
Abstract
Burkholderia glumae is a Gram-negative phytopathogenic bacterium known as the causative agent of rice panicle blight. Strain B. glumae PG1 is used for the production of a biotechnologically relevant lipase, which is secreted into the culture supernatant via a type II secretion pathway. We have comparatively analyzed the genome sequences of B. glumae PG1 wild type and a lipase overproducing strain obtained by classical strain mutagenesis. Among a total number of 72 single nucleotide polymorphisms (SNPs) identified in the genome of the production strain, two were localized in front of the lipAB operon and were analyzed in detail. Both mutations contribute to a 100-fold overproduction of extracellular lipase in B. glumae PG1 by affecting transcription of the lipAB operon and efficiency of lipase secretion. We analyzed each of the two SNPs separately and observed a stronger influence of the promoter mutation than of the signal peptide modification but also a cumulative effect of both mutations. Furthermore, fusion of the mutated LipA signal peptide resulted in a 2-fold increase in secretion of the heterologous reporter alkaline phosphatase from Escherichia coli.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Burkholderia glumae (formerly known as Pseudomonas glumae) belongs to the genus Burkholderia within the subphylum of the β-proteobacteria (Yabuuchi et al. 1992). B. glumae is a moderate rice pathogen (Ham et al. 2011), which also affects several other plants (Jeong et al. 2003). Until now, just a single case of B. glumae isolated from an immunodeficient patient was reported (Weinberg et al. 2007). All B. glumae strains studied so far infect rice panicles and produce a phytotoxin called toxoflavin (Jung et al. 2011; Kim et al. 2004; Suzuki et al. 2004; Vial et al. 2007), whose production is regulated by a LuxR-LuxI-type quorum sensing (QS) system (Chun et al. 2009; Chung et al. 2011; Goo et al. 2010; Kang et al. 2008; Kim et al. 2012; Kim et al. 2007). Biotechnological applications of Burkholderia species mainly comprise their use as biofertilizers or bioremediation agents (Chiarini et al. 2006; Paganin et al. 2011; Suarez-Moreno et al. 2012). Furthermore, the production of an extracellular lipase (Boekema et al. 2007; Santambrogio et al. 2013) and of rhamnolipid biosurfactants (Costa et al. 2011) were described, but detailed studies regarding further biotechnological applications are missing.
Lipases represent the third largest group within the worldwide enzyme market, which is estimated to increase with a 6.3 % rate per year to reach US$6.9 billion in 2017 (Casas-Godoy et al. 2012; Freedonia 2014; Hasan et al. 2006). The production of biotechnologically relevant lipases is a well-known feature of bacteria belonging to the genera Pseudomonas and Burkholderia. These and other microbial lipases (triacylglycerol hydrolases, EC 3.1.1.3) belong to the family of α/β hydrolases and catalyze the hydrolysis of triglycerides to glycerol and fatty acids. They are the most frequently used biocatalysts in organic chemistry (Jaeger et al. 1999; Sharma and Kanwar 2014) as they are readily available at low production costs, do not require cofactors, and usually show a broad substrate specificity and high enantioselectivity as well as high stability in non-aqueous media such as ionic liquids, supercritical fluids, and organic solvents. Under non-aqueous reaction conditions, lipases can catalyze the synthesis of various esters by esterification, interesterification, and transesterification (Aravindan et al. 2007; Gandhi et al. 2000; Gupta et al. 2004; Jaeger et al. 1999; Jaeger and Eggert 2002; Jaeger et al. 1994; Jaeger and Reetz 1998; Krishna and Karanth 2002; Nagarajan 2012; Yahya et al. 1998). Additional fields of lipase application include the production of food and feed ingredients as well as intermediates for pharmaceuticals (Casas-Godoy et al. 2012; Jaeger and Eggert 2002) and, more recently, also the production of biodiesel (Narwal and Gupta 2013; Santambrogio et al. 2013). Research over the last decades focused on the development of new methods to improve enzymes by directed evolution, rational design and computational methods (Bornscheuer et al. 2012; Drepper et al. 2006). However, efficient expression and preferably also secretion of lipases are still problematic, and many biotechnologically interesting lipases, e.g., those produced by Pseudozyma aphidis (formerly Candida antarctica) or various Pseudomonas species, can be produced but not efficiently secreted in Escherichia coli thus requiring optimization of homologous expression strains (Liu et al. 2006; Omori et al. 2005). Efficient secretion of enzymes into the culture medium is favored for most applications because it facilitates down-stream processing and lowers costs. P. aeruginosa is a well-studied Gram-negative bacterium for which a wealth of molecular biological and biochemical methods are available (Filloux and Ramos (Eds.) 2014), and it also produces and secretes biotechnologically relevant compounds including lipases and rhamnolipid biosurfactants (Dusane et al. 2010; Rosenau and Jaeger 2000). However, as an opportunistic human pathogen, P. aeruginosa will not be used for the majority of industrial applications. The company BASF SE discovered that a lipase similar to the one produced by P. aeruginosa is secreted by B. glumae PG1 and can be used to produce enantiopure alcohols and amines as intermediates in the synthesis of pharmaceuticals (Balkenhohl et al. 1997; Boekema et al. 2007). Classical mutagenesis methods were applied to construct the lipase overproducing strain B. glumae LU8093 derived from the PG1 wild type (R. Braatz, R. Kurth, E. Menkerl-Conen, H. Rettenmaier, T. Friedrich, and T. Subkowski, 1992, patent application WO93/00924 A1 and (Boekema et al. 2007)). Further mutagenesis resulted in strain B. glumae LU2023, which showed improved activity of another biotechnological relevant enzyme, a butyneol I esterase (T. Friedrich, B. Hauer, C. Nuebling, R. Stuermer, 2001, patent application WO2002018560 A2).
The extracellular lipase LipA is encoded in an operon together with a second gene lipB (or lif) encoding a lipase-specific foldase (Frenken et al. 1993a; Frenken et al. 1993b; Frenken et al. 1992). The N-terminal signal peptide of LipA mediates its transport through the inner membrane via the Sec secretion system (Frenken et al. 1992). In the periplasm, the steric chaperone LipB interacts with the lipase (Frenken et al. 1993b; Rosenau and Jaeger 2000) resulting in the conversion of the enzymatically inactive so-called “near-native” state into an active conformation (El Khattabi et al. 2000; Pauwels et al. 2012). Secretion through the outer membrane is subsequently achieved via the type II secretion system formed by the so-called “secreton” (or “main terminal branch” of the general secretory pathway) (Filloux 2004).
In this study, two mutations in the lipase overproducing strain B. glumae LU8093, one inside and one in front of the lipase operon lipAB, were studied in detail to unravel their contribution to lipase overproduction. Furthermore, we demonstrated that increased secretion by the modified LipA signal peptide can be transferred to the secretion of the reporter enzyme PhoA.
Material and methods
Bacterial strains and growth conditions
E. coli strains DH5α (Grant et al. 1990) and S17-1 (Simon et al. 1983) were cultivated in LB medium (Carl Roth, Karlsruhe, Germany) at 37 °C. B. glumae LU8093 ((Balkenhohl et al. 1997) and R. Braatz, R. Kurth, E. Menkerl-Conen, H. Rettenmaier, T. Friedrich, and T. Subkowski, 1992, patent application WO 93/00924 A1), B. glumae PG1 wild type ((Frenken et al. 1992), and its lipAB deficient derivate B. glumae PG1ΔlipAB (Knorr 2010) were cultivated in LB medium at 30 °C. For analysis of lipase activities and transcript-level determination, B. glumae strains were cultivated for 14 h at 150 rpm. Standard cloning experiments were performed in E. coli DH5α. Plasmids were stabilized by using appropriate concentrations of chloramphenicol (50 μg/ml for E. coli and 200 μg/ml for B. glumae). Expression of the lipAB operon from plasmid pBBR-lipAB harboring its natural promoter was defined as native expression level. The wild type strain B. glumae PG1 is deposited as strain no. CBS 322.89 at the Centraalbureau voor Schimmelcultures, P.O.Box 85167, NL-3508 AD Utrecht, The Netherlands, and the closed genome sequence of B. glumae PG1 is deposited at the DDBJ/EMBL/GenBank under the accession CP002580 (chromosome 1) and CP002581 (chromosome 2) (Voget et al. 2015).
Genome sequencing and SNP analysis of B. glumae LU8093
Genomic DNA of B. glumae LU8093 was isolated with the Masterpure DNA purification Kit (Epicentre, Madison, USA). Genome sequencing was carried out with a hybrid approach using the 454 GS-FLX system with Titanium chemistry (Roche Life Science, Mannheim, Germany) and the Genome Analyzer IIx (Illumina, San Diego, CA). Sequencing results in 437,363 and 3,998,786 reads, respectively. In order to identify SNPs, sequence reads of LU8093 were mapped onto the B. glumae PG1 reference genome (Voget et al. 2015) with the GS Reference Mapper (Roche Life Science, Mannheim, Germany). All candidate SNP positions were then manually verified by PCR-amplifying corresponding genome regions and re-sequencing these fragments. Manual editing steps were performed using the GAP4 software package v4.6 (Staden 1996).
Recombinant DNA techniques
Standard DNA techniques were performed as described (Sambrook et al. 1989). PCR Extender System (5 Prime, Hilden, Germany) was used for amplification of DNA fragments. Other DNA-modifying enzymes were obtained from Thermo Scientific (St. Leon-Rot, Germany) using the manufacturer’s instructions. Plasmid isolation from E. coli DH5α was performed with innuPREP Plasmid Mini Kit (Analytic Jena, Jena, Germany). Genomic DNA from B. glumae PG1 (wild type) and B. glumae LU8093 was isolated using DNeasy® Blood & Tissue Kit (Qiagen, Hilden, Germany).
The lipAB wild type operon (GenBank accession number: AJK49931.1 and AJK49932.1) and the lipAB operon that harbors the mutations in the promoter region and the region coding for the LipA signal peptide were amplified using the isolated genomic DNAs from both strains as template and the primer pair “PG1 lipAB up/dn” (5′-ATA TAT A TC TAG A AT TCA CCG GAT CGA TCG-3′/5′-ATA TAT AAG CTT ACC CGT TCG AAG CAC T-3′). The PCR products include 249 bp upstream of the lipA startcodon with the predicted promoter sequence. The resulting DNA fragments harboring primer introduced restriction sites were hydrolyzed with XbaI and HindIII, and the resulting 2444-bp fragments were ligated into XbaI-HindIII-treated plasmid pBBR1-MCS (Kovach et al. 1994). The resulting plasmids were named pBBR-lipAB and pBBR-lipAB-3, respectively. Plasmid pBBR-lipAB was used as template for overlap-extension PCRs (Higuchi et al. 1988) to introduce single mutations. For the mutation in the promoter region, the primer pair “OLE PCR 1/2” (5′-CCT GTC TAC AAT CAG ACG GCC G-3′/5′-CGG CCG TCT GAT TGT AGA CAG G-3′) was used whereas the pair “OLE PCR 3/4” (5′-GGA ACG CAT CAA TCT GAC CAT G-3′/5′-CAT GGT CAG ATT GAT GCG TTC C-3′) was used for the mutation in the region coding for the signal peptide. The primer pair “PG1 lipAB up/dn” was used as flanking primers, and the resulting 2463 bp amplicon was then treated as described above. The resulting plasmids were named pBBR-lipAB-1 (mutation in the promoter region) and pBBR-lipAB-2 (mutation in the signal sequence).
Transformation and conjugation
E. coli strains were transformed with plasmid DNA by heat shock transformation (Hanahan 1983). B. glumae strains were transformed by biparental mating with E. coli S17-1 as follows: For conjugation, 1 ml overnight culture of B. glumae was mixed with 2 ml of E. coli S17-1 in the exponential growth phase (O.D.580nm = 0.6–0.8) containing the plasmid of interest. After centrifugation (1 min, 21,000×g), the cell pellet was washed with 0.5 ml LB medium, resuspended in 50 μl LB medium and dropped onto a membrane filter (M24, Whatman) placed on an LB agar-plate. Cells were washed off from the filter with LB medium after 6 h at 30 °C, and the cell suspension was plated in appropriate dilutions on MME (Vogel and Bonner 1956) agar plates containing antibiotics and 0.5 % (w/v) glucose.
Western blot analysis
Proteins from cell-free supernatants were precipitated with sodium deoxycholate and trichloroacetic acid (TCA) as described (Peterson 1977). After washing with 1/2 volume 80 % (v/v) acetone, the pellet was suspended with 2× SDS-sample puffer (50 mM Tris-HCl, 4 % (w/v) SDS, 10 % (v/v) glycerol, 10 % (v/v) 2-mercaptoethanol, 0.03 % (w/v) bromophenol blue). Proteins were separated by SDS-PAGE with a 12 % polyacrylamide gel (Laemmli 1970). Western blot analysis of LipA and LipB was performed using specific antibodies (kindly provided by Jan Tommassen, University of Utrecht, The Netherlands). A goat-anti-rabbit IgG (H + L)-HRP conjugate (BioRad, Munich, Germany) was used as secondary antibody. Specific antibody-protein interactions were detected using the ECL Western Blotting Detection system (Amersham Pharmacia, Buckinghamshire, GB) and the luminescence detector Stella (raytest, Straubenhardt, Germany).
Lipase assay
Lipase activity in whole cell extracts and supernatants was measured with para-nitrophenyl palmitate (p-NPP) as the substrate (Winkler and Stuckmann 1979) at 410 nm in microtiter plates using a SpectraMax 250 photometer (Molecular Devices, Ismaning/München, Germany). Relative lipase activity was correlated to cell density (O.D.580nm) and calculated as U/ml, with 1 U (unit) defined as the amount of lipase that releases 1 mmol of para-nitrophenol per minute (molar absorption coefficient 15 μMol−1 × cm−1).
Transcript level determination
Two milliliters of culture were centrifuged (1 min, 21,000×g) and washed once with TE buffer (100 mM Tris-HCl pH 7.5, 20 mM EDTA). The cell pellet was then treated with RNeasy Mini Kit (Qiagen) according to the protocol for the isolation of bacterial RNA. DNaseI digestion was performed both, “on column” with RNase-free DNase Set (Qiagen) and after RNA elution with DNaseI (RNase-free) from Ambion® (Life Technologies, Darmstadt, Germany) according to the manufacturer’s instructions. The reverse transcription of isolated RNA into cDNA was carried out with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems™, Foster City, USA) according to the instruction manual. For subsequent real time qPCRs, 250 ng RNA were transcribed per reaction. In a separate reaction, each sample was also treated without reverse transcription to exclude DNA contaminations. The analysis of transcriptional levels of lipA and lipB was performed with real time qPCR (35 cycles) using the ΔΔCT-method (Livak and Schmittgen 2001; Schmittgen and Livak 2008). Here, the cDNA was used as template in a real time 7900HT Fast Real-Time PCR System with Power SYBR® Green PCR Master Mix (both Applied Biosystems™), and specific primers for lipA (5′-CTA TCC GGT GAT CCT CGT C-3′/5′-GAG AGA TTC GCG ACG TAC AC-3′), lipB (5′-GTG GCA GAC GCG CTA TCA AG-3′/5′-CGT GAA AGT CTG CTG CCT GAG-3′) and the constitutively expressed gene rpoD (5′-GAT GAC GAC GCA ACC CAG AG-3′/5′-GAA CGC TTC CTT CAG CAG CA-3′) as a reference. Primers were designed using Primer3 (Untergasser et al. 2012). The amount of PCR product was calculated as CT value by the Sequence Detection System (Version 2.3, Applied Biosystems™). PCR efficiencies were determined with the tool LinRegPCR (Ruijter et al. 2009). The CT values obtained for lipA and lipB were then related to those of the reference gene rpoD leading to the ΔCT value (ΔCT = CT(gene) − CT(rpoD)). By comparing the ΔCT values of a certain strain to its reference strain, the resulting ΔΔCT (ΔΔCT = ΔCT(strain) − ΔCT(reference strain)) value reflects the differences in the transcript amount of a certain gene between these two strains. Calculations were performed and statistically analyzed with REST© software (Pfaffl et al. 2002). All observed transcript exchanges are significantly different from the control sample (p < 0.05, calculated with REST©).
Results
Comparison of B. glumae wild type and the lipase production strain LU8093
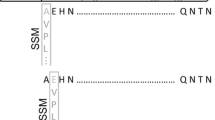
The extracellular lipase LipA produced by B. glumae PG1 is used by BASF SE for the production of enantiopure building blocks (Liese et al. 2006). Therefore, the lipase production strain B. glumae LU8093 was constructed from B. glumae PG1 by repeated rounds of random mutagenesis and subsequent assays for increased extracellular lipase production R. Braatz, R. Kurth, E. Menkerl-Conen, H. Rettenmaier, T. Friedrich, and T. Subkowski, 1992, patent application WO 93/00924 A1). We compared the genome sequences of B. glumae PG1 (Voget et al. 2015) and B. glumae LU8093 and identified in the production strain 72 SNPs of which 51 were located on chromosome 1, with 29 non-synonymous, 16 synonymous and six intergenic ones. From 21 SNPs found on chromosome 2, 13 were non-synonymous, five synonymous and three intergenic. Among the 72 SNPs identified in the B. glumae LU8093 chromosomes, two were localized within the lipase operon on chromosome 2; one in the putative promoter region and the second in the region encoding the LipA signal peptide (Fig. 1).

Two mutations were identified by comparative genome sequencing and localized to the lipAB operon of the production strain B. glumae LU8093. The first mutation is located in the lipAB promoter region (P lipAB ) and is present in the constructed variant lipAB-1; the second mutation located in the LipA signal peptide coding sequence is present in the constructed variant lipAB-2; variant lipAB-3 contains both mutations. Two putative binding sites for δ54 transcription factors and the transcription start (+1) are underlined in the DNA sequence shown below (Beselin 2005). Coding triplets no. 1–7 of lipA are translated into amino acid sequence, and mutations identified in B. glumae LU8093 are marked with asterisks. The amino acid exchange resulting from mutation lipAB-2 is indicated
We first determined lipase activity and protein amount in cell extracts and culture supernatants obtained from B. glumae PG1 wild type and the production strain LU8093 demonstrating that the production strain produced more lipase than the wild type (Fig. 2a). Previous studies using fusions of the lipase promoter with GFP indicated an increased transcription rate of lipA in the production strain B. glumae LU8093 (Boekema et al. 2007). Thus, we quantified the transcription levels of lipA by qPCR and determined a 100-fold increase in the production strain (Fig. 2b).

Lipase production of B. glumae PG1 wild type (PG1) and production strain B. glumae LU8093. a Relative lipase activity in the supernatant (SN) and cell extract (CE). LipA was detected in culture supernatants (SN LipA) and LipB in cell extract (CE LipB) by Western blotting after SDS-PAGE. Samples of 10 μl were loaded into each lane corresponding to a cell density of O.D.580nm = 5 for cell extracts and O.D.580nm = 50 for supernatants. b Relative change of lipA and lipB transcript levels in B. glumae LU8093 compared to the wild type B. glumae PG1 (arbitrarily set as 1). Error bars show standard deviations derived from examination of three biological replicates. All changes in transcript level are significant (see “Material and methods” section)
Two mutations localized within the lipase operon lipAB increase lipase production and secretion
The mutations identified in front of the lipAB operon were analyzed both separately and in combination by expression of the respective genes in a lipAB-deficient B. glumae PG1 strain (PG1ΔlipAB) to avoid basal expression of genome-encoded lipAB. To ensure that extracellular lipase activities were not caused by cell lysis, we determined cytoplasmic β-lactamase activities in cell-free culture supernatants. These activities were always less than 10 % of the overall activities for all strains tested (Fig. S1) indicating that the observed effects of the mutations on extracellular lipase levels were not caused by significant cell lysis. As shown in Fig. 3a, the mutation in the promoter region of lipAB (lipAB-1) resulted in a 38-fold increased lipase activity in the supernatant (~2.68 compared to ~0.07 U/ml) and 42-fold in the cell extract (~0.168 compared to ~0.004 U/ml). The mutation in the signal peptide (lipAB-2) led to a slight (~4–7-fold) increase of lipase activity in the supernatant and the cell extract, whereas the combination of both mutations (lipAB-3) resulted in ~100-fold increased activity in the supernatant (~6.87 U/ml) and ~140-fold increased activity (~0.57 U/ml) in the whole cell extract. It should be noted here that lower lipase activities of B. glumae PG1 wild type and B. glumae LU8093 as shown in Fig. 2a can be attributed to the fact that these strains harbor just one chromosomal copy of the lipAB operon. The increased lipolytic activity of B. glumae PG1ΔlipAB expressing plasmid-encoded lipase variants corresponded to increased production and secretion as determined by Western blot analysis of LipA in cell-free supernatants (Fig. 3a, bottom). Remarkably, a significantly increased amount of LipB was detected only in the strains harboring the promoter mutation (lipAB-1 and lipAB-3).

Expression of different lipase operons in B. glumae PG1ΔlipAB. a Relative lipase activity in cell-free supernatants (SN) and cell extracts (CE). LipA in supernatants (SN LipA) and LipB in cell extracts (CE LipB) were detected by Western blotting after SDS-PAGE with each lane containing 10 μl sample corresponding to a cell density of O.D.580nm = 5 for cell extracts and O.D.580nm = 50 for supernatants. b Relative change of lipA transcript levels in strains harboring a mutated lipAB operon (lipAB-1 to −3) compared to the wild type operon lipAB (arbitrarily set as 1). Error bars show standard deviations derived from examination of three biological replicates. All changes in transcript level are significant (see “Material and methods” section)
A mutation in the lipAB promoter increases the transcript level of lipA
Next, we analyzed the influence of these mutations on the lipA transcript level by qPCR (see Fig. 3b). Whereas the mutation in the signal sequence (lipAB-2) exhibited just a slight effect at the transcript level, the promoter mutation (lipAB-1 and lipAB-3) led to an increase by a factor of 16. Interestingly, we observed that the transcript levels of lipA and lipB were differentially affected by the two mutations. While the lipA transcript level was increased by the promoter mutation, the amount of lipB transcript remained unaffected (data not shown). This may be explained by a faster degradation of the lipB transcript as already suggested by Frenken et al. (Frenken et al. 1993a). This assumption is further supported by the observation that more LipB was detected by Western blot analysis in strains harboring a lipase operon with the promoter mutation than in strains with the wild type operon (see Fig. 3a, bottom). Apparently, more lipB transcript could be produced and translated, but may be degraded faster than lipA transcript.
A signal peptide mutation in LipA improves secretion in B. glumae PG1
The second mutation identified in the lipAB operon results in an exchange of serine to leucine at position 4 of the LipA signal peptide. This mutation has almost no effect on lipA transcription rate, but caused a remarkable increase of extracellular lipase amount (see lipAB-2 in Fig. 3). The replacement of a polar serine by a hydrophobic leucine residue increases the hydrophobicity of the LipA signal peptide and may thus facilitate its interaction with the Sec-machinery thereby accelerating transport of LipA through the bacterial inner membrane (Driessen and Nouwen 2008). This hypothesis was tested by construction of alkaline phosphatase PhoA fusions to wild type and mutant LipA signal peptides and determination of PhoA activities in B. glumae PG1 and PG1ΔlipAB. PhoA shows enzymatic activity only after transport across the inner membrane and is therefore used as secretion reporter (Manoil et al. 1990). The LipA signal peptide derived from B. glumae LU8093 carrying mutation S4L resulted in a 2-fold increased PhoA activity (Fig. S2).
In summary, these results indicate that the combination of both mutations in the lipAB operon (see lipAB-3 in Fig. 3) results in an increased transcription rate as well as in increased lipase secretion. In addition, we did not observe any growth defects of B. glumae PG1ΔlipAB expressing plasmid-encoded lipAB-1 or lipAB-2 compared to the wild type lipAB operon or the empty vector control, respectively. Expression of lipAB-3 led to a slightly decreased cell density in the stationary growth phase after 24 h (O.D.580nm = 1.2 compared to 1.7 for wild type lipAB), which was, however, not observed upon comparing growth of B. glumae LU8093 with the wild type strain PG1 (data not shown).
Discussion
In this study, we analyzed the increased lipase production of the industrial production strain B. glumae LU8093 (Fig. 2) which is a derivate of the wild type strain B. glumae PG1. The comparison of the genome sequences revealed 72 SNPs introduced in the production strain by classical mutagenesis methods with two of them being mainly responsible for increased lipase production and secretion (Fig. 3). One of these two mutations is located in the putative lipA promoter region (Fig. 1). A previous study determined the transcriptional start site located 78 bases upstream of the lipA start codon and the presence of two putative δ54-dependent promoters (Beselin 2005) with the first one located at a conserved distance of −24/−12 bp upstream of the transcriptional start (Barrios et al. 1999), and the second one in a distance of −63/−51 bp. This second putative promoter site fits perfectly with the δ54 consensus motif GG-N8-TTGC (Barrios et al. 1999). The promoter mutation analyzed in this study changes this motif from −TTGC to −TTGT (see Fig. 1). One would expect that this C-to-T transition decreases the lipA transcription rate, but surprisingly, it causes an increase in lipA transcript level. The reasons are presently unknown; however, Boekema et al. have demonstrated that lipase expression in B. glumae PG1 but not in LU8093 is prone to catabolite repression (Boekema et al. 2007). The promoter mutation may thus affect binding of not only δ54 but also of other, so far unknown, transcriptional regulators. A likely candidate could be the cAMP receptor protein (CRP) which was shown to repress the P. putida δ54-promoter Pu in a cAMP-dependent manner (Zhang et al. 2014). In B. glumae LU8093, CRP binding affinity to the mutated promoter may be diminished resulting in missing catabolic repression and correspondingly in an overall increased transcription rate. As lipase expression in B. glumae is also quorum sensing regulated (Devescovi et al. 2007), the promoter SNP may additionally uncouple lipase gene expression from quorum sensing regulation. The second mutation is located in the lipA sequence coding for the 32 amino acid long signal peptide of LipA and changes the polar residue serine with a hydropathy value of −0.8 to a more hydrophobic leucine with a hydropathy value of +3.8 (Kyte and Doolittle 1982) thereby increasing lipase secretion (Fig. 3). This observation agrees with prior studies that showed improved protein secretion by signal peptide modifications (Yoon et al. 2010). Additive effects of additional modifications, as for example observed for heterologous protein secretion in Lactococcus lactis (Ng and Sarkar 2013), could further increase LipA secretion in B. glumae. Notably, we could demonstrate that this same mutated signal peptide also resulted in a 2-fold increased secretion of PhoA (Fig. S2), a well-established secretion reporter protein (Manoil et al. 1990). Nevertheless, it should be noted that additional 70 SNPs were identified in the production strain B. glumae LU8093 which may also contribute to and further increase lipase production. Interestingly, none of the SNPs is located within or close to genes known to be involved in quorum sensing or lipase secretion.
The fact that B. glumae PG1 secretes a lipase of biotechnological interest which is indeed used in industrial applications (Balkenhohl et al. 1997; Liese et al. 2006) raises the question if this strain possesses additional features which could be interesting for biotechnological applications. A very recent study dealing with the capacity of Burkholderia to adapt to different environments revealed certain differences between B. glumae PG1 and other members of the plant pathogenic Burkholderia group (Seo et al. 2015). The most striking difference is the absence of the toxoflavin biosynthesis and transport gene cluster in B. glumae PG1. Toxoflavin is a phytotoxin and a major virulence factor for phytopathogenic B. glumae strains in rice (Ham et al. 2011; Jeong et al. 2003). B. glumae PG1 could serve as an alternative host for the production of biotechnological relevant compounds like rhamnolipids (Costa et al. 2011). We also identified 25 putative secondary metabolite clusters in the genome of B. glumae PG1 (see Table S1). This further underlines the biotechnological potential of B. glumae PG1 not only as a lipase producer, which was demonstrated in this study, but also as a prolific source for known and new secondary metabolites.
References
Aravindan R, Anbumathi P, Viruthagiri T (2007) Lipase applications in food industry. Indian J of Biotech 6:141–158
Balkenhohl F, Ditrich K, Hauer B, Ladner W (1997) Optically active amines via lipase-catalyzed methoxyacetylation. J Prak Chem-Chem Ztg 339:381–384. doi:10.1002/prac.19973390166
Barrios H, Valderrama B, Morett E (1999) Compilation and analysis of σ54-dependent promoter sequences. Nucleic Acids Res 27:4305–4313. doi:10.1093/nar/27.22.4305
Beselin A (2005) Optimization of lipase production in Burkholderia glumae. Ph.D. thesis, Heinrich-Heine-University Duesseldorf
Boekema BKHL, Beselin A, Breuer M, Hauer B, Koster M, Rosenau F, Jaeger K-E, Tommassen J (2007) Hexadecane and tween 80 stimulate lipase production in Burkholderia glumae by different mechanisms. Appl Environ Microb 73:3838–3844. doi:10.1128/Aem.00097-07
Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K (2012) Engineering the third wave of biocatalysis. Nature 485:185–194. doi:10.1038/Nature11117
Casas-Godoy L, Duquesne S, Bordes F, Sandoval G, Marty A (2012) Lipases: an overview. Methods Mol Biol 861:3–30. doi:10.1007/978-1-61779-600-5_1
Chiarini L, Bevivino A, Dalmastri C, Tabacchioni S, Visca P (2006) Burkholderia cepacia complex species: health hazards and biotechnological potential. Trends Microbiol 14:277–286. doi:10.1016/j.tim.2006.04.006
Chun H, Choi O, Goo E, Kim N, Kim H, Kang Y, Kim J, Moon JS, Hwang I (2009) The quorum sensing-dependent gene katG of Burkholderia glumae is important for protection from visible light. J Bacteriol 191:4152–4157. doi:10.1128/Jb.00227-09
Chung J, Goo E, Yu S, Choi O, Lee J, Kim J, Kim H, Igarashi J, Suga H, Moon JS, Hwang I, Rhee S (2011) Small-molecule inhibitor binding to an N-acyl-homoserine lactone synthase. Proc Natl Acad Sci U S A 108:12089–12094. doi:10.1073/pnas.1103165108
Costa SGVAO, Deziel E, Lepine F (2011) Characterization of rhamnolipid production by Burkholderia glumae. Lett Appl Microbiol 53:620–627. doi:10.1111/j.1472-765X.2011.03154.x
Devescovi G, Bigirimana J, Degrassi G, Cabrio L, LiPuma JJ, Kim J, Hwang I, Venturi V (2007) Involvement of a quorum-sensing-regulated lipase secreted by a clinical isolate of Burkholderia glumae in severe disease symptoms in rice. Appl Environ Microb 73:4950–4958. doi:10.1128/Aem.00105-07
Drepper T, Eggert T, Hummel W, Leggewie C, Pohl M, Rosenau F, Wilhelm S, Jaeger K-E (2006) Novel biocatalysts for white biotechnology. Biotechnol J 1:777–786. doi:10.1002/biot.200600059
Driessen AJ, Nouwen N (2008) Protein translocation across the bacterial cytoplasmic membrane. Annu Rev Biochem 77:643–667. doi:10.1146/annurev.biochem.77.061606.160747
Dusane DH, Zinjarde SS, Venugopalan VP, McLean RJ, Weber MM, Rahman PK (2010) Quorum sensing: implications on rhamnolipid biosurfactant production. Biotechnol Genet Eng Rev 27:159–184. doi:10.1080/02648725.2010.10648149
El Khattabi M, Van Gelder P, Bitter W, Tommassen J (2000) Role of the lipase-specific foldase of Burkholderia glumae as a steric chaperone. J Biol Chem 275:26885–26891. doi:10.1074/jbc.M003258200
Filloux A (2004) The underlying mechanisms of type II protein secretion. Bba-Mol Cell Res 1694:163–179. doi:10.1016/j.bbamcr.2004.05.003
Filloux A, Ramos (Eds.) JL (2014) Pseudomonas methods and protocols vol 1149. Methods Mol Biol. Springer, New York, NY
Freedonia (2014) World Enzymes Market. http://www.reportlinker.com/p0747897/World-Enzymes-Industry.html. Accessed July 2014
Frenken LGJ, Bos JW, Visser C, Muller W, Tommassen J, Verrips CT (1993a) An accessory gene, lipB, required for the production of active Pseudomonas glumae lipase. Mol Microbiol 9:579–589. doi:10.1111/j.1365-2958.1993.tb01718.x
Frenken LGJ, Degroot A, Tommassen J, Verrips CT (1993b) Role of the lipB gene-product in the folding of the secreted lipase of Pseudomonas glumae. Mol Microbiol 9:591–599. doi:10.1111/j.1365-2958.1993.tb01719.x
Frenken LGJ, Egmond MR, Batenburg AM, Bos JW, Visser C, Verrips CT (1992) Cloning of the Pseudomonas glumae lipase gene and determination of the active-site residues. Appl Environ Microb 58:3787–3791
Gandhi NN, Patil NS, Sawant SB, Joshi JB, Wangikar PP, Mukesh D (2000) Lipase-catalyzed esterification. Catal Rev 42:439–480. doi:10.1081/Cr-100101953
Goo E, Kang Y, Kim H, Hwang I (2010) Proteomic analysis of quorum sensing-dependent proteins in Burkholderia glumae. J Proteome Res 9:3184–3199. doi:10.1021/Pr100045n
Grant SGN, Jessee J, Bloom FR, Hanahan D (1990) Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc Natl Acad Sci U S A 87:4645–4649. doi:10.1073/pnas.87.12.4645
Gupta R, Gupta N, Rathi P (2004) Bacterial lipases: an overview of production, purification and biochemical properties. Appl Microbiol Biotechnol 64:763–781. doi:10.1007/s00253-004-1568-8
Ham JH, Melanson RA, Rush MC (2011) Burkholderia glumae: next major pathogen of rice? Mol Plant Pathol 12:329–339. doi:10.1111/j.1364-3703.2010.00676.x
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580. doi:10.1016/S0022-2836(83)80284-8
Hasan F, Shah AA, Hameed A (2006) Industrial applications of microbial lipases. Enzyme Microb Tech 39:235–251. doi:10.1016/j.enzmictec.2005.10.016
Higuchi R, Krummel B, Saiki RK (1988) A general-method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res 16:7351–7367. doi:10.1093/nar/16.15.7351
Jaeger K-E, Dijkstra BW, Reetz MT (1999) Bacterial biocatalysts: molecular biology, three-dimensional structures, and biotechnological applications of lipases. Annu Rev Microbiol 53:315–351. doi:10.1146/annurev.micro.53.1.315
Jaeger K-E, Eggert T (2002) Lipases for biotechnology. Curr Opin Biotechnol 13:390–397. doi:10.1016/S0958-1669(02)00341-5
Jaeger K-E, Ransac S, Dijkstra BW, Colson C, van Heuvel M, Misset O (1994) Bacterial lipases. FEMS Microbiol Rev 15:29–63. doi:10.1111/j.1574-6976.1994.tb00121.x
Jaeger K-E, Reetz MT (1998) Microbial lipases form versatile tools for biotechnology. Trends Biotechnol 16:396–403. doi:10.1016/S0167-7799(98)01195-0
Jeong Y, Kim J, Kim S, Kang Y, Nagamatsu T, Hwang I (2003) Toxoflavin produced by Burkholderia glumae causing rice grain rot is responsible for inducing bacterial wilt in many field crops. Plant Dis 87:890–895. doi:10.1094/PDIS.2003.87.8.890
Jung WS, Lee J, Kim MI, Ma J, Nagamatsu T, Goo E, Kim H, Hwang I, Han J, Rhee S (2011) Structural and functional analysis of phytotoxin toxoflavin-degrading enzyme. PLoS One 6:e22443. doi: 10.1371/journal.pone.0022443
Kang Y, Kim J, Kim S, Kim H, Lim JY, Kim M, Kwak J, Moon JS, Hwang I (2008) Proteomic analysis of the proteins regulated by HrpB from the plant pathogenic bacterium Burkholderia glumae. Proteomics 8:106–121. doi:10.1002/pmic.200700244
Kim H, Goo E, Kang Y, Kim J, Hwang I (2012) Regulation of universal stress protein genes by quorum sensing and RpoS in Burkholderia glumae. J Bacteriol 194:982–992. doi:10.1128/Jb.06396-11
Kim J, Kang Y, Choi O, Jeong Y, Jeong JE, Lim JY, Kim M, Moon JS, Suga H, Hwang I (2007) Regulation of polar flagellum genes is mediated by quorum sensing and FlhDC in Burkholderia glumae. Mol Microbiol 64:165–179. doi:10.1111/j.1365-2958.2007.05646.x
Kim J, Kim JG, Kang Y, Jang JY, Jog GJ, Lim JY, Kim S, Suga H, Nagamatsu T, Hwang I (2004) Quorum sensing and the LysR-type transcriptional activator ToxR regulate toxoflavin biosynthesis and transport in Burkholderia glumae. Mol Microbiol 54:921–934. doi:10.1111/j.1365-2958.2004.04338.x
Knorr J (2010) Physiologie eines industriellen Produktionsstammes: Proteinsekretion, Regulation und Produktion von Biotensiden in Burkholderia glumae. Ph.D. thesis, Heinrich-Heine-University Duesseldorf
Kovach ME, Phillips RW, Elzer PH, Roop 2nd RM, Peterson KM (1994) pBBR1MCS: a broad-host-range cloning vector. Biotechniques 16:800–802
Krishna SH, Karanth NG (2002) Lipases and lipase-catalyzed esterification reactions in nonaqueous media. Catal Rev 44:499–591. doi:10.1081/Cr-120015481
Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157:105–132. doi:10.1016/0022-2836(82)90515-0
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi:10.1038/227680a0
Liese A, Seelbach K, Buchholz A, Haberland J (2006) Processes. In: Liese A, Seelbach K, Wandrey C (eds) Industrial biotransformations—second, completely revised and, Extended edn. Weinheim, WILEY-VCH, pp. 273–446
Liu D, Schmid RD, Rusnak M (2006) Functional expression of Candida antarctica lipase B in the Escherichia coli cytoplasm—a screening system for a frequently used biocatalyst. Appl Microbiol Biotechnol 72:1024–1032. doi:10.1007/s00253-006-0369-7
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402–408. doi:10.1006/meth.2001.1262
Manoil C, Mekalanos JJ, Beckwith J (1990) Alkaline phosphatase fusions: sensors of subcellular location. J Bacteriol 172:515–518
Nagarajan S (2012) New tools for exploring “old friends-microbial lipases”. Appl Biochem Biotech 168:1163–1196. doi:10.1007/s12010-012-9849-7
Narwal SK, Gupta R (2013) Biodiesel production by transesterification using immobilized lipase. Biotechnol Lett 35:479–490. doi:10.1007/s10529-012-1116-z
Ng DT, Sarkar CA (2013) Engineering signal peptides for enhanced protein secretion from Lactococcus lactis. Appl Environ Microbiol 79:347–356. doi:10.1128/AEM.02667-12
Omori K, Isoyama-Tanaka J, Ihara F, Yamada Y, Nihira T (2005) Active lactonizing lipase (LipL) efficiently overproduced by Pseudomonas strains as heterologous expression hosts. J Biosci Bioeng 100:323–330. doi:10.1263/jbb.100.323
Paganin P, Tabacchioni S, Chiarini L (2011) Pathogenicity and biotechnological applications of the genus Burkholderia. Cent Eur J Biol 6:997–1005. doi:10.2478/s11535-011-0072-2
Pauwels K, Sanchez Del Pino MM, Feller G, Van Gelder P (2012) Decoding the folding of Burkholderia glumae lipase: folding intermediates en route to kinetic stability. PLoS One 7::e36999. doi:10.1371/journal.pone.0036999
Peterson GL (1977) A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem 83:346–356. doi:10.1016/0003-2697(77)90043-4
Pfaffl MW, Horgan GW, Dempfle L (2002) Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30:e36. doi: 10.1093/nar/30.9.e36
Rosenau F, Jaeger K-E (2000) Bacterial lipases from Pseudomonas: regulation of gene expression and mechanisms of secretion. Biochimie 82:1023–1032. doi:10.1016/S0300-9084(00)01182-2
Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ, Moorman AF (2009) Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 37:e45. doi: 10.1093/nar/gkp045
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, US
Santambrogio C, Sasso F, Natalello A, Brocca S, Grandori R, Doglia SM, Lotti M (2013) Effects of methanol on a methanol-tolerant bacterial lipase. Appl Microbiol Biotechnol 97:8609–8618. doi:10.1007/s00253-013-4712-5
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108. doi:10.1038/nprot.2008.73
Seo YS, Lim JY, Park J, Kim S, Lee HH, Cheong H, Kim SM, Moon JS, Hwang I (2015) Comparative genome analysis of rice-pathogenic Burkholderia provides insight into capacity to adapt to different environments and hosts. BMC Genomics 16:349. doi:10.1186/s12864-015-1558-5
Sharma S, Kanwar SS (2014) Organic solvent tolerant lipases and applications. TheScientificWorldJOURNAL 2014:625258. doi:10.1155/2014/625258
Simon R, Priefer U, Puhler A (1983) A broad host range mobilization system for in vivo genetic-engineering—transposon mutagenesis in Gram-negative bacteria. Bio-Technol 1:784–791. doi:10.1038/nbt1183-784
Staden R (1996) The Staden sequence analysis package. Mol Biotechnol 5:233–241
Suarez-Moreno ZR, Caballero-Mellado J, Coutinho BG, Mendonca-Previato L, James EK, Venturi V (2012) Common features of environmental and potentially beneficial plant-associated Burkholderia. Microb Ecol 63:249–266. doi:10.1007/s00248-011-9929-1
Suzuki F, Sawada H, Azegami K, Tsuchiya K (2004) Molecular characterization of the tox operon involved in toxoflavin biosynthesis of Burkholderia glumae. J Gen Plant Pathol 70:97–107. doi:10.1007/s10327-003-0096-1
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3-new capabilities and interfaces. Nucleic Acids Res 40:e115. doi: 10.1093/nar/gks596
Vial L, Groleau MC, Dekimpe V, Deziel E (2007) Burkholderia diversity and versatility: an inventory of the extracellular products. J Microbiol Biotechn 17:1407–1429
Vogel HJ, Bonner DM (1956) Acetylornithinase of Escherichia coli—partial purification and some properties. J Biol Chem 218:97–106
Voget S, Knapp A, Poehlein A, Vollstedt C, Streit WR, Daniel R, Jaeger K-E (2015) Complete genome sequence of the lipase producing strain Burkholderia glumae PG1. J Biotechnol 204:3–4. doi:10.1016/j.jbiotec.2015.03.022
Weinberg JB, Alexander BD, Majure JM, Williams LW, Kim JY, Vandamme P, LiPuma JJ (2007) Burkholderia glumae infection in an infant with chronic granulomatous disease. J Clin Microbiol 45:662–665. doi:10.1128/Jcm.02058-06
Winkler UK, Stuckmann M (1979) Glycogen, hyaluronate, and some other polysaccharides greatly enhance the formation of exolipase by Serratia marcescens. J Bacteriol 138:663–670
Yabuuchi E, Kosako Y, Oyaizu H, Yano I, Hotta H, Hashimoto Y, Ezaki T, Arakawa M (1992) Proposal of Burkholderia genus and transfer of 7 species of the genus Pseudomonas homology group II to the new genus, with the type species Burkholderia cepacia (Palleroni and Holmes 1981). Microbiol Immunol 36:1251–1275. doi:10.1111/j.1348-0421.1992.tb02129.x
Yahya ARM, Anderson WA, Moo-Young M (1998) Ester synthesis in lipase-catalyzed reactions. Enzyme Microb Tech 23:438–450. doi:10.1016/S0141-0229(98)00065-9
Yoon SH, Kim SK, Kim JF (2010) Secretory production of recombinant proteins in Escherichia coli. Recent Patents on Biotechnology 4:23–29. doi:10.2174/187220810790069550
Zhang YT, Jiang F, Tian ZX, Huo YX, Sun YC, Wang YP (2014) CRP-cyclic AMP dependent inhibition of the xylene-responsive σ54-promoter Pu in Escherichia coli. PLoS One 9:e86727. doi: 10.1371/journal.pone.0086727
Acknowledgments
This work was funded by the German Federal Ministry of Education and Research as project No. 0315594 entitled “Microbes for production: A genomics-based approach to engineer novel industrial production strains” (MiPro). Work in the laboratory of Karl-Erich Jaeger was funded by the Deutsche Forschungsgemeinschaft (DFG) through the Excellence Cluster EXC 1028.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by the German Federal Ministry of Education and Research (BMBF, grant no. 0315594) and the Deutsche Forschungsgemeinschaft (DFG, grant- No. EXC 1028).
Conflict of Interest
The authors declare that they have no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Andreas Knapp and Sonja Voget contributed equally to this work.
Electronic supplementary material
ESM 1
(PDF 453 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Knapp, A., Voget, S., Gao, R. et al. Mutations improving production and secretion of extracellular lipase by Burkholderia glumae PG1. Appl Microbiol Biotechnol 100, 1265–1273 (2016). https://doi.org/10.1007/s00253-015-7041-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-7041-z