
Abstract
Plant disease caused by fungal pathogens results in vast crop damage globally. Microbial communities of soil that is suppressive to fungal crop disease provide a source for the identification of novel enzymes functioning as bioshields against plant pathogens. In this study, we targeted chitin-degrading enzymes of the uncultured bacterial community through a functional metagenomics approach, using a fosmid library of a suppressive soil metagenome. We identified a novel bacterial chitinase, Chi18H8, with antifungal activity against several important crop pathogens. Sequence analyses show that the chi18H8 gene encodes a 425-amino acid protein of 46 kDa with an N-terminal signal peptide, a catalytic domain with the conserved active site F175DGIDIDWE183, and a chitinase insertion domain. Chi18H8 was expressed (pGEX-6P-3 vector) in Escherichia coli and purified. Enzyme characterization shows that Chi18H8 has a prevalent chitobiosidase activity with a maximum activity at 35 °C at pH lower than 6, suggesting a role as exochitinase on native chitin. To our knowledge, Chi18H8 is the first chitinase isolated from a metagenome library obtained in pure form and which has the potential to be used as a candidate agent for controlling fungal crop diseases. Furthermore, Chi18H8 may also answer to the demand for novel chitin-degrading enzymes for a broad range of other industrial processes and medical purposes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Disease of plants caused by fungal pathogens contributes to extensive loss of crops important for food and energy production globally. Moreover, the norm of monoculture practice, further increases opportunities for the invasion of phytopathogens (Herrera-Estrella and Chet 1999). As a consequence, the use of synthetic fungicides, many which are toxic, is extensive and evidently results in costs for public and ecosystem health (Mullin et al. 2010). A more environmentally sustainable approach to the use of toxic chemicals is microbiological control of fungal disease employing bacteria that exhibit antifungal action (Herrera-Estrella and Chet 1999). There are soils that are naturally suppressive toward plant diseases and microorganisms in these soils are proposed to be involved in the suppressiveness (Borneman and Becker 2007; Steinberg et al. 2007). As a result, several bacterial species have been isolated and commercially introduced for biocontrol purposes (Steinberg et al. 2007 and references therein). However, given the inherent limitations in the use of living organisms, such as relatively short shelf life of the products or inconsistent performance in the field, their application is confined (Neeraja et al. 2010). Alternative solutions are formulations with enzymes possessing antiphytopathogenic activity.
Promising candidates for this purpose are bacterial chitinases, as these degrade chitin, one of the main constituents of the fungal cell wall (Herrera-Estrella and Chet 1999; Zhang et al. 2001). Chitinolytic enzymes are also generally attractive for their potential use in a broad range of biotechnological applications, for example in biofuel production or bioconversion processes on shellfish waste to obtain value-added products, such as chitosan and chitooligosaccharides for the pharmaceutical market (Bhattacharya et al. 2007; Li and Greene 2010). In an ecological perspective, bacterial chitinases are crucial in the global biogeochemical re-cycling of carbon and nitrogen through the hydrolyzation of chitin. After cellulose, chitin is the most abundant biopolymer in nature, widely distributed within exoskeletons invertebrates, fungal cell walls, marine diatoms, crustaceans, and zooplankton (Gooday 1990). It is otherwise rather resistant to degradation and would without bacterial chitinases be trapped in biomass as insoluble in nature (for reviews, see Karlsson and Stenlid 2009; Keyhani and Roseman 1999). Degradation of chitin enables bacterial utilization of the end products, chitobiose, and N-acetylglucosamine compounds, as an energy-, carbon-, and/or nitrogen source (Gooday 1990).
Most of the bacterial chitinases are glycosyl hydrolases of family 18 (Henrissat and Bairoch 1993), which can be further classified into subfamilies A to C based mainly on amino acid sequence similarities of the catalytic domain and a conserved consensus motif of the catalytic site (Henrissat and Bairoch 1993; Karlsson and Stenlid 2009; Suzuki et al. 2002). Depending on the catalytic specificity as a result of enzyme structure, chitinases may show either endo- or exo-activity (Henrissat and Davies 2000). To date, bacterial chitinase genes have been identified, by conventional molecular screening approaches, in bacterial isolates or uncultured bacteria within both aquatic and soil environments (for example, Hobel et al. 2005; Ikeda et al. 2007; LeCleir et al. 2004; Metcalfe et al. 2002; Ramaiah et al. 2000; Terahara et al. 2009; Uchiyama and Watanabe 2006). Only a few studies, however, have used metagenomics tools to identify novel bacterial chitinase genes (Cottrell et al. 1999; LeCleir et al. 2007). The advantage of metagenome-based approaches is the complete access to the entire community genetic makeup without the need of microbial cultivation (for review, see Sjöling et al. 2007). To our knowledge, none of these studies has yet resulted in the isolation and characterization of novel biologically active chitinases.
As chitin degradation is such an important environmental function, we investigated, in a previous study, the effect of chitin amendment to a suppressive field soil on the bacterial community structure (Hjort et al. 2007). We could show using terminal restriction fragment length polymorphism and denaturing gradient gel electrophoresis analyses—that chitin amendment to the soil dramatically increased the relative abundances of known chitin-degrading genera, such as Oerskovia, Kitasatospora, and Streptomyces. These organisms became dominant also among the actively growing bacteria in the community (Hjort et al. 2007). Given that the soil bacterial community of the suppressive field responded to chitin amendment (Hjort et al. 2007), the active community contained taxa that typically are chitinolytic (Hjort et al. 2007) and that a number of isolates with antifungal and chitinase activity previously were obtained from this soil (Adesina et al. 2007), we sought to investigate this soil metagenome for novel chitinolytic enzymes with biocontrol capacity, suitable for more environmentally sustainable applications in agricultural processes. In this study, we used a targeted screening approach based on genetic and functional metagenomics with the aim to identify novel chitinolytic enzymes through heterologous expression. Our aim was to characterize further isolated enzymes for the purpose of identifying a promising candidate for phytopathogen biocontrol.
Materials and methods
Metagenomic library
A soil characterized as suppressive to club-root disease of cabbage (Worku and Gerhardson 1996) was sampled from a field at the Swedish University of Agricultural Sciences in Uppsala, Sweden (Hjort et al. 2007), and a fosmid metagenomic library was constructed from extracted high molecular weight soil DNA as previously described in Hjort et al. (2010) and Hårdeman and Sjöling (2007).
Screening of the metagenomic library for chitinase genes
Clones of the fosmid library, i.e., 7,800 fosmids, were replica plated into 150 μL of Luria-Bertani (LB) medium supplemented with 12.5 μg/mL chloramphenicol and cultured over night at 37 °C. The clones were induced into high copy number with Fosmid Induction Solution (Epicentre, Nordic Biolabs, Täby, Sweden) for 5 h at 37 °C. For screening, aliquots from the 96-clone cultures of every microtitre plate were pooled, making a total of 78 superpools, representing the whole metagenomic library. Fosmid DNA was extracted from 150 μL of the 78 superpool cultures with R.E.A.L. Prep 96 Plasmid Kit (Qiagen, Hilden, Germany), following the manufacturer's instructions.
The metagenomic library, i.e., the super pools, was screened for family 18 chitinase genes by PCR using degenerate primers ChiA_F2 (5′-CGT GGA CAT CGA CTG GGA RTW YCC-3′) and ChiA_R2 (5′-CCC AGG CGC CGT AGA RRT CRT ARS WCA-3′) (Hobel et al. 2005), designed on the basis of the conserved regions of the catalytic domain of glycoside hydrolase family 18. The PCR reactions were set up according to Hobel et al. (2004) with a few modifications as described in Hjort et al. (2010). Depooling was performed with a second round of PCR screening to identify the single fosmid clone containing a chitinase gene. For activity-based screening, super pools were analyzed for chitinolytic activity using the activity assay with 4-methylumbelliferyl-N-N′-chitobiose as substrate, as described below.
Sequence analysis of the chi18H8 gene
The complete gene sequence of the identified chitinase chi18H8 was obtained by primer walking technique (Macrogen Inc., Seoul, Korea) from the primers described above, towards the downstream and upstream insert/fosmid junctions. A putative signal peptide in the Chi18H8 amino acid sequence was identified by SignalP 4.0 server (Petersen et al. 2011) and InterProScan 4.8 (Zdobnov and Apweiler 2001). Sequence similarity comparisons were performed with InterProScan and Prosite (Sigrist et al. 2010) softwares. Phylogenetic comparison by amino acid sequence alignment of the identified putative chitinase with homologous chitinases was performed with protein BLAST (Altschul et al. 1990). A phylogenetic tree using PhyML was constructed based on amino acid sequence alignments of the catalytic domain of Chi18H8 with that of representatives of the eight groups of bacterial glycosyl hydrolases according to Karlsson and Stenlid (2009).
Subcloning of the chi18H8 gene
The chi18H8 gene was cloned into the expression vector pGEX-6P-3 (GE Healthcare, Uppsala, Sweden), by first amplifying the gene with the primers Chi_18H8_F (5′)-GGG CCC G AAT TCC ATG CGC CAG CTC ACG CTT CTC and Chi_18H8_R (5′)-GCG CGC CTC GAG CTA TCA ATT GCC CCT ATG CAG ACT, with the positive fosmid clone DNA as template. For the correct orientation of the gene into the vector, restriction enzyme sites EcoRI and XhoI were introduced, respectively, into the primers Chi_18H8_F and Chi_18H8_R, as underlined. A standard PCR reaction (Sambrook and Russell 2001) at 68 °C annealing temperature was performed with High Fidelity PCR Enzyme (Fermentas, Thermo Fisher Scientific Inc, VWR International, Stockholm, Sweden). The amplified fragment, double digested with EcoRI and XhoI was then ligated with T4-DNA ligase (New England Biolabs, Ipswich, England) into the expression vector pGEX-6P-3 (GE Healthcare) treated with EcoRI and XhoI and including the protein tag glutathione S transferase (GST). The resulting plasmid, pGEX-6P-3::chi18H8 was transformed into Escherichia coli TOP-10 (Invitrogen Life Technologies, Stockholm, Sweden) and sequenced to make sure that the gene was correctly inserted (Macrogen Inc). For overexpression, the construct pGEX-6P-3::chi18H8 was transformed into E. coli BL21 (DE3) cells (Invitrogen).
Expression and purification of the Chi18H8
For GST-Chi18H8 fusion protein overexpression conditions in E. coli BL21 (DE3), see the Electronic supplementary material. For protein purification, a single colony of E. coli pGEX-6P-3::chi18H8 was inoculated into 5 mL of LB medium with 50 mg/L ampicillin (Sigma-Aldrich, St. Louis, MO, USA) and incubated at 37 °C for 2 h at 200 rpm. The culture was inoculated into 300 mL Erlenmeyer flasks 50 mL malt extract (ME) medium (6 g/L malt extract, 1.8 g/L maltose, 6 g/L dextrose, and 1.2 g/L yeast extract: all medium components from Sigma-Aldrich) supplemented with 50 mg/L of ampicillin and incubated overnight at 25 °C and 150 rpm; 25 mL of the overnight culture was then inoculated into 2 L flasks with 375 mL ME medium and 25 mg/L ampicillin and incubated at 20 °C and at 150 rpm. At an O.D.600 nm of 0.4, isopropyl-β-D-thiogalactopyranoside (IPTG, Sigma-Aldrich) was added to a final concentration of 0.1 mM and the incubation continued for 48 h at 16 °C. Cells were harvested by centrifugation at 8,000 g for 15 min and then sonicated (6 cycles of 30 s each, with a 1 min interval using a Branson Sonifier 250, Danbury, USA) in phosphate-buffered saline (PBS; 2 mL/g cells) at pH 7.4 (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4), containing 1 mM phenylmethylsulfonylfluoride (Sigma-Aldrich), 0.7 μg/mL of pepstatin A (Sigma-Aldrich) and 10 μg/mL deoxyribonuclease I (Sigma-Aldrich). The soluble fraction was separated from the insoluble fraction by centrifugation at 18,000×g for 1 h at 4 °C. The GST-Chi18H8 fusion protein was purified from the soluble fraction by loading on a GSTrap FF column 1 mL (GE Healthcare) using PBS at pH 7.4. For the on-column cleavage of the GST tag, PreScission buffer (50 mM TrisHCl at pH 7.0, 0.15 M NaCl, 1 mM ethylenediaminetetraacetic acid, and 1 mM dithiothreitol) containing 30 U of PreScission protease (GE Healthcare) was loaded. The column was then incubated for 15 h at 4 °C, followed by 2 h at room temperature. The cleaved Chi18H8 was eluted by PreScission buffer. The GST tag and the PreScission protease bound to the column were eluted by 50 mM TrisHCl at pH 8.0 containing 15 mM reduced l-glutathione, according to the manufacturer's protocol.
Protein and zymogram analyses
Protein samples from soluble and insoluble E. coli cell fractions and from fractions collected during purification were loaded onto 10 % (v/v) sodium dodecyl sulfate (SDS)–polyacrylamide gel for electrophoresis (PAGE). For the insoluble fractions, pellets obtained after sonication and centrifugation of E. coli cells, were resuspended in an appropriate volume of Laemmli sample buffer. Protein bands were visualized in gels by staining with Coomassie brilliant blue (Laemmli 1970). For molecular mass determination, PageRuler™ Prestained Protein Ladder (Thermo Scientific, Milan, Italy) was used. Protein concentration was estimated by densitometric analysis of SDS gel bands using the Quantity One analysis software (Bio Rad, Hercules, CA, USA) and by the use of the extinction coefficient at 280 nm (77,015 M−1 cm−1) determined by urea denaturation compared with the theoretical extinction coefficient based on the amino acid sequence.
Zymogram was used to detect chitinolytic activity, using 10 % (v/v) polyacrylamide gel containing 0.7 mg/mL carboxymethyl-chitin–Remazol brilliant violet (CM-chitin-RBV; Loewe Biochemica, Sauerlach, Germany). The purified Chi18H8 was diluted in an appropriate sample buffer volume lacking any reducing agent and incubated for 10 min at room temperature. Electrophoresis was conducted at 4 °C in Tris–glycine–SDS running buffer according to the standard running conditions. The gel was then rinsed twice in 2.5 % (v/v) Triton X-100 for 30 min at room temperature to remove SDS and equilibrated in 0.1 M sodium acetate buffer (pH 5) at 37 °C. Clear zones indicating chitinolytic activity appeared after overnight incubation at 37 °C.
Chitinase activity assay
Enzymatic activity of the metagenomic library, fosmid clone crude extract, and purified enzyme was assayed fluorometrically by measuring the release of 4-methylumbelliferone (MU) from the chitooligosaccharide analogs 4-methylumbelliferyl-N-N′-N″-chitotriose (MU-NAG3), 4-methylumbelliferyl-N-N′-chitobiose (MU-NAG2), and 4-methylumbelliferyl-N-acetyl-d-glucosamine (MU-NAG). In the case of metagenome library screening of the pooled fosmid clones, 100 μL of cell suspensions previously grown in LB medium supplemented with 12.5 μg/mL chloramphenicol, and induced by Fosmid Induction Solution (Epicentre) according to the manufacturer's protocol, were added in black microtitre plates with 10 μL 10-mM MU-NAG2. The plates were incubated at 20 °C for up to 7 days. Fluorescence was monitored daily using a FLUOstar (BMG LABTECH, Offenburg, Germany) plate reader with an excitation wavelength at 320 nm and an emission at 460 nm. For assaying enzyme extracts, the standard reaction mixture (50 μL) contained: 0.4 mM of the chitooligosaccharide in 50 mM sodium acetate buffer pH 5 and 5 μL of protein sample. The reaction was stopped after 30 min at 37 °C by the addition of 150 μL of 400 mM sodium carbonate solution. Fluorescence was monitored as described above. All measurements were performed at least in triplicate. One unit of enzyme activity was defined as the amount of enzyme that hydrolyses 1 μmole of MU of the substrate per minute at pH 5.0 and 37 °C (McCreath and Gooday 1992; Di Maro et al. 2010).
The optimum pH for Chi18H8 activity was determined using the fluorescent assay described above with the following buffers (50 mM) at respective pH: sodium acetate (pH 4.0 and 5.0), sodium phosphate (pH 6.0, 7.0, and 8.0), and glycine–sodium hydroxide (pH 9.0) (Pearse 1980). The optimum temperature for Chi18H8 activity was determined by the fluorescent assay incubating the reaction mixture at various temperatures (5–70 °C).
Antifungal activity assay
Antifungal activity of Chi18H8 was estimated by a growth inhibition assay on a set of eight plant pathogenic fungi, listed in Table 1. An agar plug, harboring an actively growing fungal culture, was placed onto the middle of a ME agar plate, pH 5, supplemented with 50 mg/L ampicillin and 0.1 mM IPTG. Subsequently, cell suspensions of E. coli BL21(DE3) pGEX-6P-3::chi18H8 were stamped onto the agar plate in a circular perimeter around the plug. Expression of the chi18H8 gene from the expression plasmid pGEX-6P-3::chi18H8 was induced by the IPTG added into the agar media. E. coli BL21(DE3) pGEX-6P-3 was used as negative control. The plates were incubated at 25 °C and growth of fungal hyphaea was monitored using a microscope. The following fungal strains were all generously obtained as a gift from Prof. J. Schnürer's research group, Department of Microbiology, Agricultural University of Sweden: Colletotrichum gloeosporioides CR21, Fusarium graminearum IBT 1958 (from Center of Microbial Biotechnology, Department of Systems Biology, Technical University of Denmark), Fusarium oxysporum FRR3414 (from Commonwealth Scientific and Industrial Research Organization, Food and Nutritional Sciences, North Ryde Sydney, NSW, Australia), Penicillium chrysogenum ATCC9179, and Trichoderma harzianum CBS226.95. Furthermore, the following strains from Schnürer's research group's own fungal collection were used: Alternaria alternata J414, Aspergillus niger J8, and Rhizopus stolonifer J45.
Sequence database deposition
The nucleotide sequence of the chitinase chi18H8 gene was deposited in the GenBank database under the accession number KC763366.
Results
Identifying a novel chitinase in the metagenomic library
The fosmid library, previously generated from a field soil reported to be suppressive for fungal phytopathogens, was screened by combining activity screening with a “genetic sieving” utilizing degenerate PCR primers for the glycoside hydrolases of family 18 group A. For this purpose, a pooling strategy was successfully applied which made screening of the library more efficient. Those clones expressing chitinolytic activity in the presence of the synthetic chitin surrogate substrate oligosaccharide MU-NAG2 were considered as positive hits.
Among the 78 superpools screened, four pools returned a PCR product of the expected size (270 nucleotides). Each clone within the four positive super pools was further analyzed individually and 25 positive clones were identified in a second PCR run. From one of the clones (plate 18 row H8) that showed both strong chitinase activity and PCR signal, we identified the complete open-reading frame of a novel chitinase by primer walking technique. The gene, named chi18H8, consists of 1,275 nucleotides encoding a protein of 425-amino acid residues with a predicted molecular weight of 46 kDa. In silico sequence analysis identified a protein motif for a signal peptide of 20 amino acid residues at the N-terminal, followed by a putative catalytic domain (amino acid residues, 37 to 408) (Fig. 1a). The catalytic domain includes the consensus motif for the active site consisting of the amino acid residues FDGIDIDWE (amino acid residues, 175 to 183) with the motif DxDxE conserved among all the family 18 chitinase members. It also contains the so-called chitinase insertion domain (CID) between amino acid residues 310 and 379, identified by the conserved residues YxR. Chitin has been suggested to insert at this site to reach the active site, and it consists of a small α+β domain which is the main structural feature of subfamily A of family 18 chitinases (Li and Greene 2010).

a Protein motifs identified within the novel chitinase Chi18H8 protein sequence include a signal peptide (amino acidic residues, 1–20), catalytic domain (aa, 37–408) with the active site (aa, 175–183), and chitinase insertion (CID; aa, 310–379), and indicate that it belongs to the family 18 of glycoside hydrolases. Sequences were analyzed using SignalP, InterProScan, and Prosite. b Phylogenetic tree of the bacterial glycosyl hydrolases family 18 based on protein sequence alignment of the catalytic domains, according to Karlsson and Stenlid (2009), demonstrating that the chitinase Chi18H8 belongs to group II. The tree was generated using PhyML and only bootstrap values above 70 % are included in the tree
A phylogenetic tree based on sequence alignments of the catalytic domain of Chi18H8 with that of representatives of the eight groups of bacterial glycosyl hydrolases (according to Karlsson and Stenlid 2009), shows that Chi18H8 belongs to the group II of chitinase family 18. Chi18H8 clusters with representatives of divergent phyla, such as proteobacteria (Saccharophagus) and actinobacteria (Streptomyces) (Fig. 1b). Protein sequence comparison of the full-length protein sequence showed highest amino acid identity (45 %) to a chitinase from an uncultured bacterium (Uchiyama and Watanabe 2006). The closest match to a protein sequence from an identified organism was with the following myxobacteria: Chondromyces apiculatus (41 % identity), Corallococcus coralloides (40 % identity), and Myxococcus xanthus (39 % identity).
Purification of the novel chitinase Chi18H8
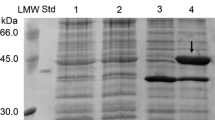
Heterologous expression of the chi18H8 gene, which was cloned into a pGEX-6P-3 vector in E. coli gave the fusion protein GST-Chi18H8 of an estimated molecular mass of 73 kDa (Fig. 2a) and with a pI of 6.39. This expression system was selected as we successfully used it previously for purifying metagenomically sourced lipases and esterases (Hårdeman and Sjöling 2007) and others have used it for expressing bacterial chitinases (Tsujibu et al. 2000). The GST moiety of the vector adds at the amino terminus of the recombinant protein to facilitate on column purification via affinity chromatography as the GST-tag binds to the column and then can by cleaved off using a PreScission protease, allowing final recovery of the native protein (Makrides 1996).

a SDS-PAGE analysis of Chi18H8 purification. Lanes 1 and 2, E. coli pGEX-6P-3::chi18H8 insoluble and soluble fractions; lane 3, elution with reduced glutathione of the GST-chi18H8 fusion protein; lane 4, Chi18H8 after on-column cleavage of GST-tag; lane 5, elution with reduced glutathione of the GST-tag and PreScission protease; lane 6, PreScission buffer for the on-column cleavage. LMW Marker (PageRuler™ Prestained Protein Ladder, Fermentas); lane S standard Rhodotorula gracilis His6-d amino acid oxidase, 36 kDa, kindly provided by Prof. Pollegioni, University of Insubria. b Zymogram analysis of the purified GST-Chi18H8 (lane 1) and Chi18H8 (lane 2) with CM-chitin-RBV as substrate
Results from experiments conducted on heterologous fusion protein production by varying different cultivation conditions are reported in the Electronic supplementary material and shown in Figs. S1 and S2. In these experiments, enzymatic activity assay (i.e., the fluorimetric assay with MU-NAG analogs as substrates, Fig. S1) and detection of protein accumulation by SDS-PAGE (Fig. S2) revealed that the fusion protein maintained the chitinolytic activity, but most of it accumulated into insoluble fractions, from which protein recovery is known to be challenging (Huang et al. 2012). To facilitate purification, different expression trials (Electronic supplementary material) were conducted to increase the protein production in the soluble form. At the most optimal conditions we achieved so far (inducing cells growing in ME at early exponential phase, with 0.1 mM IPTG and harvesting the cells after 48 h at 16 °C), only 10 % of the chitobiosidase activity initially detected in the soluble fraction (34 U) was recovered in the purified fraction, starting from 2.3 g of cell paste. After the on-column PreScission protease cleavage, 30 μg high-purity Chi18H8 per liter of culture (21 μg/g cells) was obtained (Fig. 2a).
Enzyme characterization of Chi18H8
Notwithstanding the limited amount of the purified enzyme, characterization of the Chi18H8 protein was possible. Zymogram on carboxymethyl chitin confirmed the chitinolytic activity of the purified Chi18H8 (Fig. 2b), both with (lane 1) and without (lane 2) the GST-tag. Substrate specificity analyses of Chi18H8 against three different-length analogs of chitooligosaccharides showed an activity of 113 U/mg protein for MU-NAG2 as substrate, and of 39 U/mg protein on MU-NAG3, but no activity against MU-NAG. These substrates are commonly used for detecting chitobiosidase, endochitinase, and N-acetylglucosaminidase activities, respectively (Howard et al. 2003). Our results indicated that the Chi18H8 enzyme has a prevalent chitobiosidase activity.
The optimum temperature for Chi18H8 activity with both MU-NAG2 (Fig. 3a) and MU-NAG3 was 35 °C. The enzymatic activity was also tested at different pH ranging from 3 to 9. With a pH optimum between 3 and 5, more than 80 % of the Chi18H8 activity remained at acidic and sub-acidic conditions. At pH above 6, the hydrolytic activity drastically decreased (Fig. 3b).

Enzymatic properties of the chitinase Chi18H8, using 0.4 mM MUF-NAG2 as substrate. a Temperature influence on chitobiosidase activity. Assays were performed in 50 mM sodium acetate buffer pH 5. In both experiments, enzymatic activities are expressed relative to the maximal recorded activity: 125.5 and 121.5 U/mg protein, respectively. b pH influence on chitobiosidase activity. Assays were performed using 50 mM sodium acetate buffer (pH 4.0 and 5.0), 50 mM sodium phosphate buffer (pH 6.0, 7.0, and 8.0), and 50 mM glycine–sodium hydroxide buffer (pH 9.0)
Chi18H8 has antifungal activity
The antifungal activity of Chi18H8 was determined by co-cultivating E. coli pGEX-6P-3::chi18H8 or pGEX-6P-3 strains, with the phytopathogen fungi listed in Table 1. The recombinant bacterial growth, surrounding the fungal plug, inhibited the growth of C. gloeosporioides, F. graminearum, and F. oxysporum as shown in Fig. 4, and A. alternata. No growth inhibition was observed in the negative control with the bacterial suspension harboring the plasmid without chi18H8.

Chi18H8 inhibiting growth of the fungal plant pathogens a C. gloeosporioides CR21, b Fusarium oxosporum FRR3414, and c F. graminearum IBT1958. The chitinase was heterologously expressed after IPTG induction and the E. coli cells containing the chi18H8 gene in an expression vector (left in each picture) were stamped in a ring shape around the center with fungal inoculum. E. coli cells with the expression vector but without the chi18H8 gene were used as controls (right in each picture)
Discussion
Access to the functional capacities of the bacterial community without limits of cultivation is important in the discovery of novel biocatalysts, not the least when the demand for more environmentally friendly alternatives is strong. We used a genetic and functional metagenomics approach to identify a novel chitinase with antifungal activity from a Swedish field soil, previously characterized as suppressive toward phytopathogens.
The novel chitinase Chi18H8 only shows 45 %, or less, amino acid sequence identity to any known chitinase but contains the conserved consensus sequences of family 18 chitinases including the conserved active site motif, DxDxE of the catalytic domain. Consistently with what has been shown for the catalytic domain of the family 18 chitinases (Suzuki et al. 2002), structure prediction showed that Chi18H8 has a triosephosphate isomerase fold (TIM barrel) that consists of an (α/β)8-barrel fold. Furthermore, the Chi18H8 catalytic domain contains a CID with the conserved amino acid residues YxR that have been shown to interact with the substrate (Li and Greene 2010). This small domain that inserts into the TIM barrel is present in chitinases of subfamily A, but absent in the subfamily B (Suzuki et al. 2002). The CID forms a wall alongside the TIM barrel's substrate-binding cleft, making the substrate-binding cleft deeper (Li and Greene 2010). As a result, family 18 chitinases with both TIM domain and CID can bind long-chain substrates. Hence, the structure of the substrate-binding cleft has importance for the substrate specificity and correlates with the endo- or exo-activity of family 18 glycoside hydrolases (Hurtado-Guerrero and van Aalten 2007). Subfamily A chitinases possessing the deep substrate binding cleft, such as Serratia marcescens ChiA and ChiB, are classified as exochitinases (Horn et al. 2006), while subfamily B chitinases, such as S. marcescens ChiC, have a shallow substrate binding cleft as they lack the CID, and have endochitinase activity (Suzuki et al. 2002). Exochitinases are suggested to hydrolyze off oligosaccharide dimers or chitobiosidase from the polysaccharide by “sliding” the substrate through the substrate-binding cleft (Horn et al. 2006). Results both from structure prediction and substrate specificity analyses suggest that Chi18H8 is an exochitinase. Chi18H8 has a preference for the dimer substrate MUF-NAG2 over the trimer substrate analog MUF-NAG3, but it is not active on the monomer MUF-NAG, indicating a chitobiosidase (exo-N,N′-diacetyl-glucosaminidase) activity. According to studies on ChiA in S. marcescens (Zees et al. 2009), the presence of the CID is suggested to enhance the chitobiosidase activity as well as the processivity during degradation of the polysaccharide chains, facilitating the accessibility to crystalline and insoluble substrate, such as chitin.
The metagenome-sourced Chi18H8 shows optimal activity and stability at acidic pH and mesophilic temperatures, consistently with what has been reported for family 18 chitinases isolated from cultured microorganisms (Bhattacharya et al. 2007). Unlike other family 18 chitinases, however, the Chi18H8 seems to exert antagonistic selective activity against four important plant pathogens, coherently with the suppressive nature of the soil from which it was isolated. To date, antifungal activity has been reported among the family 19 of glycosyl hydrolases (Gherbawy et al. 2012). Family 19 chitinases differ from those belonging to family 18 in amino acid sequence, three-dimensional protein structure, molecular mechanism of catalytic region, and enzymology and are thus considered to have different evolutionary origin (Kawase et al. 2006). Even if further investigation should be conducted on the antifungal spectrum of the Chi18H8 by using the pure protein product, to our knowledge, this is the first report of a family 18 chitinase possessing an antifungal activity.
Further investigations on the mechanism of action and on the antifungal activity of Chi18H8 will be possible by obtaining a higher yield of the pure protein though improving the host-expression system. Low purification yield is a persistent bottleneck in heterologous expression in model hosts of metagenomically derived genes of unknown origin (Schmeisser et al. 2007; Sjöling et al. 2007). Limitations in availability of suitable alternative host systems have so far made E. coli based expression the most common for metagenomic screening and as a result most of the metabolic diversity present in environmental samples is less likely to be efficiently accessed. Success of protein expression depends on the host's ability to transcribe the metagenomic DNA efficiently, translate the mRNA to form a functional protein, fold, and localize the protein correctly. In our case, detection of Chi18H8 in the E. coli hosted metagenomic library was facilitated by the high sensitivity of the chitobiosidase fluorimetric assay probably in combination with the occurrence of cell lysis during the incubation period. Indeed, Chi18H8 purification from E. coli was hampered by its accumulation within the insoluble fraction, i.e., inclusion bodies and /or membranes. Our recent trials (Berini et al., unpublished results) on cloning the chi18H8 gene into another E. coli vector (pET24b+, Novagen, Milan, Italy) providing only a histidine hexamer at the C terminus of the protein product (C-His6-Chi18H8), indicate that most of the protein accumulates into the insoluble fraction as in case of the GST-Chi18H8 fusion protein. Thus, we could exclude that protein accumulation is caused by the length and the position of the GST-tag in the pGEX-6P-3 expression system. However, the chitinase was expressed in both systems with its putative signal peptide, which may, through its hydrophobic nature, facilitate aggregation into inclusion bodies and/or sticking to E. coli membranes. A previous paper from other authors (Hoster et al. 2005) reported that a tenfold higher heterologous production of a Streptomyces chitinase in E. coli was achieved when the gene was cloned without the region encoding the putative signal sequence. Alternatively, the development of solubilisation and refolding protocols (Huang et al. 2012) or the use of expression systems alternative to E. coli are being investigated to allow larger scale production of Chi18H8 required for further detailed protein characterization and for its in-field evaluation as a novel biocontrol agent.
To our knowledge, this is the first active chitinase identified, produced, and purified using a metagenomic approach. This study confirms that accessing the metabolic functions of a suppressive soil by functional metagenomics can provide a novel chitinase with antifungal activity that has the potential application as a biocontrol compound against crop fungal pathogens and may offer a more environmentally sustainable alternative to the otherwise often toxic chemical fungicides.
References
Adesina MF, Lembke A, Costa R, Speksnijder A, Smalla K (2007) Screening of bacterial isolates from various European soils for in vitro antagonistic activity towards Rhizoctonia solani and Fusarium oxysporum: site-dependent composition and diversity revealed. Soil Biol Biochem 39:2818–2828
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Bhattacharya D, Nagpure A, Gupta RK (2007) Bacterial chitinases: properties and potential. Crit Rev Biotech 27:21–28
Borneman J, Becker JO (2007) Identifying microorganisms involved in specific pathogen suppression in soil. Annu Rev Phytopathol 45:153–172
Cottrell MT, Moore JA, Kirchman DL (1999) Chitinases from uncultured marine microorganisms. Appl Environ Microb 65:2553–2557
Dehestani A, Kazemitabar K, Ahmadian G, Jelodar NB, Salmanian AH, Seyedi M, Rahimian H, Ghasemi S (2010) Chitinolytic and antifungal activity of a Bacillus pumilus chitinase expressed in Arabidopsis. Biotechnol Lett 32:539–546
Di Maro A, Terracciano I, Sticco L, Fiandra L, Ruocco M, Corrado G, Parente A, Rao R (2010) Purification and characterization of a viral chitinase active against plant pathogens and herbivores from transgenic tobacco. J Biotechnol 147:1–6
Gooday GW (1990) The ecology of chitin degradation. Adv Microb Ecol 11:387–430
Gherbawy Y, Elhariry H, Altalhi A, El-Deeb B, Khiralla G (2012) Molecular screening of Streptomyces isolates for antifungal activity and family 19 chitinase enzymes. J Microbiol 50:459–468
Hassan F, Meens J, Jacobsen HJ, Kiesecker H (2009) A family 19 chitinase (Chit30) from Streptomyces olivaceoviridis ATCC 11238 expressed in transgenic pea affects the development of T. harzianum in vitro. J Biotechnol 143:302–308
Hårdeman F, Sjöling S (2007) Metagenomic approach for the isolation of a novel low-temperature-active lipase from uncultured bacteria of marine sediment. FEMS Microbiol Ecol 59:524–534
Henrissat B, Bairoch A (1993) New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 293:781–788
Henrissat B, Davies G (2000) Glycoside hydrolases and glycosyltransferases: families, modules and implications for genomics. Plant Physiol 124:1515–1520
Herrera-Estrella A, Chet I (1999) Chitinases in biological control. In: Jollès P, Muzzarelli RAA (eds) Chitin and chitinases EXS 87. Birkhäuser Verlag, Basel, pp 171–184
Hjort K, Lembke A, Speksnijder A, Smalla K, Jansson JK (2007) Community structure of actively growing bacterial populations in plant pathogen suppressive soil. Microb Ecol 53:399–413
Hjort K, Bergström M, Adesina MF, Jansson JK, Smalla K, Sjöling S (2010) Chitinase genes revealed and compared in bacterial isolates, DNA extracts and a metagenomic library from a phytopathogen-suppressive soil. FEMS Microbiol Ecol 71:197–207
Hobel CFV, Marteinsson VT, Hauksdottir S, Fridjonsson OH, Skirnisdottir S, Hreggvidsson GO, Kristjansson JK (2004) Use of low nutrient enrichments to access novel amylase genes in silent diversity of thermophiles. World J Microb Biot 20:801–809
Hobel CF, Marteinsson VT, Hreggvidsson GO, Kristjansson JK (2005) Investigation of the microbial ecology of intertidal hot springs by using diversity analysis of 16S rRNA and chitinase genes. Appl Environ Microb 71:2771–2776
Horn SJ, Sørbotten A, Synstad B, Sikorski P, Sørlie M, Vårum KM, Eijsink VG (2006) Endo/exo mechanism and processivity of family 18 chitinases produced by Serratia marcescens. FEBS J 273:491–503
Hoster F, Schmitz JE, Daniel R (2005) Enrichment of chitinolytic microorganisms: isolation and characterization of a chitinase exhibiting antifungal activity against phytopathogenic fungi from a novel Streptomyces strain. Appl Microbiol Biotechnol 66:434–442
Howard MB, Nathan A, Ronald M, Weiner E, Steven WH (2003) Detection and characterization of chitinases and other chitin-modifying enzymes. J Ind Microbiol Biot 30:627–635
Huang C, Lin H, Yang X (2012) Industrial production of recombinant therapeutics in Escherichia coli and its recent advancements. J Ind Microbiol Biot 39:383–399
Hurtado-Guerrero R, van Aalten DMF (2007) Structure of Saccharomyces cerevisiae chitinase 1 and screening based discovery of potent inhibitors. Chem Biol 14:589–599
Ikeda S, Ytow N, Ezura H, Minamisawa K, Miyashita K, Fujimura T (2007) Analysis of molecular diversity of bacterial chitinase genes in the maize rhizosphere using culture-independent methods. Microbes Environ 22:71–77
Karlsson M, Stenlid J (2009) Evolution of family 18 glycoside hydrolases: diversity, domain structures and phylogenetic relationships. J Mol Microbiol Biotechnol 16:208–223
Kawase T, Yokokawa S, Saito A, Fujii T, Nikaidou N, Miyashita K, Watanabe T (2006) Comparison of enzymatic and antifungal properties between family 18 and 19 chitinases from S. coelicolor A3(2). Biosci Biotech Bioch 70:988–998
Keyhani NO, Roseman S (1999) Physiological aspects of chitin catabolism in marine bacteria. Biochim Biophys Acta 1473:108–122
Laemmli UK (1970) Cleavage of structural proteins during the assemblage of the head of bacteriophage T4. Nature 277:680–685
LeCleir GR, Buchan A, Hollibaugh JT (2004) Chitinase gene sequences retrieved from diverse aquatic habitats reveal environment-specific distributions. Appl Environ Microb 70:6977–6983
LeCleir GR, Buchan A, Maurer J, Moran MA, Hollibaugh JT (2007) Comparison of chitinolytic enzymes from an alkaline, hypersaline lake and an estuary. Environ Microbiol 9:197–205
Lee YS, Park IH, Yoo JS, Chung SY, Lee YC, Cho YS, Ahn SC, Kim CM, Choi YL (2007) Cloning, purification, and characterization of chitinase from Bacillus sp. DAU101. Biores Technol 98:2734–2741
Li H, Greene LH (2010) Sequence and structural analysis of the chitinase insertion domain reveals two conserved motifs involved in chitin binding. PLoS ONE 5:e8654
Makrides SC (1996) Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol Rev 90:512–538
McCreath KJ, Gooday GW (1992) A rapid and sensitive microassay for determination of chitinolytic activity. J Microbiol Meth 14:229–237
Metcalfe AC, Krsek M, Gooday GW, Prosser JI, Wellington EM (2002) Molecular analysis of a bacterial chitinolytic community in an upland pasture. Appl Environ Microb 68:5042–5050
Mukherjee G, Sen SK (2006) Purification, characterization and antifungal activity of chitinase from Streptomyces venezuelae P10. Curr Microbiol 53:265–269
Mullin CA, Frazier M, Frazier JL, Ashcraft S, Simonds R (2010) High levels of miticides and agrochemicals in north american apiaries: implications for honey bee health. PLoS ONE 5:e9754
Neeraja C, Anil K, Purushotham P, Suma K, Sarma PVSRN, Moerschbacher BM, Podile AR (2010) Biotechnological approaches to develop bacterial chitinases as a bioshield against fungal diseases of plants. Crit Rev Biotechnol 30:231–241
Pearse AGE (1980) Histochemistry: theoretical and applied, 4th edn. Churchill-Livingstone, London
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786
Prapagdee B, Kuekulvong C, Mongkolsuk S (2008) Antifungal potential of extracellular metabolites produced by Streptomyces hygroscopicus against phytopathogenic fungi. Int J Biol Sci 4:330–337
Ramaiah N, Hill RT, Chun J, Ravel J, Matte MH, Straube WL, Colwell RR (2000) Use of a chiA probe for detection of chitinase genes in bacteria from the Chesapeake Bay1. FEMS Microbiol Ecol 34:63–71
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schmeisser C, Steele H, Streit WR (2007) Metagenomics, biotechnology with non-culturable microbes. Appl Microbiol Biot 75:955–962
Sigrist CJA, Cerutti L, de Castro E, Langendijk-Genevaux PS, Bulliard V, Bairoch A, Hulo N (2010) PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res 38:161–166
Singh A, Kirubakaran SI, Sakthivel N (2007) Heterologous expression of new antifungal chitinase from wheat. Protein Express Purif 56:100–109
Sjöling S, Stafford W, Cowan DA (2007) Soil metagenomics: Exploring and exploiting the soil microbial gene pool. In: van Elsas JD, Jansson J, Trevors JT (eds) Modern soil microbiology, 2nd edn. CRC Press, Boca Raton, pp 409–434
Steinberg C, Edel-Hermann V, Alabouvette C, Lemanceau P (2007) Soil suppressiveness to plant diseases. In: van Elsas JD, Jansson J, Trevors JT (eds) Modern soil microbiology, 2nd edn. CRC Press, Boca Raton, pp 455–501
Suzuki K, Sugawara N, Suzuki M, Uchiyama T, Katouno F, Nikaidou N, Watanabe T (2002) Chitinases A, B, and C1 of Serratia marcescens 2170 produced by recombinant Escherichia coli: enzymatic properties and synergism on chitin degradation. Biosci Biotech Bioch 66:1075–1083
Terahara T, Ikeda S, Noritake C, Minamisawa K, Ando K, Tsuneda S, Harayama S (2009) Molecular diversity of bacterial chitinases in arable soils and the effects of environmental factors on the chitinolytic bacterial community. Soil Biol Biochem 41:473–480
Tsujibu H, Okamoto T, Hatano N, Miyamoto K, Watanabe T, Mitsutomi M, Inamuri Y (2000) Family 19 chitinases from Streptomyces thermoviolaceus OPC-520: molecular cloning and characterization. Biosci Biotechnol Biochem 64:2445–2453
Uchiyama T, Watanabe K (2006) Improved inverse PCR scheme for metagenome walking. Biotechniques 41:183–188
Wang S, Shao B, Fu H, Rao P (2009) Isolation of thermostable legume chitinase and study on the antifungal activity. Appl Microbiol Biot 85:313–321
Worku Y, Gerhardson B (1996) Suppressiveness to clubroot, pea root and Fusarium wilt in Swedish soils. J Phytopathol 144:143–146
Xiao L, Xie CC, Cai J, Zj L, Chen YH (2009) Identification and characterization of a chitinase produced bacillus showing significant antifungal activity. Curr Microbiol 58:528–533
Yan R, Hou J, Ding D, Guan W, Wang C, Wu Z, Li M (2008) In vitro antifungal activity and mechanism of action of chitinase against four plant pathogenic fungi. J Basic Microbiol 48:293–301
Zdobnov EM, Apweiler R (2001) InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17:847–848
Zees AC, Pyrpassopoulos S, Vorgias CE (2009) Insights into the role of the (α+β) insertion in the TIM-barrel catalytic domain, regarding the stability and the enzymatic activity of Chitinase A from Serratia marcescens. Biochim Biophys Acta 1794:23–31
Zhang Z, Yuen GY, Sarath G, Penheiter AR (2001) Chitinases from the plant disease biocontrol agent, Stenotrophomonas maltophilia C3. Phytopathol 91:204–211
Acknowledgments
We are grateful to Prof. Johan Schnürer and Dr. Su-Lin Leong at the Department of Microbiology at the Swedish Agricultural University in Uppsala for the fungal strains. This study was funded by the EU Metaexplore project (KBBE-222625; SS and FM) and the Foundation for Baltic and East European Studies (SS). Support from Ministero Italiano della Ricerca e della Università and Consorzio Interuniversitario per le Biotecnologie (CIB) is also acknowledged (IP).
Author information
Authors and Affiliations
Corresponding author
Additional information
Karin Hjort and Ilaria Presti equally contributed to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 1.69 MB)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Hjort, K., Presti, I., Elväng, A. et al. Bacterial chitinase with phytopathogen control capacity from suppressive soil revealed by functional metagenomics. Appl Microbiol Biotechnol 98, 2819–2828 (2014). https://doi.org/10.1007/s00253-013-5287-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5287-x