
Abstract
We developed two independent approaches for separation and quantitation of 24 oxygenated polycyclic aromatic hydrocarbons (OPAHs) using both liquid chromatography-atmospheric pressure chemical ionization/mass spectrometry (LC-APCI/MS) and gas chromatography-electron impact/mass spectrometry (GC-EI/MS). Building on previous OPAH research, we examined laboratory stability of OPAHs, improved existing method parameters, and compared quantification strategies using standard addition and an internal standard on an environmental sample. Of 24 OPAHs targeted in this research, 19 compounds are shared between methods, with 3 uniquely quantitated by GC-EI/MS and 2 by LC-APCI/MS. Using calibration standards, all GC-EI/MS OPAHs were within 15 % of the true value and had less than 15 % relative standard deviations (RSDs) for interday variability. Similarly, all LC-APCI/MS OPAHs were within 20 % of the true value and had less than 15 % RSDs for interday variability. Instrument limits of detection ranged from 0.18 to 36 ng mL−1 on the GC-EI/MS and 2.6 to 26 ng mL−1 on the LC-APCI/MS. Four standard reference materials were analyzed with each method, and we report some compounds not previously published in these materials, such as perinaphthenone and xanthone. Finally, an environmental passive sampling extract from Portland Harbor Superfund, OR was analyzed by each method using both internal standard and standard addition to compensate for potential matrix effects. Internal standard quantitation resulted in increased precision with similar accuracy to standard addition for most OPAHs using 2-fluoro-fluorenone-13C as an internal standard. Overall, this work improves upon OPAH analytical methods and provides some considerations and strategies for OPAHs as focus continues to expand on this emerging chemical class.

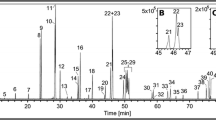
OPAH chromatograms from GC-EI/MS and LC-APCI/MS methods for NIST SRM 1650b (diesel particulate matter)
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Scientific interest for oxygenated polycyclic aromatic hydrocarbons (OPAHs), also known as oxy-PAHs, has increased in the last decade because of environmental presence and concern over potential toxicity [1]. OPAHs consist of one or more oxygen atoms attached to an aromatic ring structure that may also contain other chemical groups [2]. Formations of these compounds can derive from petrogenic and pyrogenic sources, specifically through chemical oxidation, photo-oxidation, or biological transformation of the unsubstituted PAHs [1]. Ongoing research has described several processes of incomplete biodegradation leading to production of substituted PAHs, including OPAHs [3–6]. Determinations of individual and mixed OPAH toxicities are active areas of research, but there is increasing evidence that some OPAH compounds are more toxic than the unsubstituted PAH analog [1, 7–11]. Additionally, it has been observed that OPAHs are persistent in the environment as opposed to other transient organic compounds, which contributes to increased attention of this chemical class [1, 4, 6]. Other concerns are that OPAHs are not routinely monitored, accumulate at PAH-contaminated sites [1, 3], or are potentially formed from bio-remediation strategies [9].
Although some analytical methods include OPAHs that contain hydroxylated or carboxylated compounds [12–16], our focus is on OPAHs that have at least one ketone group. Ketone-containing OPAHs include those previously reported in environmental samples, such as 9,10-anthraquinone and 9-fluorenone [14, 17] or those that have shown potential for mutagenicity, such as perinaphthenone, benzanthrone, and benzo[cd]pyrenone [18]. Quantitative analysis is challenging for these OPAHs because of wide ranges in solubility. Considering the diverse physiochemical properties, adequate solvation and stability become key factors in successful analytical method development. Both gas (GC) [13, 14, 17, 19–22] and liquid (LC) [15, 16, 23–26] chromatographic systems have been effectively used for OPAH analyses, but only four of the abovementioned papers target more than ten ketone-containing OPAHs. Of those papers, only one has quantitated and separated more OPAHs than this work using GC mass spectrometry (MS) [19], but it utilizes only a single response factor with a deuterated PAH of similar retention time to quantitate several OPAHs. Response factors ranged from 0.09 to 0.74 [19], indicating that PAHs and OPAHs might differ dramatically in analytical response regardless of retention time. Although this strategy would be useful at the time of publication, deuterated OPAHs have become increasingly available in recent years, and the following work highlights OPAHs that could benefit from closely matched laboratory surrogates or internal standards. Liquid chromatographic methods using atmospheric pressure chemical ionization (APCI), or LC-APCI-MS, have also been used successfully to quantitate 15–17 individual OPAHs [16, 23], but both papers use both positive and negative modes which can lead to losses in sensitivity or requires running each sample twice to quantify all target compounds. Given the large variability in physicochemistry of OPAHs (log K ow, 0.2–5.31), ionization of a target compound may or may not be optimal for a single chromatography system; it may be necessary to use multiple ionization methods to quantify larger sets of environmentally relevant OPAHs.
To develop methods for quantifying as many OPAHs as possible, 24 target OPAHs were analyzed on both GC and LC systems. To facilitate analysis on either GC or LC, we developed a final extraction preparation that employed a single solvent (ethyl acetate) that is both LC and GC compatible. We also discovered significant response differences with solvents, inlet temperatures, inlet liners, and OPAH stability not previously reported for this compound class. Our initial objective was to optimize and validate each method for increased sensitivity, accuracy, and precision for as many OPAHs as each system could quantify. Our second objective was to demonstrate each method using real environmental matrices, including National Institute of Standards and Technology (NIST) Standard Reference Materials (SRMs). Our third objective was to illustrate differences between instruments because of any potential matrix effects and compare the effectiveness of standard addition over our choice of an internal standard quantitation using an aqueous sample from a National Priority List Superfund site. In this research, we detail the identification and quantification of 24 ketone-containing OPAHs on two independent methods, providing a foundation of analytical investigation as new OPAHs are identified, become commercially available, or targeted through ongoing environmental research.
Experimental section
Chemicals and reagents
Analytical grade standards (purity, ≥97 %) were obtained from several vendors including: 9,10-anthraquinone-D8 (9,10-ANTQ-D8), 9-fluorenone-D8 (9-FLUO-D8), 1,4-naphthoquinone-D6, 2-methyl-1,4-naphthalenequinone-D8 (2me-1,4-NQ-D8), and 2-fluoro-fluorenone-13C (2F-FLUO-13C) from CDN Isotopes (Pointe-Claire, Quebec, Canada); 6H-benzo[cd]pyrenone (B[cd]PYRO), 1,4-phenanthrenedione (1,4-PHED), and 1,4-benzo[c]phenanthrenequinone (1,4-B[c]PHEQ) from Chiron (Trondheim, Norway); 9-fluorenone (9-FLUO), 9,10-anthraquinone (9,10-ANTQ), and 1,9-benzanthrone (BANO) from Fluka (part of Sigma-Aldrich, St. Louis, MO); benzo[a]pyrene-1,6-dione (1,6-B[a]PYRD) and benzo[a]pyrene-7,8-dione (7,8-B[a]PYRD) from NCI (Bethesda, MD); 1,4-benzoquinone (1,4-BQ), chromone (CHRO), 9,10-phenanthrenequinone (9,10-PHEQ), 5,12-naphthacenequinone (5,12-NAPQ), benz[a]anthracene-7,12-dione (7,12-B[a]ANCQ), xanthone (XAN), 1,2-napthoquinone (1,2-NQ), 1,4-naphthoquinone (1,4-NQ), perinaphthenone (PNAPO), pyrene-4,5-dione (4,5-PYRD), aceanthrenequinone (AANEQ), benzo[a]fluorenone (B[a]FLUO), 1,2-acenaphthenequinone (1,2-ANAPQ), 2-ethyl-9,10-anthraquinone (2-ethANTQ), and cyclopenta[def]phenanthrenedione (CP[def]PHED) from Sigma-Aldrich. In total, 29 compounds were acquired including 24 nonlabeled OPAHs and 5 labeled OPAHs (Fig. 1).

Structures, abbreviations, and CAS numbers for OPAHs used in the described methods
All solvents used were at least Optima grade (Fisher Scientific, Pittsburg, PA) or equivalent. Whenever possible, nonchlorinated solvents were chosen preferentially to reduce chlorinated waste. However, attempts at using only nonchlorinated solvents for initial stock solutions resulted in incomplete dissolution for some OPAHs even after sonication. Therefore, all OPAH stock solutions were prepared from neat at approximately 130–1,100 μg mL−1 in a mixture of ethyl acetate/dichloromethane, 95:5 (v/v), to ensure solvation. Further dilutions and mixtures used only ethyl acetate. Owing to solubility constraints at low temperatures, all solutions were brought to room temperature and sonicated for at least 15 min prior to any dilution or further use. This step is crucial as some initial stock compounds recrystallized at 4 °C. No additional peaks were identified as co-eluters with other compounds of similar ion mass/charge ratios, so any impurities in OPAH standards were deemed negligible. Finally, 2-fluoro-fluorenone-13C was chosen as an instrumental internal standard for target OPAHs because of similar physicochemistry, excellent response on each method, and certified stability. Laboratory surrogates (deuterated OPAHs other than 2F-FLUO-13C) were used only in environmental samples, and data reported in this paper is not corrected for any potential laboratory losses unless otherwise stated.
Method parameters and optimization
LC-APCI/MS
For LC analysis, we used an Agilent (Agilent, Santa Clara, CA) 1100 liquid chromatography binary pump stack coupled to a single quadrupole MS (Agilent/1956B) with an APCI source (Agilent/G1947A). Each OPAH was optimized individually for MS acquisition parameters by performing flow injection analysis for fragmentor voltages ranging from 50 to 200 V with a step increase between injections of 10 V. The largest response was used for each respective compound. Target ions and fragmentor voltages used for extracted ion chromatograms (EICs) are listed in Table 1. Other final MS parameters used in this study include: gas temperature at 350 °C, vaporizer temperature at 400 °C, drying gas flow at 5 L min−1, nebulizer pressure at 50 psi, capillary voltage at 4,500 V, and the corona current at 10 μA (negative mode).
The nonaqueous mobile phase (B) consisted of dichloromethane/methanol (1:99, v/v), and the aqueous phase (A) was filtered through a Barnstead D7389 (Dubuque, IA). Mobile phases passed through the system at a flow rate of 0.5 mL min−1, for a total of 55 min. Initially, the percentage of A/B was 95:5 and increased to 70:30 in the first 7 min, ramped to 60:40 in the next 8 min, then to 25:75 in the next 10 min, and finally increased to 10:90 over 15 min. The final percentage of the nonaqueous phase was then held for 10 min and ramped back down to 95:5 over 5 min. Similar to a previous method [16], a phenyl column (150 × 3 mm length and diameter, particle size 3 μm, Agilent) was chosen to achieve separation for most of the OPAHs on the LC (Table 1). Column temperature was kept above ambient at 42 °C, and 20 μL was used for each sample injection.
GC-EI/MS
Experiments to improve previous GC-EI/MS OPAH methods were performed on an Agilent 7890A gas chromatograph coupled to a 5975C mass spectrometer (Agilent) under electron ionization (70 eV). Oven temperatures were evaluated and modified for a wider range of OPAHs from a previous study [17]. Briefly, temperature was ramped from 60 to 180 °C at 10 °C min−1, to 290 °C at 5 °C min−1, and to 310 °C at 25 °C min−1 and held at maximum temperature for 2 min for a total run time of 37.8 min. A DB5-MS column (30 m length, 0.25 mm inner diameter, 0.25 μm film thickness, Agilent) was used to separate target OPAHs, with a 1-mL min−1 flow rate using helium (>99.99 %) as a carrier gas. MS temperatures included the thermal auxiliary control set at 280 °C, the MS source at 230 °C, and the MS quadrupole at 150 °C. Quantitation ions are listed in Table 2, as well as qualification ions used to help identify OPAHs in complex matrices.
Inlet parameters were optimized to 3 mL min−1 septum purge, a purge to split flow of 20 mL min−1 at 0.75 min, an injection pulse pressure of 35 psi until 0.05 min, and an injection temperature of 300 °C. Sample volume was 1 μL. Initially, deactivated glass wool was used in 4-mm injection liners (Restek, Bellefonte, PA) to reduce nonvolatile components from environmental samples getting on the column. Over several consecutive runs with composite calibration solutions, it was observed that 14 compounds had relative standard deviations (RSDs) over 15 %, and 1,2-NQ was no longer identifiable. Further experiments using other inlet liners, glass wool, and glass liners with no filter in place were evaluated.
Method Calibration and Validation
Each method was calibrated using EICs for each OPAH. The calibration curve ranged from 5–5,000 ng mL−1 and included nine calibration points. To determine instrument detection limits (IDLs) and the limit of quantitation (LOQ), the lowest calibration standard that resulted in a signal to noise ratio greater than 3:1 was found for each compound and for each method. The lowest calibration standard was repeatedly analyzed (n ≥ 4), and a standard deviation was calculated for each compound. IDLs were determined by multiplying the resulting standard deviation estimates with the Student’s t value corresponding to the appropriate degree of freedom and 99 % confidence interval [27]. Tables 1 and 2 report the resulting IDLs for each compound and instrument method. LOQs were calculated by multiplying the IDL by five which was considered as a conservative estimate for reasonable quantitation.
Validation for each method utilized two test series using 500 ng mL−1 concentrations. First, several ethyl acetate aliquots spiked only with the internal standard 2F-FLUO-13C, served as blank controls. A second series of composite solutions with all target OPAHs including deuterated compounds was used to explore accuracy and repeatability as shown in Tables 1 and 2. To examine interday accuracy and precision, the set of calibration standards were run on at least 2 days.
OPAH stability
All deuterated compounds included in this study were certified as viable for a 3-year period, but because of limited commercial availability and novelty of OPAHs, not all target compounds had known expiration dates when purchased. To examine OPAH stability, a set of 15 aliquots at 500 ng mL−1 in ethyl acetate was kept at approximately 4 °C (±2 °C) with all native OPAHs listed in Tables 1 and 2. At the beginning of the experiment, 2F-FLUO-13C was added into each sample as an internal standard. A set of three aliquots were analyzed on days 0, 14, 32, 67, and 111 (LC-APCI/MS) or 116 (GC-EI/MS). Area counts were corrected for the internal standard, but analytes were not quantitated as calibration curves over the course of the study potentially changed with the aliquots, and if degradation had occurred, quantitating responses would have masked any temporal changes.
Method demonstration using environmental matrices
Environmental extracts were spiked with 2F-FLUO-13C at 500 ng mL−1 before instrumental analyses. Calibration check standards were run before and after each set of samples, and were considered successful if native OPAHs quantitated at ±30 % the true value for 90 % of compounds in each method. Qualitative analytes as noted Tables 1 and 2 were not included in the 90 % criteria. At a minimum, all extracts were run in triplicate on both methods.
NIST SRM
Four different environmental extracts were examined on each method as verification that OPAHs could be quantitated successfully in environmental matrices. Multiple matrices were chosen to exemplify a range of complexity including: urban dust, river sediment, diesel extract, and diesel particulate matter (NIST SRMs: 1649b [28], 1944 [29], 1975 [30], and 1650b [31], respectively). Extracts were analyzed from SRM material extracted and reported elsewhere [17]. In the original extraction, 9-FLUO-D8 and ANTQ-D8 were spiked as surrogates in each SRM extract and were solvent exchanged to ethyl acetate from hexane and stored until this analysis at 4 °C. Accelerated solvent extraction (ASE) and solid phase extraction cleanup of these SRMs were described in the original work [17]. The purpose of analyzing this set of extracts was not to characterize or compare previously reported values of NIST SRM material as previously described [17], but to demonstrate the applicability of our two independent instrumental methods for environmental samples, and to compare values between the LC and GC generated data.
Standard addition on Portland Harbor superfund passive sampler extract
To reduce uncertainty of matrix effects between instruments and to examine our choice for internal standard, we quantified an environmental extract from Portland Harbor superfund, OR using both internal standard and standard addition quantitation. A silicone strip was cut to approximately 3.2 × 99 cm from purchased commercial material (Stockwell Elastomerics Inc., Philadelphia, PA) and was exposed for 27 days in the Willamette River, mile 3.5 West, within Portland Harbor superfund, OR from 3 to 30 September 2010. Once recovered, the silicone was rinsed two times with ultra-pure water, and then with isopropyl alcohol to remove excess water and stored at −20 °C until extraction. To track potential losses in the laboratory, the sample was spiked with 2me-1,4-NQ-D8 and 9-FLUO-D8 at 500 ng mL−1, extracted twice with ethyl acetate, and finally reduced under filtered nitrogen to 1 mL. Extractions were performed on an S-500 orbital shaker (VWR, Radnor, PA) for a total period of approximately 14 h, and solvent reduction was performed by using closed cell reduction devices (Zymark, Hopkinton, MA). Each sample was stored at 4 °C until analysis.
Standard addition samples consisted of four dilutions. In each dilution, 100 μL of Portland Harbor extract was placed into a 250-μL chromatography vial. For the initial dilution, an additional 100 μL of ethyl acetate was added to make a total volume of 200 μL. In each subsequent addition, 10, 20, or 30 μL of a 1,000-ng mL−1 stock of target OPAHs was added, corresponding to an equivalent of 50, 100, or 150 ng mL−1, respectively. The addition scheme was no more than ten times the average response of OPAHs in either method, with most responses within a factor of 3. Estimates of variability for standard addition values incorporated the standard deviation of both the slope and intercept of each regression model as described in Bader 1980 [32].
Results and discussion
Method optimization
LC-APCI/MS
APCI was found to be sensitive for both ketone and diketone OPAHs, whereas some compounds produced no ions with electron ionization similar to a detailed investigation of HPLC-MS ionization sources [15]. Once initial MS parameters were set, methanol and water were used as mobile phases similar to other papers [15, 16, 23], but some OPAHs had lower than expected responses. Therefore, several dopants were assessed including formic acid and ammonium formate. However, dramatic improvement was only observed with dichloromethane. Most notable was the effect on 5,12-NAPQ (Fig. 2a), where peak shape improved and the peak response increased about 5-fold. Enhanced responses of dichloromethane dopant in the mobile phase have been found for other structurally similar aromatic compounds as well [33]. Improvements from dichloromethane may be due to increases in solvation of some OPAHs in the LC system, or from enhancement of ionization efficiency by stabilizing the charge. Enhancement of ionization efficiency has been shown to be greatly affected by different dopants or solvents for APCI ionization [11, 33]. Although positive mode was more sensitive for a subset of the compound list, negative mode provided better responses over the entire target compound list. One benefit of running solely in negative mode is reduced analysis time compared with running samples twice in negative and positive mode [23]. Additionally, if an LC method is run with dual positive/negative mode it might suffer from reduced sensitivity depending on the number of analytes in the quantitation window. The sensitivity of our LC-APCI/MS method (2.6–26 ng mL−1) is comparable or better than OPAHs of another method with similar ionization parameters (0.10–250 ng mL−1) [23].

Results of analytical investigations of OPAHs: a) 5-fold signal improvement after adding 1 % dichloromethane (DCM) to LC-MS solvent system for 5,12-napthacenequinone, b) 400-fold enhanced peak response for 1,4-benzoquinone with an improved temperature profile in the GC-MS method, c) 2 to 6-fold reduction in the RSDs between injection liner configurations, d) stability of acenaphthenequinone responses after internal standard normalization (area of target over the area of the internal) on the GC-EI/MS. Error bars correspond to instrumental variation from Table 2
GC-EI/MS
Even with the addition of eight more target compounds compared with a similar GC-EI/MS method [17], we were able to increase resolution and sensitivity, which resulted in lower detection limits (0.18–36 compared with 0.5–50 ng mL−1). Specifically, a lower starting temperature of 60 °C (vs. 70 °C) increased the response by over 400-fold for 1,4-BQ, (Fig. 2b) while still being able to acquire slower eluters like 1,4-B[c]PHEQ, 5,12-NAPQ, 7,12-B[a]ANCQ, and B[cd]PYRO in under 40 min (Fig. 3). In addition, better separation was achieved by slowing the rate of oven temperature increase between 1,4-PHED and 9,10-ANTQ [17]. Improvements are also likely due to pulsed splitless injection over previous nonpulsed splitless injection [13, 14, 17] by getting compounds in column more efficiently. Pulsed splitless injection has been found to improve recoveries in organophosphorus pesticides with physiochemical properties similar to the OPAHs studied here [34].

Chromatograms from both instruments for NIST SRM 1650b (diesel particulate matter). The GC chromatogram is displayed as a total ion chromatogram, whereas the LC figure is displayed as EICs. The vertical breaks (dashed lines) in the LC chromatograph represent EICs from one or more ions during the run. The LC chromatogram has been scaled evenly between breaks in response (y-axis) and time (x-axis)
Glass wool was thought to be a source of variability for OPAHs due to surface chemistry between the ketone groups of our target compounds and active sites in the wool created in preparing the liner for analysis. Figure 2c shows the RSDs from five or more consecutive runs of calibration aliquots. Using no inlet packing decreased the average RSD for all OPAHs dramatically (8.5 ± 1.4 vs. 21 ± 2.5 % on 95 % confidence intervals) compared with glass wool inlets. CarboFrit™ liners improved repeatability over using no inlet packing (6.6 ± 0.7 % on 95 % confidence intervals). There was a 2- to 6-fold reduction in RSDs between CarboFrit™ liners and glass wool, but the most dramatic change occurred for 1,2-NQ. Using glass wool, we were not able to consistently identify or quantify 1,2-NQ (Fig. 2c). Similar accuracy and precision was also observed for deactivated dimpled liners with no inlet packing compared with CarboFrit™ filters, so all additional experiments were performed without glass wool.
Method validation
LC-APCI/MS
Each compound was calibrated with a linear calibration model with a 9-point correlation coefficient (R 2) of 0.99 or better. Table 1 lists the accuracy of individual compounds using the lowest detectable calibration standard as well as the IDL and LOQ calculated. Other APCI methods have slightly lower IDLs (sub-parts per billion), but use less conservative estimates based only on signal-to-noise ratios [35]. Spiked replicates show good accuracy over multiple days (n = 6 repeated over 3 days unless otherwise noted), with 18 compounds within 20 % of the true value, and 11 were within 10 % (Table 1). OPAHs outside of 20 % accuracy include: 1,4-BQ (52 %), 9-FLUO (>100 %), AANEQ (35 %), 2me-1,4-NQ-D8 (56 %), and 9-FLUO-D8 (25 %). Accuracy of our method compares well or better than that of another LC method which reported 29–87 % accuracy for nine OPAHs [35]. It is unclear why 9-FLUO has unpredictable responses on our LC-APCI/MS system despite an effective calibration, but this discrepancy underscores the necessity of validating new compounds. Concerning repeatability, 20 compounds showed acceptable variability below 20 % RSD, most of which (14 OPAHs), had RSDs below 15 % (Table 1). Our repeatability (interday RSDs, 4.3–28 %) is comparable or better than that of another similar method [15] and with a method using tandem mass spectrometry (interday RSDs, 4.1–17.7 %) [35]. Other compounds besides 9-FLUO that showed variability above 20 % RSD were CP[def]PHED (28 %), 1,4-ANTQ (28 %), and AANE (31 %). Additionally, XAN, PNAPO, and CHRO were unable to be ionized using our APCI source. It is important to note that the chromatographic separation described here should be effective for LC-APCI/MS-MS, thus expanding the ability to incorporate more OPAHs as environmental or toxicological data become available.
GC-EI/MS
One unexpected observation during GC OPAH calibration was non-linearity over concentrations within one or two orders of magnitude. Nonlinearity was observed despite three separate calibration solutions prepared by two chemists (exemplary compound, 7,12-B[a]ANTQ, shown in Fig. S1, Electronic supplementary material). As nonlinearity occurs throughout the calibration curve, detector fatigue is not responsible nor is cross-contribution likely [36] as there are no other ions detected to interfere with quantitation in clean matrices from the internal standard, 2F-FLUO-13C. Quadratic curves had an average linear coefficient (R 2) over 0.99 for all compounds including those OPAHs that were listed as not quantifiable in a previous study [17]. Variability between each calibration set is reduced by over 40 % when modeled as quadratic curves compared with linear models using the same data. Though linear models would be preferential, the range for each calibration curve would have to be reduced to just over one order of magnitude (50 to 750 ng mL−1) for the curve to be considered linear (>0.99 R 2) for 1,4-ANTQ, CP[def]PHEO, 9,10-PHEQ, and 4,5-PYRD, yet would still be below 0.99 for nearly half of the OPAHs in this method (11 out of 26). The result of such a small acceptable calibration range would make quantitation impractical for environmental samples. Therefore, quadratic calibration curves were used for all GC-EI/MS analyses.
All compounds were within ±15 % of the true value, and had less than 10 % RSD on the GC-EI/MS method except for 7,8- and 1,6-B[a]PYRD which did not ionize well under the GC-EI/MS method (Table 2). The accuracy observed is better than that of the only other GC-EI/MS method published with more ketone-OPAHs (20 % accuracy on one calibration point) [19]. Overall precision is excellent, but comparisons of precision to other methods for solvent solutions is difficult since previous GC-MS methods are either qualitative [26], or precision is based using laboratory and instrument variability that are sample specific [13, 14, 17, 19, 21, 22]. Despite 1,2-NQ, 4,5-PYRD, and 9,10-PHEQ performing well on the initial calibration and verification, these three compounds showed considerable variability over time and subsequent analyses. It is unclear why these three compounds show either reduced or no response, but evidence from repeated analyses supports active surface chemistry in the injection port as a potential source of variability. As variability for these compounds typically only occurs with interday injections, experiments examining the cause of reduced responses of these OPAHs should take place over the course of multiple days. For this reason, 1,2-NQ, 4,5-PYRD, and 9,10-PHEQ are considered for qualitative purposes on the GC-EI/MS method for environmental demonstrations described below.
OPAH stability
During the course of the OPAH stability experiment (111 and 116 days, LC and GC, respectively), all OPAHs were stable with one possible exception, 1,2-ANAPQ, which showed evidence that responses decreased slightly over time (Fig. S2, Electronic supplementary material) or at least were variable on both instrumental methods. Subsequent analyses of several old and newly prepared stock standards showed no response decrease for 1,2-ANAPQ even after a full year (Fig. 2d). Specific causes of reduced or variable responses for 1,2-ANAPQ during the stability study remains unknown, but variability for this specific OPAH has been reported elsewhere [13]. The variability of this compound underscores the necessity of running calibration verification samples before and after each batch to monitor system stability on either instrumental method.
Method demonstration and comparison
NIST SRM instrument comparison
All standard reference materials were able to be successfully quantitated for OPAHs on both instruments and compares well to other published results (Table S1, Electronic supplementary material). Figure 3 represents chromatograms using each method for diesel particulate matter (SRM 1650b). An unexpected result from this diesel particulate sample was the large response of PNAPO, as well as the presence of XAN (Fig. 3), neither of which has been previously reported for this SRM at the time of this publication. Suggestive evidence of 1,6-/7,8-B[a]PYRD was seen in diesel and river extracts, although as the values are at or below the LOQ of 55 ng mL−1, this compound is yet to be confirmed in these samples, and is not included in Fig. 3. Reports of PNAPO, XAN, or 1,6-B[a]PYRD have been reported in other environmental samples, including aerosol particulate matter [23, 35].
Figure 4 displays the average quantitated results of comparable OPAHs in all four matrices from each chromatographic system. Concentrations are prior to any back-calculations from the weight of starting material, dilutions, or surrogate correction so that comparisons between instruments are more easily identified. For individual OPAHs, there is excellent agreement (<20 % difference) for 9-FLUO, CP[def]PHEO, B[a]FLUO, and 5,12-NAPQ between the instrumental methods across all matrices tested. Furthermore, there are less than 30 % differences for 9,10-ANTQ-D8 and 7,12-B[a]PHEQ. However, wide discrepancies exist for 2-ethANTQ, which shows poor agreement between the LC-APCI/MS and GC-EI/MS runs (>100 % difference). While overall concentrations in urban dust (SRM 1649b) differ by less than 3 % (GC-EI/MS, 2,513 ng mL−1; LC-APCI/MS, 2,435 ng mL−1), there is only reasonable agreement between the total sum of OPAHs between the two instruments (<50 % difference) because of differences with a few OPAHs as discussed above. Reasons for specific discrepancies between compounds could be due to matrix components that affect quantitation differently on each method.

Comparison of GC and LC data for SRM-extracted material. GC data are presented as the average and one standard deviation of five runs over 2 days, whereas LC data are from three runs over 2 days. Only comparable data are represented here, target compounds that were unique to each system are not shown. No data has been corrected for recovery to compare directly differences between chromatograph systems. Asterisk, extract was diluted by 1/3. Accent symbol, extract was diluted by 1/10
Evaluation of quantitation strategies using Portland Harbor superfund passive sampler extract
All OPAHs that were identified in the original extract using internal standard (IS) quantitation were able to be successfully identified in the series of standard additions (SA). In total, 12 OPAH compounds are identified between methods, with a total of 10 from the LC-APCI/MS, and 8 from the GC-EI/MS (Fig. 5). While all of the data presented in Fig. 5 is above instrumental detection limits, eight of ten standard addition values for the LC-APCI/MS method are below LOQs. No GC-EI/MS data shown in Fig. 5 are below LOQs.

Comparison of standard addition and internal standard quantitation for both methods. Standard deviation for all values are the result of replication (n = 3) on the instrument, with the addition of standard addition variability incorporating slope and intercept standard deviations from the linear regression performed for each analyte. Internal standard quantitation below LOQ is marked with a circle. If the lowest dilution of the standard addition series was below the LOQ, the final value for that regression was also marked with a circle. Abbreviations: LC liquid chromatography, GC gas chromatography, IS internal standard quantitation, SA standard addition quantitation, LOQ limit of quantitation (five times the IDL). Asterisk, 9,10-ANTQ and 1,4-PHEQ co-elute on the LC-APCI/MS method but are separated on the GC-EI/MS method. Values for the GC represent 9,10-ANTQ only
Although conclusions for LC-APCI/MS data are difficult to make considering much of the data is below the LOQ, there are interesting comparisons between quantitation methods. For BANO, both quantitation methods result in values that differ by less than 5 % (SA, 67 ng mL−1; IS, 70 ng mL−1), indicating that there is no interference issues and excellent agreement between quantitation methods. Many other OPAHs have good agreement (differ by 30 % or less) between quantification strategies include AANEQ (29 %), 9,10-ANTQ+1,4-PHED (15 %), CP[def]PHEO (22 %), B[a]FLUO (5 %), 5,12-NAPQ (29 %), and 7,12-B[a]ANCQ (1 %) even though they are at or below LOQ. B[cd]PYRO does not show good agreement between quantitation methods (SI, 13 ng mL−1; IS, 110 ng mL−1). One likely explanation for this discrepancy is matrix enhancement of the 254 m/z ion, which would make the internal standard response higher over that of standard addition. Similar LC-MS matrix enhancement has been described in previous methods [37]. Enhancement is also seen for 7,8/1,6-B[a]PYRD while suppression is shown for 2-ethANTQ, both below the LOQ (Fig. 5). The discrepancy for 2-ethANTQ could be ion suppression which is also common in LC-MS data [37]. Overall, there is excellent agreement between both quantitation strategies with seven out of ten compounds differing by less than 30 %. As matrix interferences differ from sample to sample, SA quantitation is not usually employed. In this instance, IS quantitation seems reasonable for all but a few OPAHs. Less onerous strategies to improve quantitation accuracy could employ the use of more laboratory surrogates. Recoveries of both 2me-1,4-NQ-D8 and 9-FLUO-D8 were within 30 % of the true value, suggesting that recovery correction might only account for a partial resolution of discrepancies.
For the GC-EI/MS, quantitation methods had good agreement (differed by less than 30 %) for 9,10-ANTQ (11 %), B[a]FLUO (6 %), and FLUO (15 %). CP[def]PHEO differed by 35 % between SA and IS estimates. For the other four OPAHs, larger discrepancies exist with SA estimates higher than IS values for the individual OPAHs. The consistent trend on the GM-EI/MS instrument warranted further investigation as passing calibration check standards were analyzed prior to, and after this series of samples, and no obvious signs of suppression were present. The apparent suppression could have been due to either the silicone in the passive sampling device, or from interferences from the deployment in Portland Harbor itself. After simulating silicone background by extracting a nondeployed silicone sampler, results from both standard addition and internal standard quantitation suggest that 1,4-B[c]PHEQ, 7,12-B[a]ANCQ, 5,12-NAPQ, and BANO were indeed suppressed (Fig. S3, Electronic supplementary material). Ongoing work in our laboratory is focused on reducing silicone background for passive sampling devices through additional solvent precleaning prior to deployment, and through surrogate correction experiments. Recovery of both 2me-1,4-NQ-D8 and 9-FLUO-D8 was over 90 %, so these surrogates would not have corrected for 1,4-B[c]PHEQ, 7,12-B[a]ANCQ, 5,12-NAPQ, and BANO that were suppressed because of silicone background.
Conclusions
Improved sensitivities, optimization strategies, and the successful validation of two independent methods containing a large number of OPAHs were described in this work. By utilizing both systems, 24 target OPAHs were able to be quantified in addition to 4 deuterated compounds, with 19 compounds conserved in both methods. Despite surface chemistry difficulties in the injection port with some OPAHs (especially those compounds containing vicinal quinones), the GC-MS method is preferred over the LC-MS method for those compounds that were able to be successfully quantified. Obvious benefits include the additional ions used for identification purposes on the GC-MS method, which drastically reduce the likelihood of false positives that may be present in complex mixtures using a single quadrupole LC-MS. Additionally, the interday variability of even clean standards tended to be less using our GC-MS method. Standard addition experiments showed potential suppression from the environmental sample that was later identified as coming from the silicone of a passive sampling device. Currently, there are very few labeled OPAHs commercially available, and this work highlights the need for improved laboratory surrogates for OPAHs. Ultimately, we hope separation and quantitation strategies provided in this work will provide improved sensitivity, accuracy, and reproducibility for OPAH quantitation on LC or GC mass spectrometry instrumentation.
References
Lundstedt S, White PA, Lemieux CL, Lynes KD, Lambert LB, Oberg L, Haglund P, Tysklind M (2007) Ambio 36:475–485
Wischmann H, Steinhart H (1997) Chemosphere 35:1681–1698
Andersson BE, Lundstedt S, Tornberg K, Schnurer Y, Oberg LG, Mattiasson B (2003) Environ Toxicol Chem 22:1238–1243
Cerniglia CE (1997) J Ind Microbiol Biotechnol 19:324–333
Kanaly RA, Harayama S (2000) J Bacteriol 182:2059–2067
Kochany J, Maguire RJ (1994) Sci Total Environ 144:17–31
Lampi MA, Gurska J, McDonald KIC, Xie FL, Huang XD, Dixon DG, Greenberg BM (2006) Environ Toxicol Chem 25:1079–1087
Yu HT (2002) J Environ Sci Health Part C-Environ Carcinog Ecotoxicol Rev 20:149–183
Bamforth SM, Singleton I (2005) J Chem Technol Biotechnol 80:723–736
Hartnik T, Norli HR, Eggen T, Breedveld GD (2007) Chemosphere 66:435–443
Walgraeve C, Demeestere K, Dewulf J, Zimmermann R, Van Langenhove H (2010) Atmos Environ 44:1831–1846
Gold A (1975) Anal Chem 47:1469–1472
Bandowe BAM, Wilcke W (2010) J Environ Qual 39:1349–1358
Albinet A, Leoz-Garziandia E, Budzinski H, ViIlenave E (2006) J Chromatogr A 1121:106–113
Grosse S, Letzel T (2007) J Chromatogr A 1139:75–83
Letzel T, Poschl U, Wissiack R, Rosenberg E, Grasserbauer M, Niessner R (2001) Anal Chem 73:1634–1645
Layshock JA, Wilson G, Anderson KA (2010) Environ Toxicol Chem 29:2450–2460
Durant JL, Busby WF, Lafleur AL, Penman BW, Crespi CL (1996) Mutat Res-Genet Toxicol 371:123–157
Allen JO, Dookeran NM, Taghizadeh K, Lafleur AL, Smith KA, Sarofim AF (1997) Environ Sci Technol 31:2064–2070
Manzano C, Hoh E, Simonich SLM (2012) Environ Sci Technol 46:7677–7684
Niederer M (1998) Environ Sci Pollut Res 5:209–216
Shen G, Tao S, Wang W, Yang Y, Ding J, Xue M, Min Y, Zhu C, Shen H, Li W, Wang B, Wang R, Wang W, Wang X, Russell AG (2011) Environ Sci Technol 45:3459–3465
Delhomme O, Millet M, Herckes P (2008) Talanta 74:703–710
Ahmed S, Kishikawa N, Ohyama K, Maki T, Kurosaki H, Nakashima K, Kuroda N (2009) J Chromatogr A 1216:3977–3984
Mirivel G, Riffault V, Galloo JC (2010) Anal Bioanal Chem 397:243–256
Adelhelm C, Niessner R, Poschl U, Letzel T (2008) Anal Bioanal Chem 391:2599–2608
EPA (1995) 40 CFR Part 136, Appendix B, Revision 1.11. p. 343–345
NIST (2009) Certificate of Analysis, Standard Reference Material 1649b, Urban Dust
NIST (2011) Certificate of Analysis, Standard Reference Material 1944, River Sediment
NIST (2008) Certificate of Analysis, Standard Reference Material 1975, Diesel Particulate Matter Extract
NIST (2006) Certificate of Analysis, Standard Reference Material 1650b, Diesel Particulate Matter
Bader M (1980) J Chem Educ 57:703
Takats Z, Vekey K (1998) European Mass Spectrometry 4:365–370
Wylie PL, Uchiyama K (1996) J AOAC Int 79:571–577
Lintelmann J, Fischer K, Matuschek G (2006) J Chromatogr A 1133:241–247
Chen BG, Chang CD, Wang CT, Chen YJ, Chang WT, Wang SM, Liu RH (2008) J Am Soc Mass Spectrom 19:598–608
Kruve A, Auling R, Herodes K, Leito I (2011) Rapid Commun Mass Spectrom 25:3252–3258
Acknowledgments
This project was supported in part by award number P42 ES016465 and the associated Analytical Chemistry Facility Core, P30 ES000210 and R21 ES020120 from the National Institute of Environmental Health Sciences. Steven O’Connell was supported in part by NIEHS Training Grant Fellowship T32ES007060-32 from the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIEHS or the National Institutes of Health. The authors would also like to thank Alan Bergmann for his help with the stability study.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 835 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
O’Connell, S.G., Haigh, T., Wilson, G. et al. An analytical investigation of 24 oxygenated-PAHs (OPAHs) using liquid and gas chromatography–mass spectrometry. Anal Bioanal Chem 405, 8885–8896 (2013). https://doi.org/10.1007/s00216-013-7319-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-013-7319-x