disease (Kelly et al. 2015), but supplementation with these nutrients may only support and protect cognitive health if achieved early enough (Nolan et al. 2015).
Finally, more generally, inhibiting enzyme-catalysed lipid oxidation has also proven beneficial because of the role that oxidised arachidonic acid products, the eicosanoids, play in inflammation that underpins ageing. Non-steroidal anti- inflammatory drugs (NSAIDs), that include acetic acid, salicylate, propionic acid, fenamate, oxicam, and the COX-2 inhibitor classes, have analgesic, antipyretic, and anti-inflammatory properties. They are reported to have a beneficial effect on some neurodegenerative diseases (Rubio-Perez and Morillas-Ruiz 2012).
Mitochondria-specific antioxidants as a potential therapy in ROS-related diseases
There is a need to explore the potential of mitochondria-specific antioxidants as novel therapeutic strategies targeted towards the prevention, delay or treatment of mitochondria dysfunction in diseases related to mitochondria dysfunction (Mdaki et al. 2016).
An imbalance in the activity of ETC complexes I, II and III leads to ROS generation in both the mitochondrial matrix and cytosol, resulting in glutathione depletion and increased lipid peroxidation. A mitochondrion-targeted antioxidant peptide, SS31, has been reported to protect against oxidative damage (Ma et al. 2015). The antioxidant peptide has the ability to confer protective effects against mutant mitochondria and synaptic toxicities (Hemachandra Reddy et al. 2017). Hao et al. also reported that SS31 can reduce age-related activation of NFκB in mice (Hao et al. 2016). Hemachandra et al. observed SS31 was able to cross the blood brain barrier and reached the mitochondria. SS31 was found to enhance mitochondrial biogenesis and synaptic activity by reducing Aβ production, reducing mitochondrial dysfunction and maintaining mitochondrial dynamics in AD mouse models (Reddy et al. 2017).
Mitochondria-permeable antioxidants such as edaravone, idebenone, α-lipoic acid, carotenoids, vitamin E, and coenzyme Q10, and mitochondria-targeted antioxidants such as MitoQ, SkQ and astaxanthin (a ketocarotenoid from the xanthophyll family) are of increasing interest (Zhang et al. 2016). Balietti et al. reported astaxanthin as having a gender-related effect on ageing rat brain by exerting anti-inflammatory effects differentially in male and female brains (Balietti et al. 2016). Idebenone, MitoQ and SkQ have been specifically explored for the treatment of AD (Zhang et al. 2016; Selfridge et al. 2013). Idebenone (2,3-dimethoxy-5-methyl-6-(10-hydroxydecyl)-1,4-benzoquinonenoben), a short chain benzoquinone is structurally similar to coenzyme Q10 and functions both as an antioxidant and electron carrier, however, its clinical application has been limited to date because of gastrointestinal, neurotoxic or cardiotoxic side effects (Zhang et al. 2016). MitoQ a lipophilic molecule bearing a cation moiety, passes directly through the mitochondrial membrane where it accumulates and is recognised as an effective mitochondria-targeted antioxidant (Zhang et al. 2016). Another mitochondria targeted antioxidant is cationic SkQ [10-(6′-plastoquinonyl)decyltriphenyl-phosphonium)] which also accumulates in the mitochondria. Several studies have shown that SkQ protects cells from age related diseases. However, its safety and clinical usefulness need further investigations. Neither MitoQ nor SkQ are FDA-approved (Zhang et al. 2016).
Targeting senescent cells
If senescent cells contribute to the generation of lipid peroxidation products as a consequence of altered metabolic processes, then therapeutically targeting senescent cells to reverse such metabolic abnormalities may be a viable approach for reducing their impact during ageing. No doubt many of the strategies outlined above would also be pertinent for targeting senescent cells. For example, strategies which target fatty acid oxidation might reduce their harmful impact on localised tissue. Pharmacological inhibition (Etomoxir) of carnitine palmitoyltransferase 1, the rate limiting step in mitochondrial fatty acid oxidation was shown to normalise metabolic activity, as determined by oxygen consumption in OIS fibroblasts (Quijano et al. 2012).
Alternatively, if indeed senescent cells are a major source of lipid peroxidation products that promote ageing, then one strategy may be to remove senescent cells from tissues altogether. In fact, drugs have emerged that could potentially eliminate senescent cells to improve health (see Table 2) [reviewed in (de Keizer 2017; Soto-Gamez and Demaria 2017; Schmitt 2017)]. More recently, a FOXO4 peptide was shown to specifically induce apoptosis in senescent cells and reverse some aspects of ageing (Baar et al. 2017). Nanoparticles loaded with drugs targeted towards senescent cells have also been developed (Agostini et al. 2012; Thapa et al. 2017).
Finally, rather than waiting until senescent cells are present and then tackling the problems they cause, it would be better to prevent/reduce their appearance through life-style changes. For example, it has been demonstrated in mice that exercise can prevent diet-induced cell senescence and short-term dietary restriction has been shown to reduce the number of senescent cells in tissue of mice (Schafer et al. 2016; Wang et al. 2010). It was also suggested that dietary restriction improves mitochondrial function in tissues through reduction in cell senescence.
Conclusions
Ageing and age-related diseases have been associated with mitochondrial uncoupling and elevated ROS formation. Dysfunctional mitochondria predispose to altered lipid metabolism and increased lipid peroxidation products. Mitochondrial antioxidants that can restore function and prevent pathological lipid peroxidation are showing promise in slowing biological ageing and therefore they may offer benefit for slowing the progression to age-related diseases such as neurodegeneration. In parallel, newer drug classes are providing a different strategy to delay ageing through removal of senescent cells. Using these drugs as tools offer an opportunity to increase our understanding of whether the alterations in ROS, lipid metabolism and mitochondrial lipids observed during ageing and diseases are due to the accumulation of senescent cells.
References
AbouAssi H, Connelly MA, Bateman LA, Tune KN, Huebner JL, Kraus VB, Winegar DA, Otvos JD, Kraus WE, Huffman KM (2017) Does a lack of physical activity explain the rheumatoid arthritis lipid profile? Lipid Health Dis 16(1):39. doi:10.1186/s12944-017-0427-4
Ademowo OS, Dias HKI, Milic I, Devitt A, Moran R, Mulcahy R, Howard AN, Nolan JM, Griffiths HR (2017) Phospholipid oxidation and carotenoid supplementation in Alzheimer’s disease patients. Free Radic Biol Med 108:77–85. doi:10.1016/j.freeradbiomed.2017.03.008
Agostini A, Mondragón L, Bernardos A, Martínez-Máñez R, Marcos MD, Sancenón F, Soto J et al (2012) Targeted cargo delivery in senescent cells using capped mesoporous silica nanoparticles. Angew Chem Int Edit 51(42):10556–10560. doi:10.1002/anie.201204663
Aird KM, Zhang R (2014) Metabolic alterations accompanying oncogene-induced senescence. Mol Cell Oncol 1(3):e963481. doi:10.4161/23723548.2014.963481
Akalιn FA, Baltacιoğlu E, Alver A, Karabulut E (2007) Lipid peroxidation levels and total oxidant status in serum, saliva and gingival crevicular fluid in patients with chronic periodontitis. J Clin Periodontol 34(7):558–565. doi:10.1111/j.1600-051X.2007.01091.x
Argmann C, Dobrin R, Heikkinen S, Auburtin A, Pouilly L, Cock TA, Koutnikova H, Zhu J, Schadt EE, Auwerx J (2009) Ppargamma2 is a key driver of longevity in the mouse. PLoS Genet 5(12):e1000752. doi:10.1371/journal.pgen.1000752
Asselin C, Bouchard B, Tardif JC, Des Rosiers C (2006) Circulating 4-hydroxynonenal-protein thioether adducts assessed by gas chromatography-mass spectrometry are increased with disease progression and aging in spontaneously hypertensive rats. Free Radic Biol Med 41(1):97–105. doi:10.1016/j.freeradbiomed.2006.03.011
Aufschnaiter A, Kohler V, Diessl J, Peselj C, Carmona-Gutierrez D, Keller W, Büttner S (2017) Mitochondrial lipids in neurodegeneration. Cell Tissue Res 367(1):125–140. doi:10.1007/s00441-016-2463-1
Ayala A, Muñoz MF, Argüelles S (2014) Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014:31. doi:10.1155/2014/360438
Baar MP, Brandt RMC, Putavet DA, Klein JD, Derks KW, Bourgeois BR, Stryeck S et al (2017) Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169(1):132–147. doi:10.1016/j.cell.2017.02.031
Babizhayev MA, Kasus-Jacobi A (2009) State of the art clinical efficacy and safety evaluation of N-acetylcarnosine dipeptide ophthalmic prodrug. Principles for the delivery, self-bioactivation, molecular targets and interaction with a highly evolved histidyl-hydrazide structure in the treatment. Curr Clin Pharmacol 4(1):4–37. doi:10.2174/157488409787236074
Balietti M, Giannubilo SR, Giorgetti B, Solazzi M, Turi A, Casoli T, Ciavattini A, Fattorettia P (2016) The effect of astaxanthin on the aging rat brain: gender-related differences in modulating inflammation. J Sci Food Agric 96(2):615–618. doi:10.1002/jsfa.7131
Baltacıoğlu E, Yuva P, Aydın G, Alver A, Kahraman C, Karabulut E, Akalın FA (2014) Lipid peroxidation levels and total oxidant/antioxidant status in serum and saliva from patients with chronic and aggressive periodontitis. Oxidative stress index: a new biomarker for periodontal disease? J Periodontol 85(10):1432–1441. doi:10.1902/jop.2014.130654
Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH (2014) Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol 171(8):2017–2028. doi:10.1111/bph.12468
Bittles AH, Harper N (1984) Increased glycolysis in ageing cultured human diploid fibroblasts. Biosci Rep 4(9): 751–756. http://www.ncbi.nlm.nih.gov/pubmed/6509159
Bochkov VN, Oskolkova OV, Birukov KG, Levonen A-L, Binder CJ, Stöckl J (2010) Generation and biological activities of oxidized phospholipids. Antioxid Redox Signal 12(8):1009–1059. doi:10.1089/ars.2009.2597
Burkart AM, Tan K, Warren L, Iovino S, Hughes KJ, Kahn CR, Patti ME (2016) Insulin resistance in human iPS cells reduces mitochondrial size and function. Sci Rep. doi:10.1038/srep22788
Burton DGA, Faragher RGA (2015) Cellular senescence: from growth arrest to immunogenic conversion. Age 37(2):9764. doi:10.1007/s11357-015-9764-2
Burton DGA, Krizhanovsky V (2014) Physiological and pathological consequences of cellular senescence. Cell Mol Life Sci. doi:10.1007/s00018-014-1691-3
Cai M, Li J, Lin S, Chen X, Huang J, Jiang X, Yang L, Luo Y (2015) Mitochondria-targeted antioxidant peptide SS31 protects cultured human lens epithelial cells against oxidative stress. Curr Eye Res 40(8):822–829. doi:10.3109/02713683.2014.959607
Cai X, Yan A, Fu N, Wang S (2017) In vitro antioxidant activities of enzymatic hydrolysate from Schizochytrium Sp. and its hepatoprotective effects on acute alcohol-induced liver injury in vivo. Mar Drugs. doi:10.3390/md15040115
Calabrese V, Cornelius C, Mancuso C, Lentile R, Stella AMG, Butterfield DA (2010) Redox homeostasis and cellular stress response in aging and neurodegeneration. Method Mol Biol 610:285–308. doi:10.1007/978-1-60327-029-8_17
Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K et al (2015) Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 22(1):78–83. doi:10.1038/nm.4010
Chattopadhyay M, Khemka VK, Chatterjee G, Ganguly A, Mukhopadhyay S, Chakrabarti S (2015) Enhanced ROS production and oxidative damage in subcutaneous white adipose tissue mitochondria in obese and type 2 diabetes subjects. Mol Cell Biochem 399(1–2):95–103. doi:10.1007/s11010-014-2236-7
Childs BG, Durik M, Baker DJ, van Deursen JM (2015) Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 21(12):1424–1435. doi:10.1038/nm.4000
Choksi KB, Roberts LJ, DeFord JH, Rabek JP, Papaconstantinou J (2007) Lower levels of F(2)-isoprostanes in serum and livers of long-lived ames dwarf mice. Biochem Biophys Res Commun 364(4):761–764. doi:10.1016/j.bbrc.2007.10.100
Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, Mathis D (2012) PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue treg cells. Nature 486(7404):549–553. doi:10.1038/nature11132
Cipolletta D, Cohen P, Spiegelman BM, Benoist C, Mathis D (2015) Appearance and disappearance of the mRNA signature characteristic of Treg cells in visceral adipose tissue: age, diet, and PPAR gamma effects. Proc Natl Acad Sci USA 112(2):482–487. doi:10.1073/pnas.1423486112
Correia-Melo C, Marques FDM, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM et al (2016) Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 35(7):724–742. doi:10.15252/embj.201592862
Cracowski JL (2004) Isoprostanes: an emerging role in vascular physiology and disease? Chem Phys Lipid 128(1–2):75–83. doi:10.1016/j.chemphyslip.2003.10.006
Cui H, Kong Y, Zhang H (2012) Oxidative stress, mitochondrial dysfunction, and aging. J Signal Transduct. doi:10.1155/2012/646354
Dasuri K, Nguyen A, Zhang L, Fernandez-Kim OS, Bruce-Keller AJ, Blalock BA, De Cabo R, Keller JN (2009) Comparison of rat liver and brain proteasomes for oxidative stress-induced inactivation: influence of ageing and dietary restriction. Free Radic Res 43(1):28–36. doi:10.1080/10715760802534812
Davidson MH, Maki KC, Karp SK, Ingram KA (2002) Management of hypercholesterolaemia in postmenopausal women. Drugs Aging 19(3):169–178
de Keizer PLJ (2017) The fountain of youth by targeting senescent cells? Trends Mol Med 23(1):6–17. doi:10.1016/j.molmed.2016.11.006
De Vecchi AF, Bamonti F, Novembrino C, Ippolito S, Guerra L, Lonati S, Salini S, Aman CS, Scurati-Manzoni E, Cighetti G (2009) Free and total plasma malondialdehyde in chronic renal insufficiency and in dialysis patients. Nephrol Dial Transpl 24(8):2524–2529. doi:10.1093/ndt/gfp102
Di Domenico F, Tramutola A, Butterfield DA (2016) Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic Biol Med. doi:10.1016/j.freeradbiomed.2016.10.490
Diamond JM, Porteous MK, Roberts LJ, Wickersham N, Rushefski M, Kawut SM, Shah RJ et al (2016) The relationship between plasma lipid peroxidation products and primary graft dysfunction after lung transplantation is modified by donor smoking and reperfusion hyperoxia. J Heart Lung Transplant 35(4):500–507. doi:10.1016/j.healun.2015.12.012
Dias IH, Brown CLR, Polidori-Nelles MC, Lip GY, Griffiths HR (2015) LDL-lipids from patients with hypercholesterolaemia and Alzheimer’s disease are inflammatory to microvascular endothelial cells: mitigation by statin intervention. Clin Sci. doi:10.1042/CS20150351
Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C et al (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444(7119):638–642. doi:10.1038/nature05327
Dmitriev LF, Titov VN (2010) Lipid peroxidation in relation to ageing and the role of endogenous aldehydes in diabetes and other age-related diseases. Ageing Res Rev 9(2):200–210. doi:10.1016/j.arr.2009.09.004
Elustondo P, Martin LA, Karten B (2017) Mitochondrial Cholesterol Import. Biochim Biophys Acta (BBA)—Mol Cell Biol Lipid 1862(1):90–101. doi:10.1016/j.bbalip.2016.08.012
Esterbauer H, Schaur RJ, Zollner H (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11(1):81–128. doi:10.1016/0891-5849(91)90192-6
Flaquer A, Rospleszcz S, Reischl E, Zeilinger S, Prokisch H, Meitinger T, Meisinger C et al (2015) Mitochondrial GWA analysis of lipid profile identifies genetic variants to be associated with HDL cholesterol and triglyceride levels. PLoS ONE 10(5):e0126294
Flor AC, Kron SJ (2016) Lipid-derived reactive aldehydes link oxidative stress to cell senescence. Cell Death Dis 7(9):e2366. doi:10.1038/cddis.2016.275
Flor AC, Doshi AP, Kron SJ (2016) Modulation of therapy-induced senescence by reactive lipid aldehydes. Cell Death Discov 2(July):16045. doi:10.1038/cddiscovery.2016.45
Fulop T, Le Page A, Garneau H, Azimi N, Baehl S, Dupuis G, Pawelec G, Larbi A (2012) Aging, immunosenescence and membrane rafts: the lipid connection. Longev Heal 1:6. doi:10.1186/2046-2395-1-6
Gandhi S, Abramov AY (2012) Mechanism of oxidative stress in neurodegeneration. Oxid Med Cell Longev. doi:10.1155/2012/428010
Gao D, Bailey CJ, Griffiths HR (2009) Metabolic memory effect of the saturated fatty acid, palmitate, in monocytes. Biochem Biophys Res Commun 388(2):278–282. doi:10.1016/j.bbrc.2009.07.160
Gao D, Pararasa C, Dunston CR, Bailey CJ, Griffiths HR (2012) Palmitate promotes monocyte atherogenicity via de novo ceramide synthesis. Free Radic Biol Med 53(4):796–806. doi:10.1016/j.freeradbiomed.2012.05.026
García-Flores LA, Medina S, Martínez-Hernández P, Oger C, Galano JM, Durand T, Casas-Pina T, Ferreres F, Gil-Izquierdo A (2017) Snapshot situation of oxidative degradation of the nervous system, kidney, and adrenal glands biomarkers-neuroprostane and dihomo-isoprostanes-urinary biomarkers from infancy to elderly adults. Redox Biol 11:586–591. doi:10.1016/j.redox.2017.01.008
Gaschler MM, Stockwell BR (2017) Lipid peroxidation in cell death. Biochem Biophys Res Commun 482(3):419–425. doi:10.1016/j.bbrc.2016.10.086
Gaudin M, Panchal M, Auzeil N, Duyckaerts C, Brunelle A, Laprévote O, Touboul D (2012) Choline-containing phospholipids in microdissected human Alzheimer’s disease brain senile plaque versus neuropil. Bioanalysis 4(17):2153–5159. doi:10.4155/bio.12.189
Geng H, Zhang X, Wang C, Zhao M, Chunxiao Y, Zhang B, Wang Y, Ban B, Zhao J (2015) Even mildly elevated TSH is associated with an atherogenic lipid profile in postmenopausal women with subclinical hypothyroidism. Endocr Res 40(1):1–7. doi:10.3109/07435800.2013.879166
Girotti AW (1998) Lipid hydroperoxide generation, turnover, and effector action in biological systems. J Lipid Res 39 (8): 1529–1542. http://www.jlr.org/content/39/8/1529.abstract
Goh VH, Tong TY, Mok HP, Said B (2007) Differential impact of aging and gender on lipid and lipoprotein profiles in a cohort of healthy chinese singaporeans. Asian J Androl 9(6):787–794. doi:10.1111/j.1745-7262.2007.00294.x
Greig FH, Kennedy S, Spickett CM (2012) Physiological effects of oxidized phospholipids and their cellular signaling mechanisms in inflammation. Free Radic Biol Med 52(2):266–280. doi:10.1016/j.freeradbiomed.2011.10.481
Griffiths HR, Gao D, Pararasa C (2017) Redox regulation in metabolic programming and inflammation. Redox Biology 12(February):50–57. doi:10.1016/j.redox.2017.01.023
Hall ED, Wang JA, Bosken JM, Singh IN (2016) Lipid peroxidation in brain or spinal cord mitochondria after injury. J Bioenerg Biomembr 48(2):169–174. doi:10.1007/s10863-015-9600-5
Halliwell B (2001) Free radicals and other reactive species in disease. In eLS. Wiley, Hoboken. doi:10.1002/9780470015902.a0002269.pub3
Han X, Yang J, Cheng H, Yang K, Abendschein DR, Gross RW (2005) Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry 44(50):16684–16694. doi:10.1021/bi051908a
Hao ZH, Huang Y, Wang MR, Huo TT, Jia Q, Feng RF, Fan P, Wang JH (2016) SS31 Ameliorates age-related activation of NF-κB signaling in senile mice model, SAMP8. Oncotarget 8(2):1983–1992. doi:10.18632/oncotarget.14077
Heffern CT, Pocivavsek L, Birukova AA, Moldobaeva N, Bochkov VN, Lee KYC, Birukov KG (2013) Thermodynamic and kinetic investigations of the release of oxidized phospholipids from lipid membranes and its effect on vascular integrity. Chem Phys Lipid 175–176:9–19. doi:10.1016/j.chemphyslip.2013.07.003
Heilbronn L, Smith SR, Ravussin E (2004) Failure of fat cell proliferation, mitochondrial function and fat oxidation results in ectopic fat storage, insulin resistance and type II diabetes mellitus. Int J Obes Relat Metab Disord 28(Suppl 4):S12–S21. doi:10.1038/sj.ijo.0802853
Helkin A, Stein JJ, Lin S, Siddiqui S, Maier KG, Gahtan V (2016) Dyslipidemia part 1—review of lipid metabolism and vascular cell physiology. Vasc Endovasc Surg 50(2):107–118. doi:10.1177/1538574416628654
Reddy PH, Manczak M, Kandimalla R (2017) Mitochondria-targeted small molecule SS31, a potential candidate for the treatment of Alzheimer’s disease. Hum Mol Genet. doi:10.1093/hmg/ddx052
Herrera ACS, Victorino VJ, Campos FC, Verenitach BD, Lemos LT, Aranome AM, Oliveira SR et al (2014) Impact of tumor removal on the systemic oxidative profile of patients with breast cancer discloses lipid peroxidation at diagnosis as a putative marker of disease recurrence. Clin Breast Cancer 14(6):451–459. doi:10.1016/j.clbc.2014.05.002
Hong JH, Kang JW, Kim DK, Baik SH, Kim KH, Shanta SR, Jung JH, Mook-Jung I, Kim KP (2016) Global changes of phospholipids identified by MALDI imaging mass spectrometry in a mouse model of Alzheimer’s disease. J Lipid Res 57(1):36–45. doi:10.1194/jlr.M057869
Horvath SE, Daum G (2013) Lipids of mitochondria. Prog Lipid Res 52(4):590–614. doi:10.1016/j.plipres.2013.07.002
Inglés M, Gambini J, Carnicero JA, García-García FJ, Rodríguez-Mañas L, Olaso-González G, Dromant M, Borrás C, Viña J (2014) Oxidative stress is related to frailty, not to age or sex, in a geriatric population: lipid and protein oxidation as biomarkers of frailty. J Am Geriatr Soc 62(7):1324–1328. doi:10.1111/jgs.12876
Jackson MJ, Papa S, Bolanos J, Bruckdorfer R, Carlsen H, Elliott RM, Flier J et al (2002) Antioxidants, reactive oxygen and nitrogen species, gene induction and mitochondrial function. Mol Asp Med 23(1–3):209–285
James EL, Michalek RD, Pitiyage GN, de Castro AM, Vignola KS, Jones J, Mohney RP, Karoly ED, Prime SS, Parkinson EK (2015) Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J Proteom Res 14(4):1854–1871. doi:10.1021/pr501221g
Kahle M, Schäfer A, Seelig A, Schultheiß J, Wu M, Aichler M, Leonhardt J et al (2015) High fat diet-induced modifications in membrane lipid and mitochondrial-membrane protein signatures precede the development of hepatic insulin resistance in mice. Mol Metab 4(1):39–50. doi:10.1016/j.molmet.2014.11.004
Kelly D, Coen RF, Akuffo KO, Beatty S, Dennison J, Moran R, Stack J, Howard AN, Mulcahy R, Nolan JM (2015) Cognitive function and its relationship with macular pigment optical density and serum concentrations of its constituent carotenoids. J Alzheimer’s Dis 48(1):261–277. doi:10.3233/JAD-150199
Kennedy MA, Moffat TC, Gable K, Ganesan S, Niewola-Staszkowska K, Johnston A, Nislow C et al (2016) A signaling lipid associated with Alzheimer’s disease promotes mitochondrial dysfunction. Sci Rep 6(January):19332. doi:10.1038/srep19332
Kiebish MA, Yang K, Sims HF, Jenkins CM, Liu X, Mancuso DJ, Zhao Z et al (2012) Myocardial regulation of lipidomic flux by cardiolipin synthase: setting the beat for bioenergetic efficiency. J Biol Chem 287(30):25086–25097. doi:10.1074/jbc.M112.340521
Kobayashi T, Zadravec D, Jacobsson A (2007) ELOVL2 overexpression enhances triacylglycerol synthesis in 3T3-L1 and F442A cells. FEBS Lett 581(17):3157–3163. doi:10.1016/j.febslet.2007.05.081
Kolamunne RT, Dias IH, Vernallis AB, Grant MM, Griffiths HR (2013) Nrf2 activation supports cell survival during hypoxia and hypoxia/reoxygenation in cardiomyoblasts; the roles of reactive oxygen and nitrogen species. Redox Biol 1(1):418–426. doi:10.1016/j.redox.2013.08.002
Krag SS (2011) Special issue: human and murine redox protein atlases. Biochim Biophys Acta (BBA)—Gen Subj 1810(1):1. doi:10.1016/j.bbagen.2010.11.003
Kühlbrandt W (2015) Structure and function of mitochondrial membrane protein complexes. BMC Biol 13(1):89. doi:10.1186/s12915-015-0201-x
Lawrence T, Willoughby DA, Gilroy DW (2002) Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat Rev Immunol 2(10):787–795. doi:10.1038/nri915
Li T, Zhang Z, Kolwicz SC, Abell L, Roe ND, Kim M, Zhou B et al (2017) Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab 25(2):374–385. doi:10.1016/j.cmet.2016.11.005
Liu P, Kerr BJ, Chen C, Weber TE, Johnston LJ, Shurson GC (2014a) Methods to create thermally oxidized lipids and comparison of analytical procedures to characterize peroxidation. J Anim Sci 92(7):2950–2959. doi:10.2527/jas.2012-5708
Liu Y, Zhang D, Wu Y, Ji B (2014b) Docosahexaenoic acid aggravates photooxidative damage in retinal pigment epithelial cells via lipid peroxidation. J Photochem Photobiol, B 140:85–93. doi:10.1016/j.jphotobiol.2014.07.016
Lizardo DY, Lin YL, Gokcumen O, Atilla-Gokcumen GE (2017) Regulation of Lipids Is Central to Replicative Senescence. Mol Biosyst. doi:10.1039/c6mb00842a
Ma DWL, Seo J, Switzer KC, Fan YY, McMurray DN, Lupton JR, Chapkin RS (2004) N-3 PUFA and membrane microdomains: a new frontier in bioactive lipid research. J Nutr Biochem 15(11):700–706. doi:10.1016/j.jnutbio.2004.08.002
Ma W, Zhu X, Ding X, Li T, Hu Y, Hu X, Yuan L et al (2015) Protective effects of SS31 on t-BHP induced oxidative damage in 661 W cells. Mol Med Rep 12(4):5026–5034. doi:10.3892/mmr.2015.4055
Maeda M, Scaglia N, Igal RA (2009) Regulation of fatty acid synthesis and delta9-desaturation in senescence of human fibroblasts. Life Sci 84(3–4):119–124. doi:10.1016/j.lfs.2008.11.009
Mauerhofer C, Philippova M, Oskolkova OV, Bochkov VN (2016) Hormetic and anti-inflammatory properties of oxidized phospholipids. Mol Asp Med 49:78–90. doi:10.1016/j.mam.2016.02.003
Mdaki KS, Larsen TD, Weaver LJ, Baack ML (2016) Age related bioenergetics profiles in isolated rat cardiomyocytes using extracellular flux analyses. PLoS ONE 11(2):e0149002. doi:10.1371/journal.pone.0149002
Mecocci P, Cherubini A, Beal MF, Cecchetti R, Chionne F, Polidori MC, Romano G, Senin U (1996) Altered mitochondrial membrane fluidity in AD brain. Neurosci Lett 207 (2): 129–132. http://www.ncbi.nlm.nih.gov/pubmed/8731438
Mollinedo F, Gajate C (2015) Lipid rafts as major platforms for signaling regulation in cancer. Adv Biol Regul 57(January):130–146. doi:10.1016/j.jbior.2014.10.003
Moreira PI, Duarte AI, Santos MS, Rego AC, Oliveira CR (2009) An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer’s disease. J Alzheimer’s Dis 16(4):741–761. doi:10.3233/JAD-2009-0972
Morita M, Ishida N, Uchiyama K, Yamaguchi K, Itoh Y, Shichiri M, Yoshida Y et al (2012) Fatty liver induced by free radicals and lipid peroxidation. Free Radic Res 46(6):758–765. doi:10.3109/10715762.2012.677840
Muñoz-Espín D, Serrano M (2014) Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15(7):482–496. doi:10.1038/nrm3823
Nakamura MT, Yudell BE, Loor JJ (2014) Regulation of energy metabolism by long-chain fatty acids. Prog Lipid Res 53:124–144. doi:10.1016/j.plipres.2013.12.001
Naudí A, Cabré R, Dominguez-Gonzalez M, Ayala V, Jové M, Mota-Martorell N, Piñol-Ripoll G et al (2017) Region-specific vulnerability to lipid peroxidation and evidence of neuronal mechanisms for polyunsaturated fatty acid biosynthesis in the healthy adult human central nervous system. Biochim Biophys Acta (BBA)—Mol Cell Biol Lipids 1862(5):485–495. doi:10.1016/j.bbalip.2017.02.001
Ng ML, Wadham CA, Sukocheva OA (2017) The role of sphingolipid signalling in diabetes-associated pathologies (Review). Int J Mol Med 39(2):243–252. doi:10.3892/ijmm.2017.2855
Nolan JM, Loskutova E, Howard AN, Moran R, Mulcahy R, Stack J, Bolger M et al (2014) Macular pigment, visual function, and macular disease among subjects with Alzheimer’s disease: an exploratory study. J Alzheimer’s Dis 42(4):1191–1202. doi:10.3233/JAD-140507
Nolan JM, Loskutova E, Howard A, Mulcahy R, Moran R, Stack J, Bolger M et al (2015) The impact of supplemental macular carotenoids in Alzheimer’s disease: a randomized clinical trial. J Alzheimer’s Dis 44(4):1157–1169. doi:10.3233/JAD-142265
Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y (2000) Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J 351(1):183–193. doi:10.1042/0264-6021:3510183
Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM (2004) Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res 94(1):53–59. doi:10.1161/01.RES.0000109416.56608.64
Pararasa C, Bailey CJ, Griffiths HR (2015) Ageing, adipose tissue, fatty acids and inflammation. Biogerontology 16(2):235–248. doi:10.1007/s10522-014-9536-x
Pararasa C, Ikwuobe J, Shigdar S, Boukouvalas A, Nabney IT, Brown JE, Devitt A, Bailey CJ, Bennett SJ, Griffiths HR (2016) Age-associated changes in long-chain fatty acid profile during healthy aging promote pro-inflammatory monocyte polarization via ppargamma. Aging Cell 15(1):128–139. doi:10.1111/acel.12416
Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S et al (2010) Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol 6:347. doi:10.1038/msb.2010.5
Pavón N, Gallardo JC, Hernández-Esquivel LM, El-Hafidi M, Buelna-Chontal M, Zazueta C, Rodríguez-Enríquez S, Chávez E (2011) On the properties of calcium-induced permeability transition in neonatal heart mitochondria. J Bioenerg Biomembr 43(6):757–764. doi:10.1007/s10863-011-9401-4
Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trézéguet V, Lauquin GJ, Brandolin G (2003) Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 426(6962):39–44. doi:10.1038/nature02056
Philippova M, Resink T, Erne P, Bochkov V (2014) Oxidised phospholipids as biomarkers in human disease. Swiss Med Wkly 144(January):w14037. doi:10.4414/smw.2014.14037
Phillips DC, Allen K, Griffiths HR (2002) Synthetic ceramides induce growth arrest or apoptosis by altering cellular redox status. Arch Biochem Biophys 407(1):15–24
Pizzimenti S, Ciamporcero ES, Daga M, Pettazzoni P, Arcaro A, Cetrangolo G, Minelli R et al (2013) Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front Physiol 4:242. doi:10.3389/fphys.2013.00242
Prakash A, Kumar A (2014) Implicating the role of lycopene in restoration of mitochondrial enzymes and BDNF levels in β-amyloid induced Alzheimer’s disease. Eur J Pharmacol 741(October):104–111. doi:10.1016/j.ejphar.2014.07.036
Pytel E, Jackowska P, Chwatko G, Olszewska-Banaszczyk M, Koter-Michalak M, Kubalczyk P, Broncel M (2016) Intensive statin therapy, used alone or in combination with ezetimibe, improves homocysteine level and lipid peroxidation to a similar degree in patients with coronary artery diseases. Pharmacol Rep 68(2):344–348. doi:10.1016/j.pharep.2015.08.019
Quijano C, Cao L, Fergusson MM, Romero H, Liu J, Gutkind S, Rovira II, Mohney RP, Karoly ED, Finkel T (2012) Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle 11(7):1383–1392. doi:10.4161/cc.19800
Ramana KV, Srivastava S, Singhal SS (2013) Lipid peroxidation products in human health and disease. Oxid Med Cell Longev 2013:1–3. doi:10.1155/2013/583438
Ratnayake S, Dias IH, Lattman E, Griffiths HR (2013) Stabilising cysteinyl thiol oxidation and nitrosation for proteomic analysis. J Proteom 92:160–170. doi:10.1016/j.jprot.2013.06.019
Reddy PH, Beal MF (2005) Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Res Rev 49(3):618–632. doi:10.1016/j.brainresrev.2005.03.004
Reis A (2017) Oxidative phospholipidomics in health and disease: achievements, challenges and hopes. Free Radic Biol Med. doi:10.1016/j.freeradbiomed.2017.01.014
Reynolds CA, Gatz M, Prince JA, Berg S, Pedersen NL (2010) Serum lipid levels and cognitive change in late life. J Am Geriatr Soc 58(3):501–509. doi:10.1111/j.1532-5415.2010.02739.x
Reynolds LM, Taylor JR, Ding J, Lohman K, Johnson C, Siscovick D, Burke G et al (2014) Age-related variations in the methylome associated with gene expression in human monocytes and T cells. Nat Commun 5:5366. doi:10.1038/ncomms6366
Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11(8):973–979. doi:10.1038/ncb1909
Rossiello F, Herbig U, Longhese MP, Fumagalli M, di Fagagna FD (2014) Irreparable telomeric DNA damage and persistent DDR signalling as a shared causative mechanism of cellular senescence and ageing. Curr Opin Genet Dev 26(June):89–95. doi:10.1016/j.gde.2014.06.009
Rubio-Perez JM, Morillas-Ruiz JM (2012) A review: inflammatory process in Alzheimer’s disease, role of cytokines. Sci World J. doi:10.1100/2012/756357
Santini SA, Spada C, Bononi F, Foschia F, Mutignani M, Perri V, Giardina B, Gentiloni Silveri N, Costamagna G (2003) Enhanced lipoperoxidation products in pure pancreatic juice: evidence for organ-specific oxidative stress in chronic pancreatitis. Dig Liver Dis 35(12):888–892. doi:10.1016/j.dld.2003.09.004
Sarban S, Kocyigit A, Yazar M, Isikan UE (2005) Plasma total antioxidant capacity, lipid peroxidation, and erythrocyte antioxidant enzyme activities in patients with rheumatoid arthritis and osteoarthritis. Clin Biochem 38(11):981–986. doi:10.1016/j.clinbiochem.2005.08.003
Schafer MJ, White TA, Evans G, Tonne JM, Verzosa GC, Stout MB, Mazula DL et al (2016) Exercise prevents diet-induced cellular senescence in adipose tissue. Diabetes 65(6):1606–1615. doi:10.2337/db15-0291
Schlame M, Greenberg ML (2017) Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim Biophys Acta (BBA)—Mol Cell Biol. Lipids 1862(1):3–7. doi:10.1016/j.bbalip.2016.08.010
Schmitt R (2017) Senotherapy: growing old and staying young? Pflüg Arch—Eur J Physiol. doi:10.1007/s00424-017-1972-4
Selfridge JE, Lezi E, Lu J, Swerdlow RH (2013) Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol Dis 51:3–12. doi:10.1016/j.nbd.2011.12.057
Senyilmaz D, Teleman AA (2015) Chicken or the egg: warburg effect and mitochondrial dysfunction. F1000Prime Reports 7 (April): 41. doi:10.12703/P7-41
Sepe A, Tchkonia T, Thomou T, Zamboni M, Kirkland JL (2011) Aging and regional differences in fat cell progenitors—a mini-review. Gerontology 57(1):66–75. doi:10.1159/000279755
Sheeran FL, Pepe S (2016) Posttranslational modifications and dysfunction of mitochondrial enzymes in human heart failure. Am J Physiol Endocrinol Metab 311(2):E449–E460. doi:10.1152/ajpendo.00127.2016
Shen Z, Ye C, McCain K, Greenberg ML (2015) The role of cardiolipin in cardiovascular health. Biomed Res Int 2015:12. doi:10.1155/2015/891707
Shibata T, Shimizu K, Hirano K, Nakashima F, Kikuchi R, Matsushita T, Uchida K (2017) Adductome-based identification of biomarkers for lipid peroxidation. J Biol Chem. doi:10.1074/jbc.M116.762609
Simoncini C, Orsucci D, Caldarazzo Ienco E, Siciliano G, Bonuccelli U, Mancuso M (2015) Alzheimer’s pathogenesis and its link to the mitochondrion. Oxid Med Cell Longev. doi:10.1155/2015/803942
Soto-Gamez A, Demaria M (2017) Therapeutic interventions for aging: the case of cellular senescence. Drug Discov Today. doi:10.1016/j.drudis.2017.01.004
Sousa BC, Pitt AR, Spickett CM (2017) Chemistry and analysis of HNE and other prominent carbonyl-containing lipid oxidation compounds. Free Radic Biol Med. doi:10.1016/j.freeradbiomed.2017.02.003
Sun LJ, Zhang Y, Liu JK (2014) Exercise and aging: regulation of mitochondrial function and redox system. Sheng Li Ke Xue Jin Zhan [Progress in Physiology] 45 (5): 321–26. http://www.ncbi.nlm.nih.gov/pubmed/25764789
Swerdlow RH, Khan SM (2004) A ‘mitochondrial cascade hypothesis’ for sporadic Alzheimer’s disease. Med Hypotheses 63(1):8–20. doi:10.1016/j.mehy.2003.12.045
Tanaka M, Kovalenko SA, Gong JS, Borgeld HJ, Katsumata K, Hayakawa M, Yoneda M, Ozawa T (1996) Accumulation of deletions and point mutations in mitochondrial genome in degenerative diseases. Ann NY Acad Sci 786: 102–11. http://www.ncbi.nlm.nih.gov/pubmed/8687011
Tejovathi B, Suchitra MM, Suresh V, Reddy VS, Sachan A, Srinivas Rao PVLN, Bitla AR (2013) Association of lipid peroxidation with endothelial dysfunction in patients with overt hypothyroidism. Exp Clin Endocrinol Diabetes 121(5):306–309. doi:10.1055/s-0032-1333298
Thapa RK, Nguyen HT, Jeong JH, Kim JR, Choi HG, Yong CS, Kim JO (2017) Progressive slowdown/prevention of cellular senescence by CD9-targeted delivery of rapamycin using lactose-wrapped calcium carbonate nanoparticles. Sci Rep 7:43299. doi:10.1038/srep43299
Tindale LC, Leach S, Spinelli JJ, Brooks-Wilson AR (2017) Lipid and Alzheimer’s disease genes associated with healthy aging and longevity in healthy oldest-old. Oncotarget. doi:10.18632/oncotarget.15296
Torrao RC, Bennett SJ, Brown JE, Griffiths HR (2014) Does metabolic reprogramming underpin age-associated changes in T cell phenotype and function? Free Radic Biol Med 71:26–35. doi:10.1016/j.freeradbiomed.2014.03.002
Torres-Villalobos G, Hamdan-Perez N, Diaz-Villasenor A, Tovar AR, Torre-Villalvazo I, Ordaz-Nava G, Moran-Ramos S et al (2016) Autologous subcutaneous adipose tissue transplants improve adipose tissue metabolism and reduce insulin resistance and fatty liver in diet-induced obesity rats. Physiol Rep. doi:10.14814/phy2.12909
Umeno A, Biju V, Yoshida Y (2017) In vivo ROS production and use of oxidative stress—derived biomarkers to detect the onset of diseases such as Alzheimer’s disease, Parkinson’s disease, and diabetes. Free Radic Res. doi:10.1080/10715762.2017.1315114
Vadvalkar SS, Matsuzaki S, Eyster CA, Giorgione JR, Bockus LB, Kinter CS, Kinter M, Humphries KM (2017) Decreased mitochondrial pyruvate transport activity in the diabetic heart: role of MPC2 acetylation. J Biol Chem. doi:10.1074/jbc.M116.753509
van Deursen JM (2014) The role of senescent cells in ageing. Nature 509(7501):439–446. doi:10.1038/nature13193
van Diepen JA, Berbée JF, Havekes LM, Rensen PC (2013) Interactions between inflammation and lipid metabolism: relevance for efficacy of anti-inflammatory drugs in the treatment of atherosclerosis. Atherosclerosis 228(2):306–315. doi:10.1016/j.atherosclerosis.2013.02.028
van Lier JE, Mast N, Pikuleva IA (2015) Cholesterol hydroperoxides as substrates for cholesterol-metabolizing cytochromes P450 and alternative sources of 25-hydroxycholesterol and other oxysterols. Angew Chem 54(38):11138–11142. doi:10.1002/anie.201505002
Vaziri ND, Norris K (2011) Lipid disorders and their relevance to outcomes in chronic kidney disease. Blood Purif 31(1–3):189–196. doi:10.1159/000321845
Vuppalanchi R, Ghabril M, Chalasani N, Juluri R, Bell LN, Kamendulis L, Klaunig JE, Saxena R, Agarwal D, Johnson MS (2011) Oxidative stress in chronic liver disease: relationship between peripheral and hepatic measurements. Am J Med Sci 342(4):314–317. doi:10.1097/MAJ.0b013e31821d9905
Walter M (2009) Interrelationships among HDL metabolism, aging, and atherosclerosis. Arterioscler Thromb Vasc Biol 29(9):1244–1250. doi:10.1161/atvbaha.108.181438
Wan M, Hua X, Su J, Thiagarajan D, Frostegård AG, Haeggström JZ, Frostegård JZ (2014) Oxidized but not native cardiolipin has pro-inflammatory effects, which are inhibited by annexin A5. Atherosclerosis 235(2):592–598. doi:10.1016/j.atherosclerosis.2014.05.913
Wang C, Maddick M, Miwa S, Jurk D, Czapiewski R, Saretzki G, Langie SA, Godschalk RW, Cameron K, von Zglinicki T (2010) Adult-onset, short-term dietary restriction reduces cell senescence in mice. Aging 2(9):555–566. doi:10.18632/aging.100196
Wang Y, Chang J, Liu X, Zhang X, Zhang S, Zhang X, Zhou D, Zheng G (2016) Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging 8(11):2915–2926. doi:10.18632/aging.101100
Wei Y, Qi B, Xu J, Zhou G, Chen S, Ouyang P, Liu S (2014) Age- and sex-related difference in lipid profiles of patients hospitalized with acute myocardial infarction in East China. J Clin Lipidol 8(6):562–567. doi:10.1016/j.jacl.2014.09.006
Wensaas AJ, Rustan AC, Just M, Berge RK, Drevon CA, Gaster M (2009) Fatty acid incubation of myotubes from humans with type 2 diabetes leads to enhanced release of β-oxidation products because of impaired fatty acid oxidation. Eff Tetr Acid Eicosapentaenoic Acid 58(3):527–535. doi:10.2337/db08-1043
Whiley L, Sen A, Heaton J, Proitsi P, García-Gómez D, Leung R, Smith N et al (2014) Evidence of altered phosphatidylcholine metabolism in Alzheimer’s disease. Neurobiol Aging 35(2):271–278. doi:10.1016/j.neurobiolaging.2013.08.001
Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K et al (2016) Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab 23(2):303–314. doi:10.1016/j.cmet.2015.11.011
Wong-ekkabut J, Xu Z, Triampo W, Tang IM, Tieleman DP, Monticelli L (2007) Effect of lipid peroxidation on the properties of lipid bilayers: a molecular dynamics study. Biophys J 93(12):4225–4236. doi:10.1529/biophysj.107.112565
Wood LG, Gibson PG, Garg ML (2003) Biomarkers of lipid peroxidation, airway inflammation and asthma. Eur Respir J 21(1):177–186. doi:10.1183/09031936.03.00017003a
Yao J, Diaz Brinton R (2011) Targeting mitochondrial bioenergetics for Alzheimer’s prevention and treatment. Curr Pharm Des 17 (31): 3474–3479. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4209948&tool=pmcentrez&rendertype=abstract
Yaqoob P (2003) Lipids and the immune response: from molecular mechanisms to clinical applications. Curr Opin Clin Nutr Metab Care 6(2):133–150. doi:10.1097/01.mco.0000058589.27240.d1
Yin H, Xu L, Porter NA (2011) Free radical lipid peroxidation: mechanisms and analysis. Chem Rev 111(10):5944–5972. doi:10.1021/cr200084z
Yin F, Sancheti H, Liu Z, Cadenas E (2015) Mitochondrial function in ageing: coordination with signalling and transcriptional pathways. J Physiol. doi:10.1113/JP270541
Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, Vadai E et al (2016) Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun 7. doi:10.1038/ncomms11190
Zhang H, Forman HJ (2016) 4-hydroxynonenal-mediated signaling and aging. Free Radic Biol Med. doi:10.1016/j.freeradbiomed.2016.11.032
Zhang ZW, Xu XC, Liu T, Yuan S (2016) Mitochondrion-permeable antioxidants to treat ROS-burst-mediated acute diseases. Oxid Med Cell Longev 2016:6859523. doi:10.1155/2016/6859523
Zheng J, Mutcherson Ii R, Helfand SL (2005) Calorie restriction delays lipid oxidative damage in drosophila melanogaster. Aging Cell 4(4):209–216. doi:10.1111/j.1474-9726.2005.00159.x
Zheng R, Heck DE, Mishin V, Black AT, Shakarjian MP, Kong ANT, Laskin DL, Laskin JD (2014) Modulation of keratinocyte expression of antioxidants by 4-hydroxynonenal, a lipid peroxidation end product. Toxicol Appl Pharmacol 275(2):113–121. doi:10.1016/j.taap.2014.01.001
Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK et al (2015) The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14(4):644–658. doi:10.1111/acel.12344
Zimniak P (2008) Detoxification reactions: relevance to aging. Ageing Res Rev 7(4):281–300. doi:10.1016/j.arr.2008.04.001
Acknowledgements
H. R. Griffiths gratefully acknowledges support from the Glenn Foundation. D. G. A. Burton was supported by a Marie Skłodowska-Curie Actions Individual Fellowship. H. K. I. Dias gratefully acknowledges support from Kidney Research UK. O. S. Ademowo was supported by the Aston Research Centre for Healthy Ageing.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ademowo, O.S., Dias, H.K.I., Burton, D.G.A. et al. Lipid (per) oxidation in mitochondria: an emerging target in the ageing process?. Biogerontology 18, 859–879 (2017). https://doi.org/10.1007/s10522-017-9710-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-017-9710-z