
Abstract
Alternans and arrhythmogenicity were studied in hypokalaemic (3.0 mM K+) Langendorff-perfused murine hearts paced at high rates. Epicardial and endocardial monophasic action potentials were recorded and durations quantified at 90% repolarization. Alternans and arrhythmia occurred in hypokalaemic, but not normokalaemic (5.2 mM K+) hearts (P < 0.01): this was prevented by treatment with lidocaine (10 μM, P < 0.01). Fourier analysis then confirmed transition from monomorphic to polymorphic waveforms for the first time in the murine heart. Alternans and arrhythmia were associated with increases in the slopes of restitution curves, obtained for the first time in the murine heart, while the anti-arrhythmic effect of lidocaine was associated with decreased slopes. Thus, hypokalaemia significantly increased (P < 0.05) maximal gradients (from 0.55 ± 0.14 to 2.35 ± 0.67 in the epicardium and from 0.67 ± 0.13 to 1.87 ± 0.28 in the endocardium) and critical diastolic intervals (DIs) at which gradients equalled unity (from −2.14 ± 0.52 ms to 50.93 ± 14.45 ms in the epicardium and from 8.14 ± 1.49 ms to 44.64 ± 5 ms in the endocardium). While treatment of normokalaemic hearts with lidocaine had no significant effect (P > 0.05) on either maximal gradients (0.78 ± 0.27 in the epicardium and 0.83 ± 0.45 in the endocardium) or critical DIs (6.06 ± 2.10 ms and 7.04 ± 3.82 ms in the endocardium), treatment of hypokalaemic hearts with lidocaine reduced (P < 0.05) both these parameters (1.05 ± 0.30 in the epicardium and 0.89 ± 0.36 in the endocardium and 30.38 ± 8.88 ms in the epicardium and 31.65 ± 4.78 ms in the endocardium, respectively). We thus demonstrate that alternans contributes a dynamic component to arrhythmic substrate during hypokalaemia, that restitution may furnish an underlying mechanism and that these phenomena are abolished by lidocaine, both recapitulating and clarifying clinical findings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Correlations between beat-to-beat alternations in electrocardiographic QT interval (QT or T-wave alternans) and cardiac arrhythmia have been described in classical work [13, 25] and subsequently confirmed in both clinical studies [2, 28, 38] and experimental models [7, 33, 52]. Indeed, the presence of such alternans provides a stronger predictor of arrhythmic risk than is offered by signal-averaged electrocardiography and a prediction of a similar value to that offered by invasive programmed stimulation procedures in current use [10] (but see [45]). The association between alternans and arrhythmogenicity is particularly marked at high heart rates (short baseline cycle lengths, BCLs) [15, 49] and in conditions of congenital and acquired electrocardiographic QT, and therefore action potential, prolongation [1, 34, 39, 46, 47, 64].
The relationship between heart rate and alternans has previously been analysed by constructing restitution curves which plot relationships between action potential duration (APD) and preceding diastolic interval (DI), as BCL is varied [12]. Clinical and experimental studies then associated alternans and arrhythmia observed at short BCLs with steeply sloping restitution curves with gradients greater than unity [14, 21, 24, 32]. The latter would reflect disproportionate decreases in mean APD with DI. Furthermore, manoeuvres which decrease the slopes of restitution curves, such as hyperkalaemia in a tachycardic canine endocardial preparation [22] and certain cardiotropic drugs in canine and porcine preparations [8, 31, 37], have been reported to both suppress alternans and exert anti-arrhythmic effects. In contrast, other studies have attributed such alternans to alteration in Ca2+ cycling involving the sarcoplasmic reticulum [35, 44, 54]. Whatever the underlying mechanism, alternans results in temporal variations in the wavelengths of propagating action potentials that could potentially lead to reentry and arrhythmogenesis [55].
Hypokalaemia is an important clinical cause of acquired QT prolongation and is associated with arrhythmia of various degrees of spatial organisation [12, 30, 57] suppressible by lidocaine [6, 36, 48]. The present study uses the monophasic action potential (MAP) technique [19, 23] and an established Langendorff-perfused murine model [18, 40–42] to explore the effects of hypokalaemia and of lidocaine on alternans and arrhythmogenicity for the first time in any species. These investigations of temporal heterogeneities in recovery after depolarization are performed on hearts paced at short BCLs, thereby complimenting recent work [40, 41] which contrastingly examined the contribution of spatial heterogeneities at long BCLs. This approach successfully demonstrates such phenomena in parallel with the clinical situation and proceeds for the first time to apply Fourier analysis to MAP waveforms and to recordings obtained during hypokalaemia. This analysis demonstrates evolution from a monomorphic to a polymorphic pattern as has previously been reported in other species [55, 58, 63], thereby further validating the murine model. The presence or absence of alternans and arrhythmogenicity is then related to alterations in the slopes of restitution curves. Thus, while the relationship between APD and BCL has previously been determined [20], the present study reports restitution curves relating APD to the preceding DI for the first time in a murine system. Use of this murine system further permits the comparison of findings with results obtained from genetically modified examples [26].
Taken together, these novel findings implicate a dynamic component to arrhythmic substrate under conditions of hypokalaemia for which restitution may furnish a possible underlying mechanism.
Materials and methods
Experimental animals
Mice were housed at 21 ± 1°C with 12-h light/dark cycles, were fed sterile chow (RM3 Maintenance Diet, SDS, Witham, Essex, UK) and had free access to water. Wild-type 129 Sv mice aged 3–6 months were used in all experiments. All procedures complied with the UK Animals (Scientific Procedures) Act 1986.
Solutions
Solutions were based on bicarbonate-buffered Krebs–Henseleit solution (mM: NaCl 119, NaHCO3 25, KCl 4, KH2PO4 1.2, MgCl2 1, CaCl2 1.8, glucose 10 and Na-pyruvate 2; pH adjusted to 7.4) and were bubbled with 95% O2/5% CO2 (British Oxygen Company, Manchester, UK). Hypokalaemic (3.0 mM K+) solutions were prepared by reducing the amount of KCl added. Lidocaine-containing solutions were prepared by adding lidocaine (Sigma-Aldrich, Poole, UK) to a final concentration of 10 μM.
Preparation
A Langendorff perfusion protocol previously adapted for murine hearts [3] was used. In brief, mice were killed by cervical dislocation [Schedule 1: UK Animals (Scientific Procedures) Act 1986]. Hearts were then quickly excised and placed in ice-cold bicarbonate-buffered Krebs–Henseleit solution. A short section of aorta was cannulated under the surface of the solution and attached to a custom-made 21-gauge cannula filled with the same solution using an aneurysm clip (Harvard Apparatus, Edenbridge, Kent, UK). Fresh Krebs–Henseleit solution was then passed through 200- and 5-μm filters (Millipore, Watford, UK) and warmed (37°C) using a water jacket and circulator (Techne model C-85A, Cambridge, UK) before being used for constant-flow retrograde perfusion at 2–2.5 ml/min using a peristaltic pump (Watson-Marlow Bredel model 505S, Falmouth, Cornwall, UK). Hearts were only regarded as suitable for experimentation if they regained a healthy pink colour and began to contract spontaneously on rewarming.
Electrophysiological measurements
An epicardial MAP electrode (Hugo Sachs, Harvard Apparatus) was placed against the basal left ventricular epicardium. A small access window was created in the interventricular septum to allow access to the left ventricular endocardium [5]. A custom-made endocardial MAP electrode comprising two twisted strands of high-purity Teflon-coated silver wire of 0.25-mm diameter (Advent Research Materials, UK) was constructed. The Telflon coat was removed from the distal 1 mm of the electrode, which was then galvanically chlorided to eliminate DC offset, inserted and placed against the septal endocardial surface. MAPs were amplified, band-pass-filtered (0.5 Hz to 1 kHz: Gould 2400S, Gould-Nicolet Technologies, Ilford, Essex, UK) and digitised at a sampling frequency of 5 kHz (micro1401, Cambridge Electronic Design, Cambridge, UK). Analysis of MAPs in both the time and frequency domains was performed using Spike II (Cambridge Electronic Design).
Experimental protocol
A bipolar platinum stimulating electrode (1 mm inter-pole spacing) was placed on the basal surface of the right ventricular epicardium. Square-wave stimuli (Grass S48 stimulator, Grass-Telefactor, Slough, UK) of 2-ms duration and amplitudes of twice the excitation threshold were initially applied to hearts at a constant baseline cycle length of 125 ms until MAPs showed stable baselines, rapid upstroke phases that reached consistent amplitudes and smooth repolarisation phases [19] and for at least 10 min. Hearts were then exposed to test solutions for 20 min while stimulation was continued before subsequent recordings were made.
In initial experiments, MAPs were recorded during regular stimulation at baseline cycle lengths of 130, 100 and 70 ms. Hearts were then subjected to an adapted dynamic pacing protocol [9]. This comprised cycles each consisting of 100 stimuli delivered over a range of BCLs. Steady states were consistently reached during the first 50 responses in each cycle, and thus, mean epicardial and endocardial APD90 values and DIs were calculated from the final 50 action potentials of each cycle. With each successive cycle, BCL was decremented by 5-ms steps from an initial value of 175 ms. Cycles were continued until a reproducible sequence of consistently shaped waveforms was no longer obtained. These data were then used to construct restitution curves.
All data are presented as means ± standard errors of the means and include the number of hearts studied. Comparisons between data sets used analysis of variance (significance threshold set at P ≤ 0.05). Curve fitting of particular function to data sets used a Levenberg–Marquardt algorithm (OriginPro 7.5, OriginLab, MA, USA).
Results
The experiments explored the consequences of increases in heart rate for arrhythmogenicity in hypokalaemic murine hearts, thereby extending a previous report that considered the effect of bradycardia [41]. They thus studied identical experimental groups, first exposing hearts to normokalaemic (5.2 mM K+) and hypokalaemic (3.0 mM K+) test solutions before and after addition of lidocaine (10 μM) for 20 min during regular stimulation at a BCL of 125 ms. Experiments then proceeded to study electrical activity through a range of steady state BCLs to assess propensity to spontaneous arrhythmia. Electrical traces from arrhythmic hearts were then quantitatively analysed in the frequency domain to empirically characterise their kinetics. Finally, hearts were subjected to a dynamic pacing protocol which explored the effect of varying BCLs [21] and permitted the analysis of alternans and its relationship to arrhythmogenicity.
Alternans and arrhythmic activity occur in hypokalaemic hearts paced at a reduced baseline cycle length
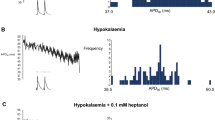
The first series of experiments recorded MAPs [23] from the epicardia of hearts paced at BCLs of 130, 100 or 70 ms over 30-min recording periods (five hearts in each case; Fig. 1). This demonstrated a tendency to both alternans, as reflected in alternating short-long-short sequences in action potential duration, and arrhythmogenesis at the shortest BCLs, corresponding to the highest heart rates, specifically in the hypokalaemic hearts and not in the other groups. Thus, MAPs obtained from normokalaemic hearts whether paced at BCLs of 130, 100 or 70 ms demonstrated consistent waveforms and stable rhythms during 30-min recording periods (five hearts in each case; Fig. 1a). In contrast, while recordings from hypokalaemic hearts paced at BCLs of 130 and 100 ms demonstrated stable rhythms (five out of five hearts), alternans leading to periods of arrhythmic activity occurred during pacing at a BCL of 70 ms in all five cases (P < 0.01 as compared to normokalaemic hearts; Fig. 1b). However, neither alternans nor arrhythmic activity occurred in either normokalaemic or hypokalaemic hearts treated with lidocaine during pacing at any BCL (five hearts in each case; P < 0.01 as compared to hypokalaemic hearts not treated with lidocaine; Fig. 1c and d, respectively), confirming the anti-arrhythmic effect of lidocaine described in earlier studies [6, 41, 42, 48].

Effect of baseline cycle length on presence of alternans and initiation of arrhythmic activity. Epicardial monophasic action potential recordings obtained from hearts exposed to normokalaemic (5.2 mM K+, a) and hypokalaemic (3.0 mM K+, b) test solutions, and normokalaemic (c) and hypokalaemic (d) test solutions containing lidocaine (10 μM) during regular stimulation at baseline cycle lengths of 130 ms (A), 100 ms (B) and 70 ms (C). Vertical lines indicate the timing of stimuli
Electrophysiological waveforms after the initiation of arrhythmic activity
When arrhythmic activity was observed in those hypokalaemic hearts paced at a BCL of 70 ms, this showed progressive degeneration from a regular monomorphic to a disorganised polymorphic pattern (four hearts), in common with earlier reports from the larger, porcine and human, hearts [16, 58, 63].
This trend was quantitatively apparent in a spectral analysis performed using a fast Fourier transform method [4] on ∼10-s sequences of MAP waveforms recorded from the epicardia and endocardia of hypokalaemic hearts to which a Hanning window had first been applied. These waveforms had been sampled at a rate of 5 kHz with low- and high-frequency filter cutoffs of 0.5 Hz and 1 kHz, respectively. Each transform was performed on 16,384 (=216) points to give a spectral resolution of 0.30 Hz up to a maximum, Nyquist, frequency of 2.5 kHz. Relative root mean square powers were then extracted from the real and imaginary parts of the transforms.
Figure 2 thus compares typical waveforms in both the time (a) and frequency (b) domains. Waveforms recorded during the first ∼5 min after the initiation of arrhythmic activity were monomorphic in appearance (A in Fig. 2a). This was confirmed by spectral analysis which similarly demonstrated a single identical dominant frequency peak of 18.9 ± 1.8 Hz in both epicardial and endocardial recordings with harmonics whose frequencies were integral multiples of a dominant fundamental frequency and any given heart showed at least two such peaks (∼38.4, ∼57.6, ∼76.8 Hz, A in Fig. 2b).

Progression from monomorphic to polymorphic arrhythmic activity. Epicardial and endocardial monophasic action potential recordings obtained from hearts exposed to hypokalaemic (3.0 mM K+) test solution 5 min (A) and 10 min (B) after initiation of arrhythmic activity in the time (a) and frequency (b) domains
In contrast, waveforms recorded ≥10 min after the initiation of arrhythmic activity were consistently polymorphic in appearance (B in Fig. 2a), with spectral analysis (B in Fig. 2b) further demonstrating dissimilar epicardial and endocardial frequency spectra. The dominant frequency peak was then at 27.6 ± 5.6 Hz in the epicardium, while that in the endocardium was 26.5 ± 5.8 Hz. Furthermore, the higher order peaks, whether recorded from the epicardium or the endocardium, occurred at irregular frequency intervals that were not multiples of the dominant frequency.
Alternans occurs in the epicardia and endocardia of hypokalaemic hearts paced at a baseline cycle length of 70 ms
As illustrated in C in Fig. 1b, hypokalaemic hearts paced at a BCL of 70 ms demonstrated alternans. Figure 3 goes on to show mean APD90 values of the alternating, odd-numbered (filled bars) and even-numbered (open bars) action potentials, and the difference between these values, giving the magnitude of any alternans (hashing), recorded from the epicardia (a) and endocardia (b) under normokalaemic (A) and hypokalaemic (B) conditions and under such conditions during treatment with lidocaine (C and D, respectively).

Alternans during stimulation at a baseline cycle length of 70 ms. Durations of successive odd (filled bars) and even (open bars) numbered action potentials (at 90% repolarisation, APD90) and the difference between these values, giving the magnitude of alternans (hashing) in the epicardia (a) and endocardia (b) of hearts exposed to normokalaemic (5.2 mM K+, A) and hypokalaemic (3.0 mM K+, B) test solutions and normokalaemic (C) and hypokalaemic (D) test solutions containing lidocaine (10 μM) during regular stimulation at a baseline cycle length of 70 ms
In normokalaemic hearts (five hearts), alternans was not observed, the difference between the mean APD90 values of odd-number and even-numbered action potentials not reaching significance (P > 0.05) in either the epicardium (4.7 ± 2.7 ms) or the endocardium (5.6 ± 1.7 ms). In contrast, alternans did occur in hypokalaemic hearts (five hearts), the difference between the mean APD90 values of odd-number and even-numbered action potentials being statistically significant (P < 0.01) in both epicardium (14.9 ± 3.8 ms) and endocardium (15.6 ± 1.8 ms). Treatment with lidocaine had no significant (P > 0.05) effect on normokalaemic hearts (five hearts): The difference between the mean APD90 values of odd-number and even-numbered action potentials remained statistically insignificant (P > 0.05) in both epicardium (2.7 ± 3.4 ms) and endocardium (2.5 ± 4.6 ms). In contrast, treatment with lidocaine eliminated alternans in hypokalaemic hearts (five hearts). Hence, the difference between the mean APD90 values of odd-number and even-numbered action potentials did not reach significance (P > 0.05) in either epicardium (4.1 ± 4.6 ms) or endocardium (2.4 ± 3.2 ms).
The dynamic pacing protocol initiates alternans in hypokalaemic hearts at short baseline cycle lengths
The above findings concerning the presence or absence of alternans at the shortest BCL studied above prompted a detailed exploration of the effect of BCL upon the alternans phenomenon. To assess this relationship, hearts were subjected to a dynamic pacing protocol [21] consisting of cycles each lasting 100 stimuli at through a range of BCLs. In these procedures, steady states were consistently reached within the first 50 responses. Accordingly, mean values of epicardial and endocardial APD90 and DI were obtained from the final 50 action potentials. BCL was decremented in 5-ms steps with each cycle from an initial value of 175 ms until a reproducible sequence of consistently shaped action potential waveforms could no longer be obtained.
Figure 4a–d shows typical recordings obtained from the epicardia of individual hearts under the four sets of experimental conditions during the dynamic pacing protocol at BCLs of 170, 130, 90, 85, 80, 75 and 70 ms when such steady states had been achieved. Figure 5a–d shows the corresponding relationships between the durations of successive odd- and even-numbered action potentials obtained from these same hearts during the dynamic pacing protocol over the full range of BCLs studied. Thus, alternans was not observed under normokalaemic conditions even at the shortest BCL studied (Fig. 4a), resulting in points falling on the line y = x (Fig. 5a). Conversely, alternans occurred under hypokalaemic conditions at short BCLs (Fig. 4b), resulting in such points falling below the line of equality (Fig. 5b). In contrast, alternans did not occur in either normokalaemic or hypokalaemic hearts during exposure to lidocaine (Fig. 4c and d), again resulting in points falling on the line of equality (Fig. 5c and d).

Effect of baseline cycle length on action potential waveforms. Epicardial monophasic action potential recordings obtained from hearts exposed to normokalaemic (5.2 mM K+, a) and hypokalaemic (3.0 mM K+, b) test solutions and normokalaemic (c) and hypokalaemic (d) test solutions containing lidocaine (10 μM) during stimulation at baseline cycle lengths of 170, 130, 90, 85, 80, 75 and 70 ms. Vertical lines indicate the timing of stimuli

Relationships between durations of successive action potentials over a range of baseline cycle lengths. Relationships between the durations of successive odd- and even-numbered epicardial monophasic action potentials (at 90% repolarisation, APD90) in the same hearts exposed to normokalaemic (5.2 mM K+, a) and hypokalaemic (3.0 mM K+, b) test solutions and normokalaemic (c) and hypokalaemic (d) test solutions containing lidocaine (10 μM). These were obtained at baseline cycle lengths decremented in 5-ms steps from 175 ms until stimulation no longer resulted in a reproducible sequence of waveforms. Filled and open circles indicate data obtained at baseline cycle lengths at which arrhythmogenesis was and was not observed, respectively. Arrow indicates departure of points from the line y = x (broken line)
Alterations in the slopes of restitution curves correlate with pro- and anti-arrhythmic effects
The findings described above prompted the construction and analysis of restitution curves relating APD90 and the preceding DI given by the difference between the BCL and the preceding APD90 [21, 29, 32, 55]. Alternans and arrhythmia have previously been associated with increases in the slopes of such restitution curves reflecting disproportionate decreases in mean APD90 with DI. Such curves (data points, left ordinate) were plotted using the APD90 (left ordinate) and DI data obtained from the epicardia (circles, Fig. 6 A) and endocardia (squares, Fig. 6 B) of hearts under these conditions (a–d, respectively).

Restitution curves obtained from epicardia and endocardia. Restitution curves plotting action potential duration (at 90% repolarization, APD90) against preceding diastolic interval (DI) obtained from the epicardia (circles, A) and endocardia (squares, B) of hearts exposed to normokalaemic (5.2 mM K+, a) and hypokalaemic (3.0 mM K+, b) test solutions and normokalaemic (c) and hypokalaemic (d) test solutions containing lidocaine (10 μM). Curves are fitted with mono-exponential growth functions obtained by least-squares fitting to the experimental values of APD90 and DI (solid lines, left ordinates). Gradients were obtained by differentiation of the fitted functions (broken lines, right axes). Shaded boxes indicate ranges of DI values at which such gradients exceed unity
Under all conditions, both epicardial and endocardial APD90 decreased as DI decreased. However, the slope of this relationship was greater under hypokalaemic (A and B in Figs. 6b) than under normokalaemic conditions (A and B in Figs. 6a). This paralleled both the alternans and arrhythmic activity that were observed under the hypokalaemic conditions. Such increases in the slopes of restitution curves were observed even when the DI was so short such that stimuli were delivered before 90% repolarisation was achieved. This would tend to result in the underestimation of APD90, and hence, the underestimation of the slopes of restitution curves.
Exposure to lidocaine had little effect on the slopes of the corresponding curves obtained under normokalaemic (A and B in Figs. 6c), but markedly decreased the slopes of curves obtained under hypokalaemic conditions (A and B in Figs. 6d), in precise agreement with its corresponding effects in suppressing alternans and arrhythmia.
Restitution curves yield parameters predictive of arrhythmogenicity
The restitution curves obtained above proved amenable to quantitative analyses that has been described on previous occasions. For example, the simplest, mono-exponential growth function that has been used to describe data of this kind [13] takes the form:
where y represents APD90, x represents DI and y 0 and A and τ are constants obtained by least squares fitting to the experimental values of APD90 and DI in each case. Such curve-fits are used merely to empirically describe the data and should not be taken to imply any particular underlying mechanistic process. The corresponding gradient is then given by:
and assumes its maximum value at the shortest BCL.
Figure 6 plots the original APD90 and DI values and superimposes optimisations of Eq. 1 to these data (solid lines, left-hand ordinates). Such optimisations gave reduced χ 2 values indicative of better fits to data points obtained under normokalaemic than hypokalaemic conditions. It also shows the corresponding gradients derived from constants obtained from the optimisations (Eq. 2: broken lines, right-hand axes). Shaded regions where present indicate the range of DIs below a critical DI (DI < critical DI) at which these gradients exceed unity, obtained from Eq. 2:
Such a phenomenon is associated with an alternans initiated by incomplete recovery of one action potential and the consequent shortening of its subsequent DI. This would result in a disproportionate shortening of the subsequent action potential which would consequently permit its full recovery and a lengthening of the DI that follows, thereby reinitiating the cycle and resulting in a progressive decrease in mean APD90 relative to mean DI with decreasing BCL [29].
The resulting alternans would assume an amplitude determined by the gradient of the restitution curve at the given BCL, whether the latter is determined by altered external pacing rates or local ventricular arrhythmic activity. A BCL that gives a gradient of >1 will then initiate a shortening not only of mean APD90 but also of mean APD90 relative to mean DI. The resulting shortening of action potential wavelength would then predispose to reentry and arrhythmogenesis.
These parameters successfully predict the pro-arrhythmic effect of hypokalaemia
Figure 7 goes on to show these maximum gradients (a) and values of the critical DI (b) derived from the computed A and τ values in epicardia (filled bars) and endocardia (open bars). Under normokalaemic conditions (Fig. 7A) A and τ took values of 56.3 ± 2.4 ms and 58.4 ± 9.8 ms in the epicardium and 73.8 ± 1.7 ms and 65.2 ± 8.4 ms in the endocardium, with reduced χ 2 values from least-squares regression of \( \chi ^{2}_{R} \) = 1.4 and 1.2, respectively. This gave maximal gradients of 0.55 ± 0.14 in the epicardium and 0.67 ± 0.13 in the endocardium and critical DI values of −2.14 ± 0.52 ms and 8.14 ± 1.49 ms, respectively. None of the experimental DI values realised the critical DI in either the epicardium or the endocardium, paralleling the absence of arrhythmic activity (Fig. 2a).

Maximum gradients and critical diastolic intervals obtained from restitution curves. Maximum gradients (a) and critical diastolic intervals at which gradients equalled unity (b) obtained from epicardial (filled bars) and endocardial (open bars) restitution curves shown in Fig. 6 under normokalaemic (5.2 mM K+, A) and hypokalaemic (3.0 mM K+, B) conditions and under normokalaemic (C) and hypokalaemic (D) conditions during exposure to lidocaine (10 μM). Asterisks indicate values that are significantly (P < 0.05) larger than those recorded in normokalaemic hearts
Hypokalaemia (Fig. 7B) significantly (P < 0.05) increased the slopes of both epicardial and endocardial curves, concurring with the arrhythmogenic findings. Thus, A and τ took values of 166.7 ± 5.63 ms and 29.3 ± 8.29 ms in the epicardium and 133.3 ± 8.0 ms and 29.8 ± 2.1 ms in the endocardium, with \( \chi ^{2}_{R} \) values of 8.2 and 2.5, respectively. This gave significantly (P < 0.05) increased maximum gradients of 2.35 ± 0.67 in the epicardium and 1.87 ± 0.28 in the endocardium and significantly (P < 0.05) increased the critical DI values of 50.93 ± 14.45 ms and 44.64 ± 5 ms, respectively. The critical DI was realised by the observed data points in both epicardium and endocardium. Furthermore, the DIs at which this occurred were in full agreement with the corresponding DIs at which alternans was observed. Thus, in the epicardium, alternans occurred during pacing at BCLs of ≤95 ms, corresponding to DIs of 46.7 ± 3.6 ms, while in the endocardium, alternans occurred during pacing at BCLs of ≤75 ms, corresponding to DIs of 44.0 ± 5.7 ms.
These parameters also predict the anti-arrhythmic effect of lidocaine
Lidocaine had no significant (P > 0.05) effect on these values when applied to normokalaemic hearts (Fig. 7C). Thus, A and τ took values of 81.1 ± 3.05 ms and 56.92 ± 10.29 ms in the epicardium and 104.47 ± 13.69 ms and 97.17 ± 36.22 ms in the endocardium, with \( \chi ^{2}_{R} \) values of 5.37 and 10.47, respectively. This gave maximum gradients of 0.78 ± 0.27 in the epicardium and 0.83 ± 0.45 in the endocardium and the critical DI values of 6.06 ± 2.10 ms and 7.04 ± 3.82 ms, respectively, which were not realised even at the shortest BCLs studied.
In contrast, treatment of hypokalaemic hearts with lidocaine (Fig. 7D) significantly (P < 0.05) reduced the slopes of both epicardial and endocardial restitution curves, in parallel with its anti-arrhythmic effect. Thus A and τ took values of 109.90 ± 11.95 ms and 37.36 ± 5.97 ms in the epicardium and 231.72 ± 44.70 ms and 27.60 ± 4.44 ms in the endocardium, with \( \chi ^{2}_{R} \) values of 6.83 and 15.68, respectively. This gave significantly decreased (P < 0.05) maximal gradients of 1.05 ± 0.30 in the epicardium and 0.89 ± 0.36 in the endocardium and significantly decreased (P < 0.05) critical DI values of 30.38 ± 8.88 ms and 31.65 ± 4.78 ms, respectively. The critical DI was realised only at the shortest BCLs studied and then only in the epicardium, in parallel with the substantial reduction in arrhythmogenicity produced by this drug.
Slopes of restitution curves thus precisely correlate with the presence or absence of alternans and arrhythmogenesis under all the conditions explored in the hypokalaemic Langendorff-perfused murine heart.
Discussion
Electrical alternans, beat-to-beat alternation in action potential duration in turn reflected in alternation in electrocardiographic QT interval (QT or T-wave alternans), is most commonly seen at high heart rates and has been associated with arrhythmogenesis in both clinical [2, 25, 28, 38] and experimental [7, 33, 52] studies. This association is especially marked in situations of preexisting action potential, and therefore electrocardiographic QT, prolongation [39, 46, 47, 49, 64] as might occur in the congenital long QT syndromes.
Alternans phenomena have classically been analysed by the construction of restitution curves relating action potential duration to the preceding diastolic interval as heart rate is varied [29]. Restitution curves of slopes greater than unity have been associated with alternations in action potential durations of progressively increasing magnitude [14, 21, 24, 29, 32]. In contrast alternans has more recently been associated with disruption of normal cellular Ca2+ homeostasis leading to alternating short-long-short pulses of release from sarcoplasmic reticular stores [35, 44, 54]. Irrespective of the underlying mechanism, any spatial discordance in either phase or magnitude of alternans between myocardial regions would lead to spatial repolarisation gradients that might cause reentry and arrhythmogenesis [53, 56]. Furthermore, any resulting decrease in action potential wavelength, given by the product of action potential duration and conduction velocity, to less than a critical value, might break up the propagating wave of excitation and lead to reentry and arrhythmogenesis [55].
Relationships between restitution curve slopes, alternans and arrhythmogenicity at high heart rates have been established in canine preparations made to model congenital long QT syndrome type 2 (LQT2) by application of the I Kr blocking agent E-4031 [27, 61]. Correspondingly, there is a known association between sudden arousal, presumably resulting in increased heart rate, and arrhythmogenesis in human LQT2 [59]. This association has also been established in clinical hypokalaemia [11]. This common and pathophysiologically important condition similarly results in decreased repolarising K+ currents, together with action potential, and therefore electrocardiographic QT, prolongation [17, 62].
The experiments reported recorded MAP technique [19, 23] from an established intact hypokalaemic murine model [18, 40–42] and explored for alternans and arrhythmogenicity and the relationship between these before proceeding to study the effect of lidocaine upon these phenomena. Such studies of temporal heterogeneities in recovery were made at short BCLs in contrast to recent work [40, 41] which examined spatial heterogeneities at long BCLs. The murine system utilised facilitates the introduction of genetic modifications [26] and may therefore permit future comparisons with models replicating congenital arrhythmic syndromes. Indeed, alternans, in association with arrhythmogenicity, has previously been demonstrated in a murine model of LQT5 [50].
While the murine heart is well established as a model for human disease, it must be noted that important interspecies differences exist. Thus, differences between humans and mice in the kinetics of the ion channels carrying the key repolarising currents (the delayed rectifier and slowly-inactivating delayed rectifier currents, I Kr and I Ks, in humans and the transient outward current, I to in mice) result in shorter ventricular AP and the absence of a plateau phase in mice [2]. However, in both species, these key repolarising currents are K+-sensitive [6, 11, 16], making the murine heart well suited to modelling clinical hypokalaemia. Furthermore, in both species, depolarisation is rapid and attributable to the same fast Na+ current (I Na,f) [4], making our model well suited to the study of Na+-channel-blocking agents such as lidocaine. While both species share similar differences in AP duration between epicardium and endocardium [7], M cells appear to be absent in the murine heart [1]. Nonetheless, relationships between APD and refractory period are similar between the two species [3, 8], and transmural conduction velocities are almost identical [5, 10]. With these caveats in mind, similarities between human and murine hearts make our murine model well suited for the study of the basic mechanisms of arrhythmogenicity.
First, we confirm that alternans is absent in control normokalaemic hearts and that this is associated with the maintenance of stable rhythms even at short BCLs. Secondly, we report alternans in association with arrhythmogenesis at short BCLs for the first time in hypokalaemic murine hearts, in full agreement with previous clinical and experimental studies in other species [2, 7, 33, 38, 52].
Thirdly, we associate such alternans with increases in the slopes of restitution curves above unity [29, 55]. Fourthly, we demonstrate for the first time in any experimental model that exposure to the class I agent lidocaine reduces the slopes of restitution curves and suppresses alternans in association with its anti-arrhythmic effect. Taken together, these observations implicate a restitution mechanism in driving alternans in these hypokalaemic preparations. Furthermore, this extends previous reports that the anti-arrhythmic effects of both class III and IV agents are associated with decreases in the slopes of restitution curves [8, 31, 37]. This contrasts with a previous report that found lidocaine to have no effect on restitution curves obtained using an extrasystolic stimulation procedure or alternans phenomena in a normokalaemic excised canine papillary muscle preparation [43].
Fifthly, we show that such alternans and steeply sloping restitution curves are associated with the temporal evolution of the normal regular pattern of cardiac excitation into arrhythmic activity. The latter, in turn, was processed by a spectral Fourier transform analysis of MAP waveforms performed for the first time in any cardiac system. Arrhythmic MAP waveforms were initially monomorphic, and their corresponding spectra contained a single dominant frequency and integral multiple harmonics identical in epicardial and endocardial recordings. Such monomorphic arrhythmic waveforms subsequently degenerated into polymorphic waveforms whose spectra showed dissimilar epicardial and endocardial dominant frequencies and larger higher order peaks that were not integral multiples of the dominant frequency in full agreement with previous results from the porcine heart [63]. Spectra with similar features have previously been obtained from volume-conducted electrograms and pseudoelectrograms (as opposed to MAPs) from murine hearts during both monomorphic and polymorphic arrhythmic activity [51].
Taken together, these results recapitulate in a murine system arrhythmogenic patterns classically described in clinical situations [16, 58] and implicate a dynamic component to arrhythmic substrate under conditions of hypokalaemia for which restitution may furnish a possible underlying mechanism. Furthermore, they disprove previous suggestions that small hearts are unable to sustain such arrhythmias [9, 60], and thus further validate the murine heart as a model for the study of clinical arrhythmia. The present findings might therefore form a basis for further explorations of the relationships between heart rate, restitution slopes, alternans and arrhythmogenicity in other states of QT prolongation, whether attributable to congenital or acquired factors.
References
Aiba T, Shimizu W, Inagaki M, Noda T, Miyoshi S, Ding WG, Zankov DP, Toyoda F, Matsuura H, Horie M, Sunagawa K (2005) Cellular and ionic mechanism for drug-induced long QT syndrome and effectiveness of verapamil. J Am Coll Cardiol 45:300–307
Armoundas AA, Osaka M, Mela T, Rosenbaum DS, Ruskin JN, Garan H, Cohen RJ (1998) T-wave alternans and dispersion of the QT interval as risk stratification markers in patients susceptible to sustained ventricular arrhythmias. Am J Cardiol 82:1127–1129, A9
Balasubramaniam R, Grace AA, Saumarez RC, Vandenberg JI, Huang CL (2003) Electrogram prolongation and nifedipine-suppressible ventricular arrhythmias in mice following targeted disruption of KCNE1. J Physiol 552:535–546
Bracewell RN (2000) The Fourier transform and its applications. McGrawHill, New York
Casimiro MC, Knollmann BC, Ebert SN, Vary JC Jr, Greene AE, Franz MR, Grinberg A, Huang SP, Pfeifer K (2001) Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange–Nielsen Syndrome. Proc Natl Acad Sci U S A 98:2526–2531
el-Sherif N, Zeiler RH, Craelius W, Gough WB, Henkin R (1988) QTU prolongation and polymorphic ventricular tachyarrhythmias due to bradycardia-dependent early afterdepolarizations. Afterdepolarizations and ventricular arrhythmias. Circ Res 63:286–305
Euler DE (1999) Cardiac alternans: mechanisms and pathophysiological significance. Cardiovasc Res 42:583–590
Garfinkel A, Kim YH, Voroshilovsky O, Qu Z, Kil JR, Lee MH, Karagueuzian HS, Weiss JN, Chen PS (2000) Preventing ventricular fibrillation by flattening cardiac restitution. Proc Natl Acad Sci U S A 97:6061–6066
Garrey W (1914) The nature of fibrillary contraction of the heart: its relation to tissue mass and form. Am J Physiol 33:397–414
Gold MR, Bloomfield DM, Anderson KP, El-Sherif NE, Wilber DJ, Groh WJ, Estes NA 3rd, Kaufman ES, Greenberg ML, Rosenbaum DS (2000) A comparison of T-wave alternans, signal averaged electrocardiography and programmed ventricular stimulation for arrhythmia risk stratification. J Am Coll Cardiol 36:2247–2253
Habbab MA, el-Sherif N (1992) TU alternans, long QTU, and torsade de pointes: clinical and experimental observations. Pacing Clin Electrophysiol 15:916–931
Helfant RH (1986) Hypokalemia and arrhythmias. Am J Med 80:13–22
Hering HE (1909) Experimentelle studien an saugethieren uber das electrocardiogram. Z Exp Pathol Ther 7:363–378
Karagueuzian HS, Khan SS, Hong K, Kobayashi Y, Denton T, Mandel WJ, Diamond GA (1993) Action potential alternans and irregular dynamics in quinidine-intoxicated ventricular muscle cells. Implications for ventricular proarrhythmia. Circulation 87:1661–1672
Kaufman ES, Mackall JA, Julka B, Drabek C, Rosenbaum DS (2000) Influence of heart rate and sympathetic stimulation on arrhythmogenic T wave alternans. Am J Physiol Heart Circ Physiol 279:H1248–1255
Kempf FC Jr, Josephson ME (1984) Cardiac arrest recorded on ambulatory electrocardiograms. Am J Cardiol 53:1577–1582
Killeen MJ, Gurung IS, Thomas G, Stokoe KS, Grace AA, Huang CL (2007) Separation of early afterdepolarizations from arrhythmogenic substrate in the isolated prefused hypokalaemic murine heart through modifiers of calcium homeostasis. Acta Physiol PMID:17524066 (in press)
Killeen M, Thomas G, Gurung I, Goddard C, Fraser J, Mahaut-Smith M, Colledge H, Grace A, Huang C (2007) Arrhythmogenic mechanisms in the isolated perfused hypokalemic murine heart. Acta Physiologica 189:33–46
Knollmann BC, Katchman AN, Franz MR (2001) Monophasic action potential recordings from intact mouse heart: validation, regional heterogeneity, and relation to refractoriness. J Cardiovasc Electrophysiol 12:1286–1294
Knollmann BC, Schober T, Petersen AO, Sirenko SG, Franz M (2007) Action potential characterization in intact mouse heart: steady-state cycle length dependence and electrical restitution. Am J Physiol 292:H614–H621
Koller ML, Riccio ML, Gilmour RF Jr (1998) Dynamic restitution of action potential duration during electrical alternans and ventricular fibrillation. Am J Physiol 275:H1635–1642
Koller ML, Riccio ML, Gilmour RF Jr (2000) Effects of [K+]o on electrical restitution and activation dynamics during ventricular fibrillation. Am J Physiol Heart Circ Physiol 279:H2665–2672
Lab MJ, Woollard KV (1978) Monophasic action potentials, electrocardiograms and mechanical performance in normal and ischaemic epicardial segments of the pig ventricle in situ. Cardiovasc Res 12:555–565
Lee JJ, Kamjoo K, Hough D, Hwang C, Fan W, Fishbein MC, Bonometti C, Ikeda T, Karagueuzian HS, Chen PS (1996) Reentrant wave fronts in Wiggers’ stage II ventricular fibrillation. Characteristics and mechanisms of termination and spontaneous regeneration. Circ Res 78:660–675
Lewis T (1911) Notes upon alternation of the heart. QJM 4:141–144
London B (2001) Cardiac arrhythmias: from (transgenic) mice to men. J Cardiovasc Electrophysiol 12:1089–1091
Moss AJ, Kass RS (2005) Long QT syndrome: from channels to cardiac arrhythmias. J Clin Invest 115:2018–2024
Nearing BD, Huang AH, Verrier RL (1991) Dynamic tracking of cardiac vulnerability by complex demodulation of the T wave. Science 252:437–440
Nolasco JB, Dahlen RW (1968) A graphic method for the study of alternation in cardiac action potentials. J Appl Physiol 25:191–196
Nordrehaug JE (1985) Malignant arrhythmia in relation to serum potassium in acute myocardial infarction. Am J Cardiol 56:20D–23D
Omichi C, Zhou S, Lee MH, Naik A, Chang CM, Garfinkel A, Weiss JN, Lin SF, Karagueuzian HS, Chen PS (2002) Effects of amiodarone on wave front dynamics during ventricular fibrillation in isolated swine right ventricle. Am J Physiol Heart Circ Physiol 282:H1063–1070
Pak HN, Hong SJ, Hwang GS, Lee HS, Park SW, Ahn JC, Moo Ro Y, Kim YH (2004) Spatial dispersion of action potential duration restitution kinetics is associated with induction of ventricular tachycardia/fibrillation in humans. J Cardiovasc Electrophysiol 15:1357–1363
Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS (1999) Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation 99:1385–1394
Platt SB, Vijgen JM, Albrecht P, Van Hare GF, Carlson MD, Rosenbaum DS (1996) Occult T wave alternans in long QT syndrome. J Cardiovasc Electrophysiol 7:144–148
Pruvot EJ, Katra RP, Rosenbaum DS, Laurita KR (2004) Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ Res 94:1083–1090
Rehnqvist N, Ericsson CG, Eriksson S, Olsson G, Svensson G (1984) Comparative investigation of the antiarrhythmic effect of propafenone (Rytmonorm) and lidocaine in patients with ventricular arrhythmias during acute myocardial infarction. Acta Med Scand 216:525–530
Riccio ML, Koller ML, Gilmour RF Jr (1999) Electrical restitution and spatiotemporal organization during ventricular fibrillation. Circ Res 84:955–963
Rosenbaum DS, Jackson LE, Smith JM, Garan H, Ruskin JN, Cohen RJ (1994) Electrical alternans and vulnerability to ventricular arrhythmias. N Engl J Med 330:235–241
Rosenbaum MB, Acunzo RS (1991) Pseudo 2:1 atrioventricular block and T wave alternans in the long QT syndromes. J Am Coll Cardiol 18:1363–1366
Sabir IN, Fraser JA, Cass TR, Grace AA, Huang C (2007) A quantitative analysis of the effect of cycle length on arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. Pflugers Arch PMID:17437126 (in press)
Sabir IN, Fraser JA, Killeen MJ, Grace AA, Huang C (2007) The contribution of refractoriness to arrhythmic substrate in hypokalaemic Langendorff-perfused murine hearts. Pflugers Arch 454:209–222
Sabir IN, Killeen MJ, Goddard CA, Thomas G, Gray S, Grace AA, Huang CL (2007) Transient alterations in transmural repolarization gradients following premature ventricular depolarizations in arrhythmogenic hypokalaemic Langendorff-perfused murine hearts. J Physiol 581:277–289
Saitoh H, Bailey JC, Surawicz B (1989) Action potential duration alternans in dog Purkinje and ventricular muscle fibres: further evidence in support of two different mechanisms. Circulation 80:1421–1431
Sato D, Shiferaw Y, Garfinkel A, Weiss JN, Qu Z, Karma A (2006) Spatially discordant alternans in cardiac tissue: role of calcium cycling. Circ Res 99:520–527
Saumarez RC, Pytkowski M, Sterlinski M, Hauer RN, Derksen R, Lowe MD, Szwed H, Huang CL, Ward DE, Camm AJ, Grace AA (2006) Delayed paced ventricular activation in the long QT syndrome is associated with ventricular fibrillation. Heart Rhythm 3:771–778
Schwartz PJ, Malliani A (1975) Electrical alternation of the T-wave: clinical and experimental evidence of its relationship with the sympathetic nervous system and with the long Q-T syndrome. Am Heart J 89:45–50
Shimizu W, Antzelevitch C (1999) Cellular and ionic basis for T-wave alternans under long-QT conditions. Circulation 99:1499–1507
Shimizu W, Tanaka K, Suenaga K, Wakamoto A (1991) Bradycardia-dependent early afterdepolarizations in a patient with QTU prolongation and torsade de pointes in association with marked bradycardia and hypokalemia. Pacing Clin Electrophysiol 14:1105–1111
Surawicz B, Fisch C (1992) Cardiac alternans: diverse mechanisms and clinical manifestations. J Am Coll Cardiol 20:483–499
Thomas G, Killeen MJ, Gurung IS, Hakim P, Balasubramaniam RN, Goddard CA, Grace AA, Huang CL (2007) Mechanisms of ventricular arrhythmogenesis in mice following targeted disruption of KCNE1 modelling long QT syndrome 5. J Physiol 578:99–114
Vaidya D, Morley GE, Samie FH, Jalife J (1999) Reentry and fibrillation in the mouse heart. A challenge to the critical mass hypothesis. Circ Res 85:174–181
Verrier RL, Nearing BD (1994) Electrophysiologic basis for T wave alternans as an index of vulnerability to ventricular fibrillation. J Cardiovasc Electrophysiol 5:445–461
Walker ML, Rosenbaum DS (2003) Repolarization alternans: implications for the mechanism and prevention of sudden cardiac death. Cardiovasc Res 57:599–614
Walker ML, Wan X, Kirsch GE, Rosenbaum DS (2003) Hysteresis effect implicates calcium cycling as a mechanism of repolarization alternans. Circulation 108:2704–2709
Weiss JN, Chen PS, Qu Z, Karagueuzian HS, Garfinkel A (2000) Ventricular fibrillation: how do we stop the waves from breaking? Circ Res 87:1103–1107
Weiss JN, Karma A, Shiferaw Y, Chen PS, Garfinkel A, Qu Z (2006) From pulsus to pulseless: the saga of cardiac alternans. Circ Res 98:1244–1253
Whelton PK (1984) Diuretics and arrhythmias in the Medical Research Council trial. Drugs 28(Suppl 1):54–65
Wiggers CJ (1930) Studies of ventricular fibrillation caused by electric shock, II: cinematographic and electrocardiographic observation of the natural process in the dog’s heart. Its inhibition by potassium and the revival of coordinated beats by calcium. Am Heart J 5:351–365
Wilde AA, Jongbloed RJ, Doevendans PA, Duren DR, Hauer RN, van Langen IM, van Tintelen JP, Smeets HJ, Meyer H, Geelen JL (1999) Auditory stimuli as a trigger for arrhythmic events differentiate HERG-related (LQTS2) patients from KVLQT1-related patients (LQTS1). J Am Coll Cardiol 33:327–332
Winfree AT (1994) Electrical turbulence in three-dimensional heart muscle. Science 266:1003–1006
Yamauchi S, Yamaki M, Watanabe T, Yuuki K, Kubota I, Tomoike H (2002) Restitution properties and occurrence of ventricular arrhythmia in LQT2 type of long QT syndrome. J Cardiovasc Electrophysiol 13:910–914
Yang T, Roden DM (1996) Extracellular potassium modulation of drug block of IKr. Implications for torsade de pointes and reverse use-dependence. Circulation 93:407–411
Zaitsev AV, Berenfeld O, Mironov SF, Jalife J, Pertsov AM (2000) Distribution of excitation frequencies on the epicardial and endocardial surfaces of fibrillating ventricular wall of the sheep heart. Circ Res 86:408–417
Zareba W, Moss AJ, le Cessie S, Hall WJ (1994) T wave alternans in idiopathic long QT syndrome. J Am Coll Cardiol 23:1541–1546
Acknowledgements
We thank the James Baird Fund, the Frank Elmore Fund, the Medical Research Council, the Wellcome Trust, the British Heart Foundation and Downing College, Cambridge for their generous support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sabir, I.N., Li, L.M., Grace, A.A. et al. Restitution analysis of alternans and its relationship to arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. Pflugers Arch - Eur J Physiol 455, 653–666 (2008). https://doi.org/10.1007/s00424-007-0327-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-007-0327-y