
Abstract
The brain tissue obtained from ninety-five cognitively unimpaired subjects, with ages ranging from 22 to 50 years upon death, were immunohistochemically assessed for neurodegenerative changes, i.e., hyperphosphorylated tau (HPτ) and β-amyloid (Aβ) pathology in predilection neuroanatomical areas. HPτ pathology was observed in the transentorhinal cortex and/or the locus coeruleus (LC) in 33% of the subjects, without any obvious risk factors known to alter the microtubule-associated protein. HPτ pathology was noted in the LC in 25 out of 83 subjects (30%), lacking concomitant cortical Aβ or transentorhinal HPτ pathology. This observation was present even when assessing only one routine section of 7 μm thickness. The recent suggestion of prion-like propagation of neurodegeneration and the finding of neurodegeneration being quite common in middle-aged persons is alarming. It is noteworthy, however, that a substantial number of neurologically unimpaired subjects even at a very old age display only sparse to modest extent of neurodegenerative pathology. Thus, only a subset of subjects with neurodegenerative changes early in life seem to progress to a symptomatic disease with ageing. This observation brings forth the notion that other, yet unknown modifying factors influence the progression of degeneration that leads to a symptomatic disorder. The known association between alterations in the LC and mood disorders, and the finding of the LC being frequently affected with HPτ pathology suggest that clinicopathological studies on young subjects both with or without mood disorders are warranted.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hyperphosphorylation of microtubule-associated protein tau is observed in primary and secondary tauopathies [22] as well as in aged, unimpaired individuals [26]. Braak and Braak [5] reported that HPτ is observed in Alzheimer’s disease (AD), a secondary tauopathy, in a hierarchical way where the first two stages (Braak stage I and II) are characterised by alteration seen in the transentorhinal cortex. Subjects with pathology in the first two stages are cognitively unimpaired, and do not display clinical symptoms of dementia [4, 20]. Brainstem nuclei such as the locus coeruleus (LC) were reported to be involved with HPτ pathology later in the progression, starting from stage II upward. Thus, HPτ is observed in the LC in some unimpaired aged individuals (Braak stages II–III), in subjects with mild cognitive impairment (MCI) and in cases with AD (Braak stages IV–VI) [8, 15].
Since the launch of the Braak-AD staging strategy, the assessment of AD-related pathology is mostly carried out on sections including, hippocampus and cortex [2]. Recently, however, Braak et al. reported that HPτ pathology was seen prior to entorhinal cortex in as many as 22 subjects out of 26 while assessing individuals who died under 29 years of age (age range at death from 4 to 29 years) [7]. Furthermore, other studies have reported that regions such as dorsal Raphe nucleus or occipital visual association cortex (area 18/19) are in some cases affected prior to the transentorhinal cortex [14, 23, 25].
The aim of the present study was to assess the prevalence of HPτ pathology in cognitively unimpaired middle-aged subjects. Furthermore, the objective was to investigate whether the LC was involved prior to the entorhinal cortex. The assessment was carried out applying routine sampling strategy and routine immunohistochemical (IHC) techniques.
Materials and methods
Case selection
A total of 1,505 subjects underwent a clinical autopsy, including a neuropathological examination during the years 1996–2008 at the Kuopio University Hospital, Finland. Ninety-five subjects fulfilled the selection criteria delineated for this study. First, the subjects should not have displayed cognitive impairment while alive, secondly the subjects’ age at death should not exceed 50 years, and finally, all subjects with infectious disease in the brain or having primary or secondary brain tumours were excluded. Demographics of the material are summarised in Table 1.
Apolipoprotein E (APOE) genotypes were determined in a subset of cases as previously described [31].
Neuropathological assessment
According to the standard dissection protocol used in Kuopio University Hospital, the brains were weighed and then immersed for at least 1 week in 10% buffered formalin solution and thereafter, cut into 1 cm coronal slices. Macroscopically detectable lesions were noted. Brain specimens were taken from 16 routinely sampled brain regions: 1, frontal (Brodmann 9); 2, temporal (Brodmann 22); 3, gyrus cinguli; 4, parietal (Brodmann 39); 5, pre- and postcentral gyrus; 6, occipital cortices; 7, anterior hippocampal formation; 8, posterior hippocampal formation; 9, striatum; 10, basal forebrain including the amygdaloid complex; 11, thalamus; 12, midbrain including substantia nigra; 13, pons including LC; 14, medulla; 15, vermis and 16, cerebellar cortex. All sections were stained with hematoxylin-eosin (HE). Cerebrovascular lesions seen in the HE stained sections were classified as previously described to acute, subacute, chronic or mixed (Table 2) [1].
Immunohistochemistry
Seven micrometer thick sections were cut from formalin fixed, paraffin embedded tissue. The sections were deparaffinised, rehydrated in graded alcohol series. Details regarding staining and the specific antibody applied are summarised in Table 3. For detection, the power vision poly HPR-IHC detection kit was used with Romulin-3-amino-9-ethylcarbazol (AEC) chromogen. It was determined whether β Amyloid (Aβ) immunoreactivity (IR) was present or absent in the cortical sections (frontal, temporal and parietal cortices). Braak HPτ stage was given as described previously by assessing distribution of HPτ in hippocampal sections (Table 2). In the section taken at the level of pons, including LC, HPτ-IR pretangles, neurites and tangles were searched for under light microscopy at ×40 magnification and noted as being present or absent. Digital images were taken using an Olympus BX46 microscope equipped with an Olympus BX46 microscope digital camera system (Olympus Optical Co., Ltd., Tokyo, Japan).
Results
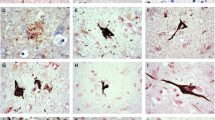
The gender distribution of the included cases was quite even (40 females, 55 males) (Table 1). The most common primary cause of death was cerebral infarct (48%) and the most common type of infarct was acute (Table 2). Seven out of 95 subjects (7%) displayed Aβ aggregates in the cortical sections whereas 31 out of 95 subjects (33%) displayed HPτ pathology in some of the assessed regions. In six out of 95 subjects (6%) lacking HPτ, Aβ pathology was seen in the cortex. Furthermore, in 30 subjects (32%) lacking Aβ, HPτ pathology was seen in the entorhinal cortex or LC. In 28 out of 95 subjects (29%), HPτ-IR lesions were seen in the LC and HPτ-IR increased with age (Tables 2, 4). All three types of lesions, i.e. pretangles, neurites, tangles, were seen in 8 subjects (Table 4; Fig. 1). In six subjects only pretangles were observed, in two others only neurites, and in two other subjects, only tangles were observed. In 3 of these 28 subjects with HPτ in the LC, HPτ pathology was also observed in the entorhinal cortex to a limited extent. Concomitant cortical Aβ-IR and HPτ-IR were observed in one of these 28 subjects.

AT8 immunoreactive (IR) (hyperphosphorylated τ) lesions in a 7 μm thick section of the locus coeruleus. a A 49-year-old unimpaired male subject with grainy IR material, pretangle, (arrow) in the neuronal cell body, b a 44-year-old unimpaired female with coarse AT8-IR structure, a tangle, (arrow) in the neuronal cell body and c a 41-year-old unimpaired female subject with thin AT8-IR fibres, dendrites, in the neuropil (arrow). Magnification ×40. Scale bar 100 μm
In 15 subjects (48%) with HPτ pathology, a concomitant cerebral vascular lesion was noted (Table 4); an incidence somewhat lower when compared with the 59% that was observed in the 64 subjects lacking HPτ pathology. In 3 of the 31 subjects with HPτ pathology, the cerebrovascular lesions were extra-axial (aneuryms), in five an acute/subacute ischemic, in one acute/subacute hemorrhagic and in the remaining six subjects, chronic or mixed infarcts were observed. In two out of 7 subjects with Aβ, concomitant cerebral vascular lesions were observed (acute hemorrhagic and acute ischemic infarct) and in one case with both Aβ and HPτ, an acute hemorrhagic infarct was detected. There was no association between the type of cerebrovascular lesions and observation of HPτ pathology.
Discussion
As indicated above, 31 out of 95 (33%) unimpaired middle-aged subjects, ranging from 22 to 50 years of age at death, manifested HPτ pathology in one or both of the assessed regions, i.e. the transentorhinal cortex and the LC, during assessments of routine diagnostic sections. Moreover, in 25 subjects out of 89 (28%) lacking HPτ pathology in the transentorhinal cortex, HPτ pathology was seen solely in the LC. Thus, our results are, in principle, in agreement with Braak et al. [7] who recently reported the LC, frequently being affected with HPτ pathology even in very young subjects. They reported that in as many as 22 out of 26 (85%) young subjects lacking HPτ pathology in the transentorhinal cortex, some HPτ pathology was seen in the LC. This percentage of 85%, in subjects ranging from 4 to 29 years of age at death, was significantly higher when compared to our results of 28%.
It is noteworthy that in our study the subjects were older in comparison with subjects studied by Braak and Del Tredici. Thus, one would have expected that the ageing-related HPτ pathology would have been observed even more frequently in our material, given that the age at death ranged from 22 to 50 years in our study. We noted in our material that indeed the incidence of HPτ pathology in the LC increases with age, being 14% in the youngest and 42% in the oldest group. There are two significant differences when comparing our study with the study reported by Braak and Del Tredici. The studies differ based on: (1) the methodology used while assessing HPτ pathology and (2) the sampling strategy of subjects.
Specifically, Braak and Del Tredici used 100 μm thick sections in comparison with our 7 μm thick sections. Consequently, cases displaying only occasional or few HPτ-IR neurites might have been missed, due to us assessing sections that were 14 times thinner. Braak and Del Tredici reported that in 12 out of 26 cases, only HPτ-IR neurites were observed in the LC (minimal pathology) whereas in 10 out of these 26 subjects, in addition to HPτ-IR neurites, IR nerve cells were also noted (moderate amount of pathology). Thus, the finding of 10 subjects out of 26 (38%) with IR neurites and nerve cells when assessing 100 μm thick sections is more in line with our results of 28%, given that we assessed sections that were 14 times thinner. While studying subjects with AD-related pathology, Busch et al. [8] reported that there is a significant difference with regard to distribution of HPτ pathology in the LC. While carrying out a topographical analysis, the dorsal and medial portions of the LC were reported to be more affected when compared with the ventral and lateral portions, emphasising the importance of neuroanatomical sampling strategy [8]. This variation in topographical distribution of pathology should not have contributed to the observed differences as the sampling strategy in our study and that of Braak and Del Tredici is comparable, i.e. the tissue block assessed was obtained at the level of pontine tegmentum. Thus, the primary difference in the presence of HPτ pathology in the LC is probably due to the thickness of the sections assessed, i.e. 7 versus 100 μm.
The selection strategy of subjects to be studied also differed. The cause of death among subjects studied by Braak and Del Tredici was quite various, including brain tumours and transplantation of organs; all diseases associated with heavy pharmaceutical treatment. Thus, in some cases hyperphosphorylation of the microtubule-associated τ protein might be due to the used pharmacological therapy. Agents such as cell cycle inhibitors, mitochondrial complex I inhibitors, anti microtubule agents as well as glucocorticoids have been reported to induce hyperphosphorylation of τ in in vivo and in vitro studies [10, 16–18, 30]. In addition, some of the subjects had suffered from head injury. An association between acute head injury and accumulation of HPτ has been reported to be seen in both experimental animal studies as well as in human studies [12, 19, 32]. Our selection strategy was more stringent. Specifically, we only included subjects who died acutely without prolonged disease or heavy pharmaceutical treatment. Thus most of our subjects, 67 out of 95, died due to an acute vascular event, i.e. cerebral or cardiac insults. Cardiovascular alterations in middle age have been suggested as being a risk factor regarding cognitive decline and dementia in later life and cardiovascular disease and AD share many common risk factors [21, 29]. However, it is noteworthy that cerebrovascular lesions were seen just as frequently, both in subjects with and also without HPτ pathology. Thus, 33% of our subjects, with age ranging from 22 to 50 years at death, displayed HPτ pathology in the transentorhinal cortex and/or locus coeruleus without any specific known causative reason.
Interestingly, while studying drug abusers, Anthony et al. [3] reported the LC to be affected with HPτ pathology in young subjects (age at death <30 years). It is noteworthy that the LC was more involved than compared to the hippocampus in these young subjects; a finding in line with both our observations, and those reported by Braak and Del Tredici. Surprisingly, in subjects >30 years of age at death, the HPτ pathology was reported to be more severe in the hippocampus than compared to the LC [3]. Overall, the HPτ pathology increased with age; a finding in line with ours. Thus, based on the above it is obvious that the LC is indeed frequently affected with HPτ pathology prior to other brain regions.
In 2004 Braak and Del Tredici stated in a commentary that the pathological process of AD, displays a simultaneous involvement of the transentorhinal cortex, magnocellular nucleus and the LC. Furthermore, in 2011 they reported that HPτ pathology in the LC is frequently seen in subjects under the age of 30 and they suggest that this pathology might be the initiation of AD or other tauopathies [6, 7]. In 2011, while assessing 889 cases fulfilling the Braak criteria of more than IV, Murray et al. identified three different distributional patterns, i.e., classical (75%), limbic predominant (14%) and hippocampal sparing (11%). This finding suggests more than one pattern of pathological progression of AD [24]. 33% of our unimpaired subjects, with age ranging from 22 to 50 years at death displayed HPτ pathology in the LC and/or entorhinal cortex. It could be interpreted from these findings that these subjects are in a preclinical stage of a tauopathy. This interpretation would further be in line with the report of tauopathy spreading from one site to another in a transgenic mice model, indicating that tauopathy might propagate in a prion-like way and thus, all those with HPτ pathology in the LC would in due time progress into a tauopathy [9, 13]. The suggestion that young and middle-aged individuals that display HPτ pathology in the LC and entorhinal cortex would progress to a symptomatic tauopathy is, however, controversial. A substantial number of neurologically unimpaired subjects even at a very old age display only sparse to modest extent of neurodegenerative pathology and only a subset of the middle-aged subjects with HP tau pathology in the LC and entorhinal cortex seem to develop a dementing disorder with aging. This observation indicates that other, yet unknown modifying mechanisms lead to the regional progression of pathology causing a symptomatic disorder.
It is noteworthy that most of our cases with HPτ-IR pathology in the LC did not display cortical Aβ aggregates. This finding is in line with results reported by Braak and Del Tredici [7]. Thus, our results and the results reported by Braak and Del Tredici are in line with the presumption that the neuronal cytoskeletal alteration is a primary lesion that might induce the extracellular deposits of Aβ. It is also important to note, however, that Wirths et al. [33] recently reported that they were able to detect intraneuronal oligomeric assemblies of N-terminally truncated Aβ peptides starting with pyroglutamate (AβpE3), not detectable with other commonly applied antibodies in an unimpaired control lacking extracellular aggregates of Aβ. Consequently subjects, even at a very young age, should be examined for oligomeric intracellular Aβ in order to assess which of the two pathologies, HPτ or oligomeric Aβ is primarily observed.
In our study, the pathology was only seen in the transentorhinal cortex in three cases. This finding of the LC being unaffected in these three cases might be due to the methodological aspects discussed above. We only stained one section, thus a minute amount of pathology could easily have been missed. Another explanation might be related to the hypothesis proposed by Braak and Del Tredici [6], that of simultaneous involvement of the transentorhinal cortex, magnocellular nucleus and the LC with HPτ pathology.
Behavioural and psychological symptoms such as depression, mood and emotional disturbances, confusion and agitation are observed in AD patients [28]. Several studies have suggested that pathological alteration in the LC and substantia nigra are probably causative regarding the depression in AD [34]. Previous assessment of the LC in subjects with mood disorders has been centred on cell count/loss. Assessment of HPτ pathology in the LC in subjects with the clinical diagnosis of pure mood disorders has not been systematically carried out to our knowledge. Association of LC pathology with depression and observation of early involvement of the LC with HPτ pathology is disturbing. First, this could suggest that a tauopathy is initiated much earlier than previously even suspected. Secondly, this finding might implicate that young subjects with depression might be at risk for developing AD or other tauopathy in later life. Rapp et al. [27] reported that the load of pathology in subjects with AD was significantly higher in those who had suffered from a lifetime history of depression, thus, suggesting an association between these two disorders. In line with the observations above, an association between AD and history of early onset of depression has been reported [11]. Thus, additional post-mortem studies assessing HPτ pathology in the brain stem of subjects of all ages with a diagnosis of a mood disorder are urgently needed to clarify this issue.
In summary, we conclude that HPτ pathology is indeed frequently seen in the LC in unimpaired middle-aged subjects without any obvious risk factors known to alter the microtubule-associated protein. HPτ pathology was noted in the LC in 25 out of 83 subjects (30%) lacking concomitant cortical Aβ or transentorhinal HPτ. This observation was obtained when assessing only one section with 7 μm thickness. The finding of frequent involvement of LC with HPτ pathology in early middle-aged subjects raises many questions and concerns. It is not clear whether this pathology is the primary stage of AD or other tauopathy or merely an insignificant alteration related to ageing. The suggestion of prion-like propagation of neurodegeneration would be in line with the former interpretation [13] whereas the observation of sparse extent of pathology in most aged subjects supports the latter. In most cases, HPτ was observed without concomitant Aβ pathology. Recent observations have shown the presence of oligomeric Aβ in aged cases without Aβ aggregates. Thus, assessment of oligomeric Aβ in individuals with sparse HPτ pathology should urgently be carried out. The association between the LC and mood disorders raises the question of whether some subjects with a mood disorder are at risk of developing a tauopathy. Clinicopathological studies on young subjects and particularly those with mood disorders are thus urgently needed.
References
Aho L, Jolkkonen J, Alafuzoff I (2006) β-amyloid aggregation in human brains with cerebrovascular lesions. Stroke 37:2940–2945
Alafuzoff I, Arzberger T, Al-Sarraj S et al (2008) Staging of neurofibrillary pathology in Alzheimer’s disease: a study of Brain Net Europe consortium. Brain Pathol 18:484–496
Anthony IC, Norrby KE, Dingwall T et al (2010) Predisposition to accelerated Alzheimer-related changes in the brains of human immunodeficiency virus negative opiate abusers. Brain 133:3685–3698
Bancher C, Braak H, Fischer P, Jellinger KA (1993) Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer’s and Parkinson’s disease. Neurosci Lett 162:179–182
Braak H, Braak E (1991) Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Del Tredici K (2004) Alzheimer’s disease: intraneuronal alterations precede insoluble amyloid-β formation. Neurobiol Aging 25:713–718
Braak H, Del Tredici K (2011) The pathological process underlying Alzheimer’s disease in individuals under 30. Acta Neuropathol 121:171–181
Busch C, Bohl J, Ohm TG (1997) Spatial, temporal and numeric analysis of Alzheimer changes in the nucleus coeruleus. Neurobiol Aging 18:401–406
Clavaguera F, Bolmont T, Crowther RA et al (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Boil 11:909–913
Conejero-Goldberg C, Townsend K, Davies P (2008) Effect of cell cycle inhibitors on tau phosphorylation in N2aTau3R cells. J Mol Neurosci 35:143–150
Geerlings MI, den Heijer T, Koudstaal PJ, Hofman A, Breteler MM (2008) History of depression, depressive symptoms, and medial temporal lobe atrophy and the risk of Alzheimer disease. Neurology 70:1258–1264
Genis L, Chen Y, Shohami E, Michaelson DM (2000) Tau hyperphophorylation in apolipoprotein E-deficient and control mice after closed head injury. J Neurosci Res 60:559–564
Goedert M, Clavaguera F, Tolnay M (2010) The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci 33:317–325
Grinberg LT, Rüb U, Ferretti REL et al (2009) The dorsal Raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer’s disease. A precocious onset? Neuropathol Appl Neurobiol 35:406–416
Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM (2007) Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol Aging 28:327–335
Guise S, Braguer D, Remacle-Bonnet M, Pommier G, Briand C (1999) Tau protein is involved in the apoptotic process induced by anti-microtubule agents on neuroblastoma cells. Apoptosis 4:47–58
Guise S, Braguer D, Carles G, Delacourte A, Briand C (2001) Hyperphosphorylation of tau is mediated by ERK activation during anticancer drug-induced apoptosis in neuroblastoma cells. J Neurosci Res 63:257–267
Höglinger GU, Lannuzel A, Khondiker ME et al (2005) The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. J Neurochem 95:930–939
Ikonomovic MD, Uryu K, Abrahamson EE et al (2004) Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol 190:192–203
Jellinger K, Braak H, Braak E, Fischer P (1991) Alzheimer lesions in the entorhinal region and isocortex in Parkinson’s and Alzheimer’s disease. Ann NY Acad Sci 640:203–209
Launer LJ (2007) Next steps in Alzheimer’s disease research: interaction between epidemiology and basic science. Curr Alzheimer Res 4:141–143
Lee VM-Y, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Ann Rev Neurosci 24:1121–1159
Mckee AC, Au R, Cabral HJ et al (2006) Visual association pathology in preclinical Alzheimer disease. J Neuropathol Exp Neurol 65:621–630
Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW (2011) Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 10:785–796
Pikkarainen M, Kauppinen T, Alafuzoff I (2009) Hyperphosphorylated tau in the occipital cortex in aged nondemented subjects. J Neuropathol Exp Neurol 68:653–660
Price JL, Morris JC (1999) Tangles and plaques in nondemented aging and preclinical Alzheimer disease. Ann Neurol 45:358–368
Rapp MA, Schnaider-Beeri M, Grossman HT et al (2006) Increased hippocampal plaques and tangles in patients with Alzheimer disease with a lifetime history of major depression. Arch Gen Psychiatry 63:161–167
Simic G, Stanict G, Mladinov M, Jovanov-Milosevic N, Kostovic I, Hof PR (2009) Does Alzheimer’s disease begin in the brain stem? Neuropathol Appl Neurobiol 35:532–554
Skoog I, Nilsson L, Persson G et al (1996) A15-year longitudinal study of blood pressure and dementia. Lancet 347:1141–1145
Sotiropoulos I, Catania C, Pinto LG et al (2011) Stress acts cumulatively to precipitate Alzheimer’s disease-like tau pathology and cognitive deficits. J Neurosci 31:7840–7847
Tsukamoto K, Watanabe T, Matsushima T et al (1993) Determination by PCR-RFLP of APOE genotype in a Japanese population. J Lab Clin Med 121:598–602
Uryu K, Chen X, Martinez D et al (2007) Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol 208:185–192
Wirths O, Erck C, Martens H et al (2010) Identification of low molecular weight pyroglutamate Aβ oligomers in Alzheimer disease. A novel tool for therapy and diagnosis. J Biol Chem 285:41517–41524
Zubenko GS, Moossy J (1988) Major depression in primary dementia. Clinical and neuropathologic correlates. Arch Neurol 45:1182–1186
Acknowledgments
We thank the medical laboratory technologist Tarja Kauppinen, Mrs. Merja Fali, Mr. Heikki Luukkonen, Monica Sternesjö and Mr. Hannu Tiainen for their skilful technical assistance. We thank Meena Strömqvist for proofreading the manuscript. This study has been authorised by the Ethics Committee of Kuopio University Hospital and the Finnish National Authority for Medicolegal Affairs. This study has been supported by Ahfad University for Women, Khartoum, Sudan and the local ALF grants at Uppsala University Hospital.
Conflict of interest
The authors report no conflicts of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Elobeid, A., Soininen, H. & Alafuzoff, I. Hyperphosphorylated tau in young and middle-aged subjects. Acta Neuropathol 123, 97–104 (2012). https://doi.org/10.1007/s00401-011-0906-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-011-0906-z