
Abstract
Plant zygotes usually undergo asymmetrical cell division, giving rise to the formation of two daughter cells with distinct developmental cell fate. The small apical cell will develop into the major part of embryo proper, whereas the larger basal cell will divide to form a transient suspensor. Thus, the apical and basal cell lineages are an excellent model to study cell fate determination in relation to zygote polarity. However, the molecular mechanism underlying the differentiation of two distinct cell lineages is not yet understood, possibly due to the technique limitations. Previously, we have established a protocol for isolating apical cell and basal cell for cDNA library construction in tobacco. However, the method for isolating tiny Arabidopsis embryos has long been considered much more difficult. Here, we present a detailed protocol for isolating early Arabidopsis proembryos and separating apical and basal cell lineages of proembryos, which allow us to establish cell lineage-specific transcriptomes of early proembryos.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Zygote is the start point of new sporophytic generation of plants, which is produced from the fusion of two gametes, a larger egg cell and a smaller sperm cell, through fertilization. The zygote divides asymmetrically to form two daughter cells with different cell sizes and distinct cell fates in the subsequent developmental process. The smaller apical cell will develop into the major parts of a mature embryo called the embryo proper, whereas the larger basal cell usually undergoes limited cell divisions to form a suspensor. The uppermost suspensor cell will differentiate into the hypophysis, i.e., the precursor to the root organizer cells, while the other suspensor cells go on to degenerate (Kawashima and Goldberg 2010; Zhao et al. 2013). Although differences in cell division pattern and cell fate of the two daughter cells of zygote have been observed for many years, the molecular mechanism regulating apical and basal cell lineage specification is still largely unknown (Jenik et al. 2007; Lau et al. 2012).
Recently, two independent reports revealed that basal cell lineage of early proembryos still has the potential of embryogenesis after the apical cell lineage was removed (Gooh et al. 2015; Liu et al. 2015). Laser disruption of the apical cell in the seed, which was labeled by a fluorescent marker, could induce cell fate conversion of the basal cell into a zygote-like cell that could divide and form a new typical embryo with embryo proper and suspensor (Gooh et al. 2015). Meanwhile, suspensor cells of early proembryo could also develop into another embryo after severing the connection between embryo proper and suspensor in the seed by destroying the fluorescent marked uppermost suspensor cell using laser ablation (Liu et al. 2015). However, the suspensor could not generate a new embryo in both Arabidopsis and tobacco when isolated from the seed (Liu et al. 2015; Qu et al. 2017), suggesting that a maternal tissue-derived signal is required to generate a second embryo. These results indicate that basal cell lineage of early proembryos still has embryonic potential in the seed that is suppressed by apical cell lineage during normal embryogenesis.
Little is known about the intrinsic molecular mechanisms directing cell fate during apical and basal lineage specification following the division of the zygote. However, by generating lineage-specific transcriptomes from the first apical and basal cell and onward, it would be possible to identify molecular pathways important for apical and basal cell lineage specification. For this purpose, it is necessary to isolate the apical and basal cell following zygote division and their subsequent descendants in the early proembryo for transcriptomic profiling. During the past decades, great efforts have been made to develop different methods to isolate embryo proper and suspensor cells or nuclei for generating lineage-specific transcriptomes in the Arabidopsis embryo (Belmonte et al. 2013; Palovaara et al. 2017; Slane et al. 2014). In these studies, different approaches such as laser capture microdissection (LCM), fluorescent-activated nuclei sorting (FANS) and isolation of nuclei tagged in specific cell types (INTACT) have been used. However, these methods have not been successfully applied to isolate the small apical and basal cell in Arabidopsis. So far, protocols for isolating living apical and basal cell have only been established in tobacco and maize (Chen et al. 2017; Ma et al. 2011; Qu et al. 2017), and an approach to isolating the apical and basal cell has not been available in Arabidopsis due to the technical difficulty. Here, we present a detailed protocol for isolating the living apical and basal cell and their descendants of the early Arabidopsis proembryo for cell lineage-specific transcriptomic profiling.
Materials and equipment
Plant material
Arabidopsis thaliana (ecotype Col-0) were cultivated in the greenhouse under long-day conditions (16 h light/8 h dark) at 22 °C.
Reagents
D-Sorbitol (Sigma), glycerol (Sinopharm Chemical Reagent Co. Ltd, China), diethyl pyrocarbonate (DEPC, Sigma), RNase inhibitor (Takara), Cellulase R-10 (Yakult Pharmaceutical Industry Co. Ltd, Japan), Macrozyme R-10 (Yakult Pharmaceutical Industry Co. Ltd, Japan), Mineral oil (Sigma), 2-(N-Morpholino) ethanesulfonic acid hydrate (MES) (Sigma), Dynabeads® mRNA DIRECT™ Micro Kit (Life Technologies), SMART-Seq® v4 Ultra® Low Input RNA Kit for Sequencing (Clontech), Agencourt AMPure purification kit (Beckman Coulter).
Equipments
AS LMD System (Leica, Germany), inverted microscope IX71 (Olympus, Japan), stereoscopic microscope (Jiangnan, China), Agilent 2100 Bioanalyzer (Agilent Technologies), Qubit Fluorometer (Life Technologies), fine tweezer (Fig. 1a), Hand-made glass needle (Fig. 1b), Hand-made capillary pipettes sealed with latex tubing at one end and a sharp tip at the other end (Fig. 1c), Slide with polyethylene terephthalate film (PET) at the bottom (Leica, Germany) (Fig. 1d), Petri dishes for microscope: diameter 3.5 cm (NEST, China).

Tools used for the manual isolation of early proembryos and laser cutting. a A fine tweezer; b hand-made glass needle; c hand-made capillary pipettes sealed with latex tubing at one end; d slide with PET at the bottom
Solutions for cell isolation
-
1.
RNase-free H2O: Add 100 µl DEPC into 100 ml deionized water. Shake the solution gently at 37 °C for 12 h, and then autoclave it to remove DEPC.
-
2.
Washing solution: 80 mM D-Sorbitol, 10% glycerol, 0.058% MES (pH = 5.8). Dissolve 0.7286 g of sorbitol and 0.029 g MES in 40 ml RNase-free H2O in a 50 ml RNase-free centrifuge tube (NEST, China). Add 5.0 ml glycerol into the centrifuge tube. Mix well with a vortex mixer and adjust the pH to 5.8. Fill up to 50 ml with RNase-free H2O. Filter–sterilize the solution with a 0.22-µm filter and store at room temperature.
-
3.
Enzyme solution: Dissolve 0.05 g Cellulase R-10 and 0.04 g Macrozyme R-10 in the 50 ml washing solution. Filter–sterilize the solution with a 0.22-µm filter and store at − 20 °C.
-
4.
Ablation solution: Add 1 μl of RNase inhibitor to 19 μl of washing solution.
Procedure
Hand emasculation and pollination
-
1.
Select plants that are blooming and remove all opened flowers with a fine tweezer.
-
2.
Select flower buds that are slightly open to only show the white petals (1 day before anthesis) and carefully remove their anthers using a fine tweezer under the stereoscopic microscope.
Note: Avoid damaging the pistils during the process of removing anthers.
-
3.
Keep the plants in the greenhouse for 1 day.
-
4.
Pollinate the emasculated mature pistils with open anthers.
Note: It is necessary to label the pistil with fine rope after pollination and record the time of pollination.
Seed collection
-
1.
After 36 h, the hand-pollinated siliques were harvested for collecting seeds and subsequent proembryo isolation.
Note: Harvesting siliques at 36 h after pollination is done to isolate two-cell proembryos. Embryos at other developmental stages can be isolated at the corresponding developmental time–point after pollination.
-
2.
Add 500 µl enzyme solution in the bottom of a 3.5-cm Petri dish to form a droplet.
-
3.
Open up the carpels and gently scrape off the seeds from the septum with a glass needle under the stereoscopic microscope.
-
4.
Transfer the seeds quickly into the enzyme solution, cover the Petri dish and maintain them for 30 min at 25 °C (Fig. 2a). Enzyme treatment will help to degrade cell wall of the seed coat cells to facilitate the dissection of the seed coat and the separation of the embryos from the seed coat.
Fig. 2 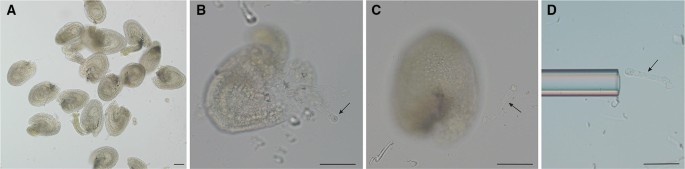
Isolating two-cell proembryo from seeds by micromanipulation. a Dissected seeds in the enzyme solution; b–c dissect two-cell proembryo from the seeds with two fine glass needles; d collect isolated two-cell proembryo by a hand-made capillary pipette. Bar = 50 µm. Arrows indicate two-cell proembryo
-
5.
Remove the enzyme solution from the bottom of the Petri dish using a pipette with a 200-µl pipette tip to suck up the solution. Add 1 ml washing solution into the Petri dish, and shake the Petri dish gently to wash the seeds. Discard the washing solution, and repeat washing step twice.
Note: Some seeds are easy to float up to the surface of the enzyme solution; hence, the enzyme solution should be removed carefully from the bottom of Petri dish.
-
6.
Two-cell proembryos can be dissected directly from the seeds with two fine glass needles under an inverted microscope by pressing the chalazal end with a fine glass needle, and dissecting the embryo from the micropylar end with another glass needle by quickly cutting on the seed coat (Figs. 2b–c, 3).
Fig. 3 
Schematic representation of the isolation of early proembryos. a Harvest seeds from the siliques; b enzyme treatment of seeds in a Petri dish; c dissect two-cell proembryos from the seeds; d–e transfer two-cell proembryos into a droplet of ablation solution
Note: Usually, two-cell proembryos can be dissected from about 20% seeds using this method. The same procedure can also be used for isolating embryos at other stages. It is much easier to isolate embryos after 32-cell embryo stages due to its relatively larger size. However, it is difficulty to isolate embryos with intact suspensor cells after the 32-cell embryo stage since the basal-most suspensor is tightly connected to the maternal tissues.
-
7.
Transfer several isolated two-cell proembryos into a droplet of 50 µl fresh ablation solution that was covered by mineral oil in a Petri dish using a hand-made capillary pipette (Figs. 2d, 3).
-
8.
Wash the isolated two-cell proembryos by gently pipetting up and down a few times with the capillary micropipette in a new 50 µl ablation solution, repeat washing step twice.
Note: It is necessary to perform extensive wash to remove seed coat tissues and endosperm contamination with the help of the pipette.


Laser-controlled separation of the apical and basal cell and their descendants
-
1.
Transfer two-cell proembryos into 10 µl ablation solution on the PET slide with the hand-made capillary pipette, and cover it with a glass coverslip.
-
3.
Set AS LMD System at the “move” mode for destroying the apical or basal cell. The energy of the laser spot is set as 40–60% of the full power (200 W), and the parameter of “specimen balance” is set at 2.
Note: The mode for laser should be set at the “move” mode since proembryos are slowly moving in the ablation solution during the process of laser ablation.
-
4.
Destroy the apical or basal cell using laser spot (Fig. 4a).
Fig. 4 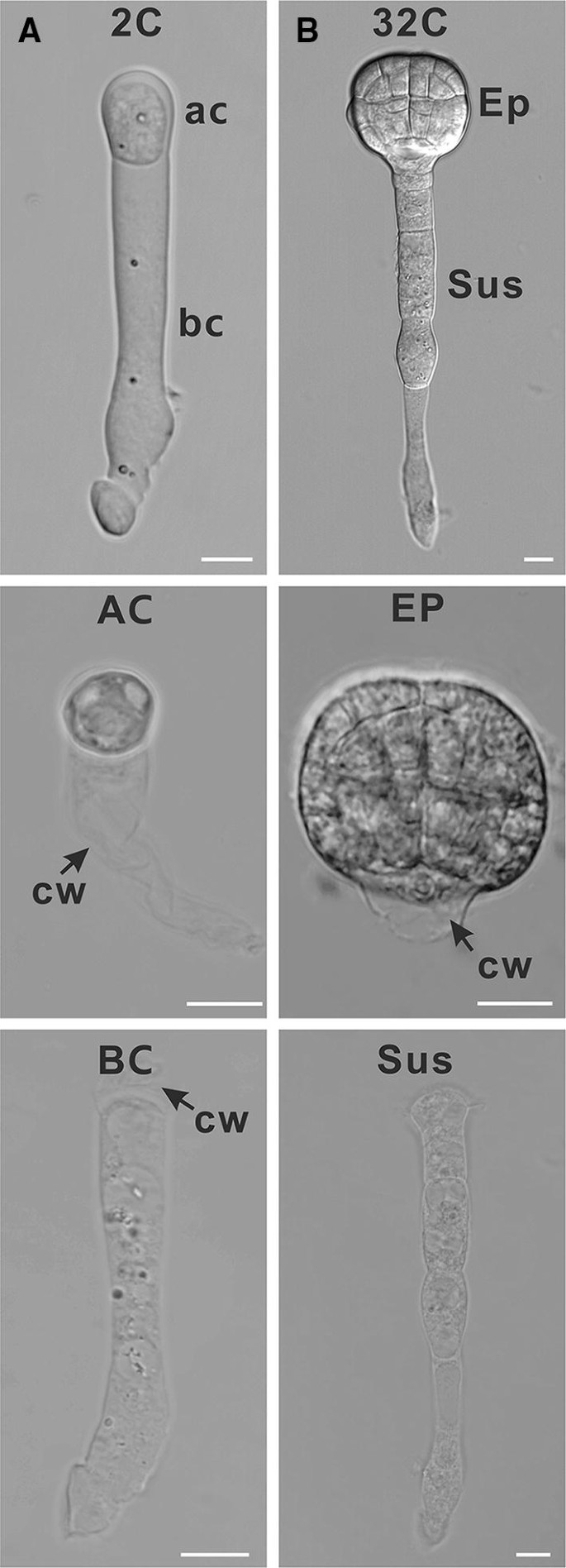
Isolation of apical or basal cell lineages of early proembryos. a Isolated two-cell proembryo, apical cell and basal cell; b isolated 32-cell embryo, embryo proper and suspensor, Bar = 10 µm. 2C, two-cell proembryo; 32C, 32-cell embryo; CW, cell wall; AC, apical cell; BC, basal cell; EP, embryo proper, SUS, suspensor
Note: To separate apical or basal cell lineage of early proembryos at other stages, destroy the uppermost suspensor cell by laser spot (Fig. 4b).
-
5.
Transfer isolated apical or basal cells with the capillary pipette into the 50 µl fresh ablation solution, gently pipette up and down a few times with the capillary micropipette, repeat washing step twice.
Note: To avoid the contamination, it is necessary to exchange the ablation solution in each washing step and perform extensive washing to remove cytoplasm from the destroyed cell.
-
6.
Transfer isolated apical cells or basal cells to a 0.2-ml RNase-free PCR tube containing 10 μl of lysis buffer (Life Technologies, USA) for mRNA isolation.
Note: Isolated cells in lysis buffer can be stored in − 80 °C for up to 1 month.

RNA extraction and cDNA preparation for RNA-seq
-
1.
mRNA is directly extracted from apical or basal cells (and their descendants) using Dynabeads mRNA DIRECT™ Micro Kit (Life technologies, USA) according to the manufacturer’s instructions.
Note: 15–30 isolated apical or basal cells for each sample are usually used for mRNA extraction and subsequent RNA-seq.
-
2.
Elute mRNA into 10 µl 10 mM Tris–HCl by incubating the beads at 70 °C for 2 min in a PCR machine.
Note: To avoid mRNA degradation, eluted mRNA should be used directly for cDNA synthesis.
-
3.
cDNA synthesis and amplification are performed with SMART-seq™ v4 Ultra™ Low Input RNA Kit for Sequencing (Clontech, USA).
-
4.
Purify PCR-amplified cDNA using an Agencourt AMPure purification kit (Beckman Coulter).
-
5.
Measure size and concentration of purified cDNA using Agilent 2100 Bioanalyzer (Fig. 5a) and Qubit Fluorometer, respectively.
Fig. 5 
Measurement of cDNA size and detection of RNA contamination in the transcriptomes of apical and basal cell. a Measurement of cDNA size distribution obtained from the amplified apical or basal cell cDNA. The profiles show a 1.0–1.5 kb peak with a small number of fragments below 500 bp. b Heat map showing the results of tissue-enrichment test of the transcriptomes of apical and basal cell. EP, embryo proper; SUS, suspensor; MCE, micropylar endosperm; PEN, peripheral endosperm; CZE, chalazal endosperm; CZSC, chalazal seed coat; GSC, general seed coat
-
6.
Library construction and illumina sequencing is performed according to the manufacturer’s instructions.
-
7.
The level of contamination in transcriptome is assessed using publicly available software (Fig. 5b) (https://github.com/Gregor-Mendel-Institute/tissue-enrichment-test) (Schon and Nodine 2017).

Discussion
Our protocol provides an alternative method to analyze the transcriptome of apical and basal cells, which will be helpful to understand the molecular mechanism regulating cell fate determination of the apical and basal cell and their descendants. Compared to the other available methods, this procedure is characterized by several unique aspects. Firstly, the procedure is designed to directly isolate living cells from early proembryos, which better preserve RNA integrity. Secondly, the laser ablation technique is employed to greatly shorten the duration for the separation of apical and basal cell or embryo proper and suspensor. Although it is also possible to isolate apical or basal cell lineages by enzymatic maceration, the whole process will last for hours since the connection between apical and basal cell lineages is very difficult to separate based on personal experience. In addition, the isolated cells are usually a protoplast after enzymatic maceration, which is easy to burst during the washing and transferring process. Thus, comparably, laser ablation technique is a better choice for this purpose. Thirdly, the procedure is superior in preventing contaminations by other cell types. Compared to LCM technique, which requires high accurate manipulation to avoid contamination during sample collection from sections of fixed material, the method reported here is easier to ensure the purity of the samples. Since the cells and embryos are isolated and collected individually under the microscope, it is easy to visually check the quality and purity of isolated cells one by one, and selective collection could monitor the purity of the final samples. For the invisible components from burst cells, which are in the solution and may be sucked up during cell collection, a three-time wash is enough to purify isolated cells. Critical technique points are to use a new micropipette in each washing step and suck up as little solution as possible when transferring isolated cells during the whole procedure. With the development of next-generation sequencing technologies in recent years, RNA input requirements, the sensitivity and accuracy are no longer the limitations for the transcriptome analysis. The quality and purity of the samples have become decisive factors for generating reliable transcriptomic data. Thus, the procedure reported here can be a useful alternative in the similar works.
References
Belmonte MF, Kirkbride RC, Stone SL, Pelletier JM, Bui AQ, Yeung EC, Hashimoto M, Fei J, Harada M, Munoz MD, Le BH, Drews GN, Brady SM, Goldberg RB, Harada JJ (2013) Comprehensive developmental profiles of gene activity in regions and subregions of the Arabidopsis seed. Proc Natl Acad Sci USA 110:E435–E444. https://doi.org/10.1073/pnas.1222061110
Chen J, Strieder N, Krohn NG, Cyprys P, Sprunck S, Engelmann JC, Dresselhaus T (2017) Zygotic genome activation occurs shortly after fertilization in maize. Plant Cell 29:2106–2125. https://doi.org/10.1105/tpc.17.00099
Gooh K, Ueda M, Aruga K, Park J, Arata H, Higashiyama T, Kurihara D (2015) Live-cell imaging and optical manipulation of Arabidopsis early embryogenesis. Dev Cell 34:242–251. https://doi.org/10.1016/j.devcel.2015.06.008
Jenik PD, Gillmor CS, Lukowitz W (2007) Embryonic patterning in Arabidopsis thaliana. Annu Rev Cell Dev Biol 23:207–236. https://doi.org/10.1146/annurev.cellbio.22.011105.102609
Kawashima T, Goldberg RB (2010) The suspensor: not just suspending the embryo. Trends Plant Sci 15:23–30. https://doi.org/10.1016/j.tplants.2009.11.002
Lau S, Slane D, Herud O, Kong J, Jurgens G (2012) Early embryogenesis in flowering plants: setting up the basic body pattern. Annu Rev Plant Biol 63:483–506. https://doi.org/10.1146/annurev-arplant-042811-105507
Liu Y, Li X, Zhao J, Tang X, Tian S, Chen J, Shi C, Wang W, Zhang L, Feng X, Sun MX (2015) Direct evidence that suspensor cells have embryogenic potential that is suppressed by the embryo proper during normal embryogenesis. Proc Natl Acad Sci USA 112:12432–12437. https://doi.org/10.1073/pnas.1508651112
Ma L, Xin H, Qu L, Zhao J, Yang L, Zhao P, Sun M (2011) Transcription profile analysis reveals that zygotic division results in uneven distribution of specific transcripts in apical/basal cells of tobacco. PLoS ONE 6:e15971. https://doi.org/10.1371/journal.pone.0015971
Palovaara J, Saiga S, Wendrich JR, Hofland NV, van Schayck JP, Hater F, Mutte S, Sjollema J, Boekschoten M, Hooiveld GJ, Weijers D (2017) Transcriptome dynamics revealed by a gene expression atlas of the early Arabidopsis embryo. Nat Plants 3:894–904. https://doi.org/10.1038/s41477-017-0035-3
Qu LH, Zhou X, Li X, Li SS, Zhao J, Zhao P, Liu Y, Sun MX (2017) The autonomous cell fate specification of basal cell lineage: the initial round of cell fate specification occurs at the two-celled proembryo stage. Plant J 91:1051–1063. https://doi.org/10.1111/tpj.13629
Schon MA, Nodine MD (2017) Widespread contamination of arabidopsis embryo and endosperm transcriptome data sets. Plant Cell 29:608–617. https://doi.org/10.1105/tpc.16.00845
Slane D, Kong JX, Berendzen KW, Kilian J, Henschen A, Kolb M, Schmid M, Harter K, Mayer U, De Smet I, Bayer M, Jurgens G (2014) Cell type-specific transcriptome analysis in the early Arabidopsis thaliana embryo. Development 141:4831–4840. https://doi.org/10.1242/dev.116459
Zhao P, Zhou XM, Zhang LY, Wang W, Ma LG, Yang LB, Peng XB, Bozhkov PV, Sun MX (2013) A bipartite molecular module controls cell death activation in the Basal cell lineage of plant embryos. PLoS Biol 11:e1001655. https://doi.org/10.1371/journal.pbio.1001655
Acknowledgements
This work was supported by National Natural Science Fund of China (31600244).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by Dolf Weijers.
A contribution to the special issue ‘Cellular Omics Methods in Plant Reproduction Research’.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhou, X., Shi, C., Zhao, P. et al. Isolation of living apical and basal cell lineages of early proembryos for transcriptome analysis. Plant Reprod 32, 105–111 (2019). https://doi.org/10.1007/s00497-018-00353-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00497-018-00353-6