
Abstract
Sequence analysis of organelle genomes and comprehensive analysis of C-to-U editing sites from flowering and non-flowering plants have provided extensive sequence information from diverse taxa. This study includes the first comprehensive analysis of RNA editing sites from a gymnosperm mitochondrial genome, and utilizes informatics analyses to determine conserved features in the RNA sequence context around editing sites. We have identified 565 editing sites in 21 full-length and 4 partial cDNAs of the 39 protein-coding genes identified from the mitochondrial genome of Cycas taitungensis. The information profiles and RNA sequence context of C-to-U editing sites in the Cycas genome exhibit similarity in the immediate flanking nucleotides. Relative entropy analyses indicate that similar regions in the 5′ flanking 20 nucleotides have information content compared to angiosperm mitochondrial genomes. These results suggest that evolutionary constraints exist on the nucleotide sequences immediately adjacent to C-to-U editing sites, and similar regions are utilized in editing site recognition.
Similar content being viewed by others
Introduction
Comprehensive analysis of RNA editing sites has been reported for several chloroplast and mitochondrial genomes (mtDNAs) of non-flowering and flowering plants. Chloroplast genomes (cpDNAs) of flowering plants typically possess about 30 C-to-U editing sites and no U-to-C editing sites (Wakasugi et al. 2001). However, cpDNAs of some non-flowering plants exhibit much higher frequencies of RNA editing. For example, the cpDNA from the hornwort, Anthoceros formosae, exhibits extensive editing with 509 C-to-U and 433 U-to-C editing sites (Kugita et al. 2003a, b), but other non-flowering plants such as Marchantia have no RNA editing sites (Oda et al. 1992). Comprehensive analysis of editing sites in the cpDNA of the fern Adiantum capillus-veneris reported 350 C-to-U and 35 U-to-C editing sites (Wolf et al. 2003, 2004). The moss Takakia lepidozioides has been partially characterized for editing sites, and 302 C-to-U and no U-to-C editing sites were reported (Yura et al. 2008). Several angiosperm mtDNAs have between 350 and 500 C-to-U editing sites (Giege and Brennicke 1999; Kubo et al. 2000; Notsu et al. 2002; Handa 2003; Mower and Palmer 2006), and the Cycas mtDNA was predicted to include over 1,000 C-to-U editing sites (Chaw et al. 2008).
Thus, the prevalence of C-to-U editing sites in the cpDNAs and mtDNAs of diverse plants offers an excellent opportunity to evaluate the nucleotide distribution in different organelle systems and diverse taxa of land plants. Several computational studies have examined RNA editing sites (Cummings and Myers 2004; Tillich et al. 2006; Mulligan et al. 2007; Jobson and Qiu 2008; Yura et al. 2008). C-to-U editing sites in angiosperm mtDNAs have similar information profiles in the 5′ and 3′ flanking regions and a similar RNA context (Mulligan et al. 2007). These results are consistent with molecular analyses that demonstrated that 5′ flanking regions are required for editing site conversion (Takenaka et al. 2004; van der Merwe et al. 2006). Edited chloroplast RNA fragments for ndhB-9 and ndhF-1 share sequence similarity around the editing site, and are specifically bound by the same chloroplast protein (Kobayashi et al. 2008). In addition, CRR4, a PPR protein required for editing in Arabidopsis chloroplasts, is a sequence-specific RNA-binding protein that binds to sequences comprised of 25 nucleotides upstream and 10 nucleotides downstream of the ndhD-1 editing site (Okuda et al. 2006).
RNA editing sites in lower plant cpDNAs have also been analyzed with computational tools. The distribution of nucleotides around editing sites in lower and higher plant chloroplasts was examined within codons, and a detailed analysis of the effects on codon changes and usage was developed (Tillich et al. 2006), and a model for the evolution of RNA editing based on conservation of codon usage was proposed. The RNA sequences flanking 302 C-to-U editing sites from Takakia cpDNA were classified into eight groups with common patterns, and these patterns could be used to predict novel editing sites (Yura et al. 2008). Recently, Jobson and Qiu (2008) examined C-to-U and U-to-C editing patterns with respect to codon position and amino acid changes in the cpDNAs and mtDNAs of plants (Jobson and Qiu 2008). Editing was reported to increase the hydrophobicity and molecular size of the amino acid side chain, was most abundant in genes of membrane proteins, and was more frequent in T-rich sequences and in genes under positive selection (Jobson and Qiu 2008).
In this paper, we report the first extensive analysis of RNA editing from the mtDNA of a gymnosperm, Cycas taitungensis (Taitung cycad) and confirm 565 editing sites in 25 mitochondrial genes. The Cycas editing sites and sequence data from known flowering and non-flowering plant mtDNAs are analyzed with informatics tools. Common features of the editing sites from these diverse taxa suggest similar mechanisms of editing site recognition and conversion.
Methods
DNA sequence analysis
Five grams of fresh Cycas leaves were frozen in liquid nitrogen and pulverized with a mortar and pestle. Total RNA was extracted and purified by RNeasy® Plant Mini Kit (Qiagen, Hilden). For reverse transcriptase-polymerase chain reaction (RT-PCR) assay, total RNA was treated with DNAse I and then extracted with phenol–chloroform to eliminate DNA contamination. RNA was reverse transcribed to synthesize cDNA with Superscript II reverse transcriptase (Invitrogen, Indianapolis) and a gene-specific primer (Additional file 2) according to the manufacturer’s protocol. cDNAs were PCR amplified with specific reverse and forward primer pairs (Additional file 2). The PCR products were purified by gel extraction (Gel-M, Viogene Inc., Taiwan) and directly sequenced with the BigDye terminator cycle sequencing kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s protocol. DNA was sequenced with Applied Biosystems ABI 3700 sequencer.
DNA and RNA sequence data
cpDNA sequences and the identification of editing sites were obtained from the following Genbank accessions and citations: Adiantum capillus-veneris, AY178864 (Wolf et al. 2003, 2004); Anthoceros formosae, NC_004543 (Kugita et al. 2003a, b); Takakia lepidozioides, AB193121, AB254134, AB299142, AB367138, AB367138 (Yura et al. 2008); Zea mays, X86563 (Maier et al. 1995); and Nicotiana tabacum, Z00044 (Shinozaki et al. 1986; Tsudzuki et al. 2001). mtDNA sequences and the identification of editing sites were obtained from the following Genbank accessions and citations: C. taitungensis, AP009381 (Chaw et al. 2008); Arabidopsis thaliana, NC001284 (Giege and Brennicke 1999); Beta vulgaris, AP006444 (Kubo et al. 2000); Brassica napus, BA000009, DQ381444–DQ381465 (Handa 2003; Mower and Palmer 2006); Oryza sativa, BA000029 (Notsu et al. 2002). Additional file 3 provides accession numbers for cDNA sequence of Cycas mitochondrial genes.
Protein-coding sequences for each genome were annotated with edited nucleotides represented by an upper case C, and these sequences are available in Additional files 4, 5, 6, and 7. Thus, edited nucleotide positions are represented as the unedited nucleotide in the sequences analyzed in this study. The sequence files were limited to known protein-coding sequences larger than 100 nucleotides, and small or uncharacterized ORFs, introns, and other non-coding sequences were not analyzed.
Computational analyses
Computational analyses were performed as previously described (Mulligan et al. 2007). Briefly, the nucleotide distribution around all edited and unedited cytidines was analyzed in a one-, two-, or three-nucleotide sliding window. Each coding sequence was scanned for edited and unedited C, and the sequence was written to an array of edited or unedited sequences. Thus, the sequences flanking all edited or unedited cytidines were aligned in a matrix. The frequency of nucleotides around the edited or unedited nucleotide (P or Q, respectively) was used to calculate the selectivity ratio (P/Q). Thus, a nucleotide or series of nucleotides with a selectivity ratio of one has the same relative frequency around edited and unedited cytidines, while a nucleotide with a selectivity ratio greater than 1 is more frequently present around an edited cytidine. Relative entropy was calculated as the Kullback–Leibler distance by the equation d = ∑P k log(P k /Q k ) over k terms (k = 4n) for the distribution of nucleotides in 1, 2, or 3 nucleotide windows.
Random editing site assignment
Random editing site assignment was used to produce coding sequences with randomly assigned editing sites. The editing site reassignment program scans each coding-sequence entry, and determines the number and codon position of each of the editing sites. This program randomly assigns a cytidine in the same codon position as an editing site and maintains the number and codon position of editing sites in a coding sequence. As a result, it is not “random”. Statistics such as mean, standard deviation, variance, and confidence intervals were determined from 1,000 iterations of random editing site reassignment.
Results
Characteristics of abundant RNA editing in Cycas mtDNA
Table 1 shows the distribution of RNA editing sites within gene sequences of the C. taitungensis mtDNA. Five hundred and sixty-five editing sites are confirmed by cDNA sequence analysis of 21 genes and partial sequences for four additional genes. Using PREP-Mt with several cutoff scores (Mower 2005), Chaw et al. (2008) predicted that the Cycas mtDNA has more than 1,000 C-to-U editing sites in the 39 protein-coding genes. Table 2 shows the distribution of editing sites within codons, and the distribution in the first, second and third codon positions are 30, 65, and 5%, respectively. This pattern of editing site distribution is very similar to that in angiosperm mtDNAs (Mulligan et al. 2007). Some start and stop codons are created by RNA editing in the Cycas mtDNA (Additional file 1). Start codons are created by editing ACG to ATG in three genes, including atp1, cox1 and sdh3, while stop codons are created by CGA to UGA conversion in atp6, atp9, ccmFC, nad4, rps12, and sdh3, and by CAA to UAA conversion for atp8, nad4L, and rps11. Table 2 shows the distribution of observed editing sites within codons in Cycas mitochondrial genome.
Informatics analysis shows high information in the 5′ flanking sequences of editing sites in the Cycas mtDNA
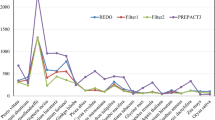
The relative entropy around Cycas editing sites is analyzed in a sliding window of 1, 2, or 3 nucleotides (Fig. 1a, b, c, respectively). The profiles show large values at nucleotides −1 and −2 and small peaks in the 5′ flanking region (−9, −6/−5/−4) and at +1. The influence of codon position is analyzed by separate analyses of editing sites in the first or second codon position, and the relative entropy of these subsets also exhibits similar information profiles (Fig. 1d). The highest relative entropy is present in the −1 position, and profile is similar in the 5′ flanking region with a peak at −5. The major difference in the relative entropy around editing sites in codon positions 1 and 2 is the large peak at the +2 position in CPA1, and a similar result was observed in angiosperm mitochondrial genomes (Mulligan et al. 2007). This position represents the first downstream wobble position, and synonymous mutations may allow optimization of the editing site for efficient editing, and would result in increased entropy at these positions. The information content around editing sites in angiosperm mtDNAs is similar with very high information immediately 5′ of the editing site, a peak at nucleotides −6/−5/−4 and +1, and relatively little information in the 3′ flanking region (Mulligan et al. 2007).

The relative entropy around C-to-U editing sites in Cycas mitochondria. The relative entropy for the distribution of nucleotides is plotted for 30 nucleotides flanking RNA editing sites in 1, 2, or 3 nucleotide sliding windows (a, b, c, respectively). Random editing site assignment is used to reassign editing sites in the same codon position, and relative entropy analysis of 1,000 editing site reassignments is used to determine a mean relative entropy value and a 95% confidence interval. d The effect of codon position on relative entropy in Cycas mitochondrial editing sites determined in a one nucleotide window for editing sites in the first or second codon position (CPA1, CPA2). The number of editing sites analyzed in the first and second codon position is 173 and 376. Only 29 editing sites are present in the third codon position and these data are not presented
RNA sequence context of C-to-U editing sites
The highest level of information around C-to-U editing sites resides in the nucleotides immediately upstream of the edited nucleotide. Table 3 compares the selectivity ratios (P/Q) for the distribution of dinucleotides in the −2/−1 position around editing sites in three mtDNAs and five cpDNAs. The analysis of C-to-U editing sites in Cycas mtDNA reveals a similar distribution in the angiosperm mtDNAs, Arabidopsis and Oryza. The dinucleotides UU, UC, CU, and AU are highly enriched at the −2/−1 position (Table 3). Dinucleotides with a purine at the −1 position were rarely observed at the −2/−1 position and exhibited very small selectivity ratios.
A scatter plot compares the selectivity ratios for dinucleotides upstream of Cycas and angiosperm mitochondrial editing sites (Fig. 2a); thus, each of the 16 points corresponds to the selectivity ratios for a specific dinucleotide. An extraordinary level of congruence exists between the selectivity ratios of the dinucleotides in the −2/−1 position of the Cycas mtDNA and the angiosperm mtDNAs. Linear regression analysis of these data indicates slope values near 1, Y-intercepts near zero, and coefficients of determination (R 2) greater than 0.9 (Fig. 2a).

Selectivity ratios around C-to-U editing sites are similar in chloroplasts and mitochondria in non-flowering and flowering plants. The selectivity ratios (P/Q) for dinucleotides in the −2/−1 window (a, b) are compared in a scatter plot. Each point represents the selectivity ratios for a specific dinucleotide in the two species. a Compares the selectivity ratios for C-to-U editing sites in Cycas and angiosperm mitochondria genomes in the −2/−1 window. Linear regression analysis for the Cycas selectivity ratios plotted against the Arabidopsis, Oryza, and Beta selectivity ratios indicates a strong congruence with slopes near 1, y-intercepts near zero, and large coefficients of determination (Arabidopsis: slope = 0.88, intercept = 0.06, R 2 = 0.91; Beta: slope = 0.96, intercept = 0.00, R 2 = 0.95; Oryza: slope = 0.0.97, intercept = 0.02, R 2 = 0.94). b Compares the selectivity ratios of C-to-U editing sites in Takakia chloroplasts with plant mitochondrial genomes in the −2/−1 window. Linear regression analysis Takakia selectivity ratios plot against the Cycas, Arabidopsis, Oryza, and Beta selectivity ratios indicates a strong congruence in the selectivity ratios (Cycas: slope = 1.14, intercept = −0.07, R 2 = 0.89; Arabidopsis: slope = 1.03, intercept = −0.03, R 2 = 0.85; Beta: slope = 1.12, intercept = −0.09, R 2 = 0.91; Oryza: slope = 1.20, intercept = −0.02, R 2 = 0.97)
The C-to-U editing sites in the cpDNAs of non-flowering plants show a similar trend in the pyrimidine-rich dinucleotides upstream of editing sites (Table 3). The selectivity ratios upstream of C-to-U editing sites in the cpDNAs of the hornwort (Anthoceros), the moss (Takakia), and the fern (Adiantum) are very similar to selectivity ratios observed in angiosperm mitochondria. Figure 2b compares the selectivity ratios of Takakia cpDNA editing sites with plant mtDNA editing sites, and the high degree of similarity is indicated by a coefficient of determination (R 2) greater than 0.85. Therefore, the distribution of nucleotides around C-to-U editing sites in non-flowering plant cpDNAs is very similar to those in plant mtDNAs, which strengthens a common origin and early evolution of RNA editing in the two organelle systems.
Discussion
C-to-U editing sites from diverse plant sources contain similar information profiles
Informatics analyses demonstrate strong similarities in the C-to-U editing sites across diverse taxa and organelle systems. The information profiles around C-to-U editing sites generally exhibit high relative entropies in the −1/−2 regions and smaller peaks in the 5′ flanking 20 nucleotides. In contrast, there is generally little information in the 3′ flanking nucleotides. Furthermore, RNA sequence context at C-to-U editing sites is very similar across these diverse taxa and both organelle systems. In plant mitochondria and in lower plant chloroplasts, pyrimidine-rich dinucleotides are highly enriched upstream of C-to-U editing sites, and a very low frequency of purines exists at −1. Thus, there is a strong selection of nucleotides immediately adjacent to C-to-U editing sites across eight organelle genomes including taxa that diverged at least 400 million years ago (mya) (Palmer et al. 2004). The conserved information profile and nucleotide context around C-to-U editing sites may result from constraints related to the editing mechanism. The peaks in the information profile suggest that similar positions are utilized in editing site recognition. These results further substantiate the model that common editing site features may exist immediately adjacent to C-to-U editing sites, and that cis-elements for individual editing sites reside in the 5′ flanking region.
The information profile around C-to-U editing sites usually exhibits small peaks in the 5′ flanking region. In contrast to the strong sequence conservation at −1/−2 positions of editing sites, relatively little sequence similarity is observed in the scatter plots in these upstream regions (data not shown). Molecular analyses of the sequences required for editing site conversion in angiosperm chloroplast and mitochondrial systems have demonstrated that the cis-element includes approximately 20 nucleotides of upstream sequence and relatively little downstream sequence (Shikanai 2006; van der Merwe et al. 2006; Hayes and Hanson 2008). Clusters of editing sites have been proposed for the recognition of editing sites in higher plant chloroplasts (Chateigner-Boutin and Hanson 2002), and some editing sites can be grouped into clusters that exhibit sequence similarity. In some cases, PPR genes are required for processing two or more editing sites, and the cis-elements share limited sequence similarity (Chateigner-Boutin 2008; Okuda et al. 2009, 2010; Zehrmann et al. 2009). The analysis of the cis-elements for 34 editing sites and 15 PPR proteins in Arabidopsis required for RNA editing indicated that the cis-elements for editing sites were not strikingly similar (Hammani et al. 2009).
Conclusions
Informatics analyses demonstrate that C-to-U editing sites share common features across diverse taxa and organelle systems. The information profiles around editing sites in these diverse systems show similar patterns. Furthermore, the nucleotides at −1/−2 show remarkable similarity across diverse taxa and different organelles systems. The conserved information profiles and nucleotide context around C-to-U editing sites across these broad taxa may be a constraint of common features of the editing mechanism.
References
Chateigner-Boutin AL, Hanson MR (2002) Cross-competition in transgenic chloroplasts expressing single editing sites reveals shared cis elements. Mol Cell Biol 22:8448–8456
Chateigner-Boutin AL, Ramos-Vega M, Guevara-Garcia A, Andres C, de la Luz Gutierrez-Nava M, Cantero A, Delannoy E, Jimenez LF, Lurin C, Small I, Leon P (2008) CLB19, a pentatricopeptide repeat protein required for editing of rpoA and clpP chloroplast transcripts. Plant J 56:590–602
Chaw SM, Shih AC, Wang D, Wu YW, Liu SM, Chou TY (2008) The mitochondrial genome of the gymnosperm Cycas taitungensis contains a novel family of short interspersed elements, Bpu sequences, and abundant RNA editing sites. Mol Biol Evol 25:603–615
Cummings MP, Myers DS (2004) Simple statistical models predict C-to-U edited sites in plant mitochondrial RNA. BMC Bioinform 5:132
Giege P, Brennicke A (1999) RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc Natl Acad Sci USA 96:15324–15329
Hammani K, Okuda K, Tanz SK, Chateigner-Boutin AL, Shikanai T, Small I (2009) A study of new Arabidopsis chloroplast RNA editing mutants reveals general features of editing factors and their target sites. Plant Cell 21:3686–3699
Handa H (2003) The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res 31:5907–5916
Hayes ML, Hanson MR (2008) High conservation of a 5′ element required for RNA editing of a C target in chloroplast psbE transcripts. J Mol Evol 67:233–245
Jobson RW, Qiu YL (2008) Did RNA editing in plant organellar genomes originate under natural selection or through genetic drift? Biol Direct 3:43
Kobayashi Y, Matsuo M, Sakamoto K, Wakasugi T, Yamada K, Obokata J (2008) Two RNA editing sites with cis-acting elements of moderate sequence identity are recognized by an identical site-recognition protein in tobacco chloroplasts. Nucleic Acids Res 36:311–318
Kubo T, Nishizawa S, Sugawara A, Itchoda N, Estiati A, Mikami T (2000) The complete nucleotide sequence of the mitochondrial genome of sugar beet (Beta vulgaris L.) reveals a novel gene for tRNA(Cys)(GCA). Nucleic Acids Res 28:2571–2576
Kugita M, Kaneko A, Yamamoto Y, Takeya Y, Matsumoto T, Yoshinaga K (2003a) The complete nucleotide sequence of the hornwort (Anthoceros formosae) chloroplast genome: insight into the earliest land plants. Nucleic Acids Res 31:716–721
Kugita M, Yamamoto Y, Fujikawa T, Matsumoto T, Yoshinaga K (2003b) RNA editing in hornwort chloroplasts makes more than half the genes functional. Nucleic Acids Res 31:2417–2423
Maier RM, Neckermann K, Igloi GL, Kossel H (1995) Complete sequence of the maize chloroplast genome: gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J Mol Biol 251:614–628
Mower JP (2005) PREP-Mt: predictive RNA editor for plant mitochondrial genes. BMC Bioinform 6:96
Mower JP, Palmer JD (2006) Patterns of partial RNA editing in mitochondrial genes of Beta vulgaris. Mol Genet Genom 276:285–293
Mulligan RM, Chang KL, Chou CC (2007) Computational analysis of RNA editing sites in plant mitochondrial genomes reveals similar information content and a sporadic distribution of editing sites. Mol Biol Evol 24:1971–1981
Notsu Y, Masood S, Nishikawa T, Kubo N, Akiduki G, Nakazono M, Hirai A, Kadowaki K (2002) The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol Genet Genom 268:434–445
Oda K, Yamato K, Ohta E, Nakamura Y, Takemura M, Nozato N, Akashi K, Kanegae T, Ogura Y, Kohchi T et al (1992) Gene organization deduced from the complete sequence of liverwort Marchantia polymorpha mitochondrial DNA. A primitive form of plant mitochondrial genome. J Mol Biol 223:1–7
Okuda K, Nakamura T, Sugita M, Shimizu T, Shikanai T (2006) A pentatricopeptide repeat protein is a site recognition factor in chloroplast RNA editing. J Biol Chem 281:37661–37667
Okuda K, Chateigner-Boutin AL, Nakamura T, Delannoy E, Sugita M, Myouga F, Motohashi R, Shinozaki K, Small I, Shikanai T (2009) Pentatricopeptide repeat proteins with the DYW motif have distinct molecular functions in RNA editing and RNA cleavage in Arabidopsis chloroplasts. Plant Cell 21:146–156
Okuda K, Hammani K, Tanz SK, Peng L, Fukao Y, Myouga F, Motohashi R, Shinozaki K, Small I, Shikanai T (2010) The pentatricopeptide repeat protein OTP82 is required for RNA editing of plastid ndhB and ndhG transcripts. Plant J 61:339–349
Palmer JD, Soltis DE, Chase MW (2004) The plant tree of life: an overview and some points of view. Am J Bot 91:1437–1445
Shikanai T (2006) RNA editing in plant organelles: machinery, physiological function and evolution. Cell Mol Life Sci 63:698–708
Shinozaki K, Ohme M, Tanaka M, Wakasugi T, Hayashida N, Matsubayashi T, Zaita N, Chunwongse J, Obokata J, Yamaguchi-Shinozaki K, Ohto C, Torazawa K, Meng BY, Sugita M, Deno H, Kamogashira T, Yamada K, Kusuda J, Takaiwa F, Kato A, Tohdoh N, Shimada H, Sugiura M (1986) The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J 5:2043–2049
Takenaka M, Neuwirt J, Brennicke A (2004) Complex cis-elements determine an RNA editing site in pea mitochondria. Nucleic Acids Res 32:4137–4144
Tillich M, Lehwark P, Morton BR, Maier UG (2006) The evolution of chloroplast RNA editing. Mol Biol Evol 23:1912–1921
Tsudzuki T, Wakasugi T, Sugiura M (2001) Comparative analysis of RNA editing sites in higher plant chloroplasts. J Mol Evol 53:327–332
van der Merwe JA, Takenaka M, Neuwirt J, Verbitskiy D, Brennicke A (2006) RNA editing sites in plant mitochondria can share cis-elements. FEBS Lett 580:268–272
Wakasugi T, Tsudzuki T, Sugiura M (2001) The genomics of land plant chloroplasts: gene content and alteration of genomic information by RNA editing. Photosynth Res 70:107–118
Wolf PG, Rowe CA, Sinclair RB, Hasebe M (2003) Complete nucleotide sequence of the chloroplast genome from a leptosporangiate fern, Adiantum capillus-veneris L. DNA Res 10:59–65
Wolf PG, Rowe CA, Hasebe M (2004) High levels of RNA editing in a vascular plant chloroplast genome: analysis of transcripts from the fern Adiantum capillus-veneris. Gene 339:89–97
Yura K, Miyata Y, Arikawa T, Higuchi M, Sugita M (2008) Characteristics and prediction of RNA editing sites in transcripts of the moss Takakia lepidozioides chloroplast. DNA Res 15:309–321
Zehrmann A, Verbitskiy D, van der Merwe JA, Brennicke A, Takenaka M (2009) A DYW domain-containing pentatricopeptide repeat protein is required for RNA editing at multiple sites in mitochondria of Arabidopsis thaliana. Plant Cell 21:558–567
Acknowledgments
This work was supported by a National Science Council grant to S.-M.C. (97-2621-B001-003-MY3), and a National Science Foundation grant to R.M.M. (MCB-0929423).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R. Bock.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Salmans, M.L., Chaw, SM., Lin, CP. et al. Editing site analysis in a gymnosperm mitochondrial genome reveals similarities with angiosperm mitochondrial genomes. Curr Genet 56, 439–446 (2010). https://doi.org/10.1007/s00294-010-0312-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-010-0312-4