
Abstract
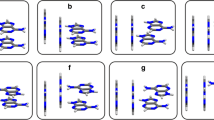
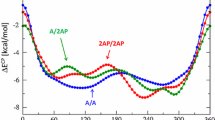
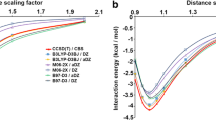
To elucidate the physical origin of the preference of nucleic acid bases for stacking over hydrogen bonding in water, Monte Carlo simulations were performed starting from Watson–Crick structures of the adenine–thymine, adenine–uracil and guanine–cytosine base pairs, as well as from the Hoogsteen adenine–thymine base pair, in clusters comprising 400 and 800 water molecules. The simulations employed a newly implemented Metropolis Monte Carlo algorithm based on the extended cluster approach. All simulations reached stacked structures, confirming that such structures are preferred over the hydrogen-bonded Watson–Crick and Hoogsteen base pairs. The Monte Carlo simulations show the complete transition from hydrogen-bonded base pairs to stacked structures in the Monte Carlo framework. Analysis of the average energies shows that the preference of stacked over hydrogen-bonded structures is due to the increased water–base interaction in these structures. This is corroborated by the increased number of water–base hydrogen bonds in the stacked structures.




Similar content being viewed by others
References
Schultze P, Smith FW, Feigon J (1994) Refined solution structure of the dimeric quadruplex formed from the Oxytricha telomeric oligonucleotide d(GGGGTTTTGGGG). Structure 2:221–233
Yakovchuk P, Protozanova E, Frank-Kamenetskii MD (2006) Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res 34:564–574
Nakano NI, Igarashi SJ (1970) Molecular interactions of pyrimidines, purines, and some other heteroaromatic compounds in aqueous media. Biochemistry 9:577–583
Ts’o POP, in: P.O.P. Ts’o (ed) (1974) Basic principles in nucleic acid chemistry. Academic Press, London, p 453
Sinnokrot MO, Sherrill CD (2004) Substituent effects in π–π interactions: sandwich and T-shaped configurations. J Am Chem Soc 126:7690–7697
Sinnokrot MO, Sherrill CD (2004) Highly accurate coupled cluster potential energy curves for the benzene dimer: sandwich, T-shaped, and parallel-displaced configurations. J Phys Chem A 108:10200–10207
Hill JG, Platts JA, Werner H-J (2006) Calculation of intermolecular interactions in the benzene dimer using coupled-cluster and local correlation methods. Phys Chem Chem Phys 8:4072–4078
Ringer AL, Sinnokrot MO, Lively RP, Sherrill CD (2006) The effect of multiple substituents on sandwich and T-shaped pi–pi interactions. Chem Eur J 12:3821–3828
Sinnokrot MO, Sherrill CD (2006) High-accuracy quantum mechanical studies of pi–pi interactions in benzene dimers. J Phys Chem A 110:10656–10668
Arnstein SA, Sherrill CD (2008) Substituent effects in parallel-displaced pi–pi interactions. Phys Chem Chem Phys 10:2646–2655
Ringer AL, Sherrill CD (2009) Substituent effects in sandwich configurations of multiply substituted benzene dimers are not solely governed by electrostatic control. J Am Chem Soc 131:4574–4575
Takatani T, Sherrill CD (2007) Performance of spin-component-scaled Møller-Plesset theory (SCS-MP2) for potential energy curves of noncovalent interactions. Phys Chem Chem Phys 9:6106–6114
Sherrill CD, Takatani T, Hohenstein EG (2009) An assessment of theoretical methods for nonbonded interactions: comparison to complete basis set limit coupled-cluster potential energy curves for the benzene dimer, the methane dimer, benzene-methane, and benzene-H2S. J Phys Chem A 113:10146–10159
Vazquez-Mayagoitia A, Sherrill CD, Apra E, Sumpter BG (2010) An assessment of density functional methods for potential energy curves of nonbonded interactions: the XYG3 and B97-D approximations. J Chem Theor Comput 6:727–734
Jurečka P, Nachtigall P, Hobza P (2001) RI-MP2 calculations wih extended basis sets—a promising tool for study of H-bonded and stacked DNA base pairs. Phys Chem Chem Phys 3:4578–4582
Hobza P, Sponer J (2002) Toward true DNA base-stacking energies: MP2, CCSD(T) and complete basis set calculations. J Am Chem Soc 124:11802–11808
Leininger ML, Nielsen IMB, Colvin ME, Janssen CL (2002) Accurate structures and binding energies for stacked uracil dimers. J Phys Chem A 106:3850–3854
Dąbkowska I, Jurecka P, Hobza P (2005) On geometries of stacked and H-bonded nucleic acid base pairs determined at various DFT, MP2, and CCSD(T) levels up to the CCSD(T)/complete basis set level. J Chem Phys 122:204322
Pitoňák M, Riley KE, Neogrády P, Hobza P (2008) Highly accurate CCSD(T) and DFT-SAPT stabilization energies of H-bonded and stacked structures of the uracil dimer. ChemPhysChem 9:1636–1644
Czyznikowska Z (2009) On the importance of electrostatics in stabilization of stacked guanine-adenine complexes appearing in B-DNA crystals. J Mol Struct (Theochem) 895:161–167
Sivanesan D, Babu K, Gadre SR, Subramanian V, Ramasami T (2000) Does a stacked DNA base pair hydrate better than a hydrogen-bonded one?: an ab initio study. J Phys Chem A 104:10887–10894
Sivanesan D, Sumathi I, Welsh WJ (2003) Comparative studies between hydrated hydrogen bonded and stacked DNA base pair. Chem Phys Lett 367:351–360
Kabeláč M, Zendlová L, Řeha D, Hobza P (2005) Potential energy surfaces of an adenine–thymine base pair and its methylated analogue in the presence of one and two water molecules: molecular mechanics and correlated ab Initio study. J Phys Chem B 109:12206–12213
Dkhissi A, Blossey R (2008) Metahybrid density functional theory and correlated ab initio studies on microhydrated adenine–thymine base pairs. J Phys Chem B 112:9182–9186
Friedman RA, Honig B (1995) A free-energy analysis of nucleic-acid base stacking in aqueous-solution. Biophys J 69:1528–1535
Danilov VI, van Mourik T, Kurita N, Wakabayashi H, Tsukamoto T, Hovorun DM (2009) On the mechanism of the mutagenic action of 5-bromouracil: a DFT study of uracil and 5-bromouracil in a water cluster. J Phys Chem A 113:2233–2235
van Mourik T, Danilov VI, Dailidonis VV, Kurita N, Wakabayashi H, Tsukamoto T (2010) A DFT study of uracil and 5-bromouracil in nanodroplets. Theor Chem Acc 125:233–244
Palafox MA, Iza N, de la Fuente M, Navarro R (2009) Simulation of the first hydration shell of nucleosides D4T and thymidine: structures obtained using MP2 and DFT methods. J Phys Chem B 113:2458–2476
Tajkhorshid E, Jalkanen KJ, Suhai S (1998) Structure and vibrational spectra of the zwitterion l-alanine in the presence of explicit water molecules: a density functional analysis. J Phys Chem B 102:5899–5913
Han W-G, Jalkanen KJ, Elstner M, Suhai S (1998) Theoretical study of aqueous N-acetyl-l-alanine N′-methylamide: structures and Raman, VCD, and ROA spectra. J Phys Chem B 102:2587–2602
Frimand K, Bohr H, Jalkanen KJ, Suhai S (2000) Structures, vibrational absorption and vibrational circular dichroism spectra of image-alanine in aqueous solution: a density functional theory and RHF study. Chem Phys 255
Jalkanen KJ, Nieminen RM, Frimand K, Bohr J, Bohr H, Wade RC, Tajkhorshid E, Suhai S (2001) A comparison of aqueous solvent models used in the calculation of the Raman and ROA spectra of image-alanine. Chem Phys 265:125–151
Jalkanen KJ, Würtz Jürgensen V, Claussen A, Rahim A, Jensen GM, Wade RC, Nardi F, Jung C, Degtyarenko IM, Nieminen RM, Herrmann F, Knapp-Mohammady M, Niehaus TA, Frimand K, Suhai S (2006) Use of vibrational spectroscopy to study protein and DNA structure, hydration, and binding of biomolecules: a combined theoretical and experimental approach. Int J Quant Chem 106:1160–1198
Jürgensen VW, Jalkanen K (2006) The VA, VCD, Raman and ROA spectra of tri-l-serine in aqueous solution. Phys Biol 3:S63
Losada M, Xu Y (2007) Chirality transfer through hydrogen-bonding: experimental and ab initio analyses of vibrational circular dichroism spectra of methyl lactate in water. Phys Chem Chem Phys 9:3127–3135
Deplazes E, van Bronswijk W, Zhu F, Barron L, Ma S, Nafie L, Jalkanen K (2008) A combined theoretical and experimental study of the structure and vibrational absorption, vibrational circular dichroism, Raman and Raman optical activity spectra of the l-histidine zwitterion. Theor Chem Acc 119:155–176
Jalkanen KJ, Degtyarenko IM, Nieminen RM, Cao X, Nafie LA, Zhu F, Barron LD (2008) Role of hydration in determining the structure and vibrational spectra of l-alanine and N-acetyl l-alanine N′-methylamide in aqueous solution: a combined theoretical and experimental approach. Theor Chem Acc 119:191–210
Mukhopadhyay P, Zuber G, Beratan DN (2008) Characterizing aqueous solution conformations of a peptide backbone using Raman optical activity computations. Biophys J 95:5574–5586
van Mourik T (2006) Density functional theory reveals an increase in the amino 1H chemical shift in guanine due to hydrogen bonding with water. J Chem Phys 125:191101
Degtyarenko IM, Jalkanen KJ, Gurtovenko AA, Nieminen RM (2007) l-Alanine in a droplet of water: a density-functional molecular dynamics study. J Phys Chem B 111:4227–4234
Sykes MT, Levitt M (2007) Simulations of RNA base pairs in a nanodroplet reveal solvation-dependent stability. PNAS 104:12336–12340
Stewart JJP (2007) Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J Mol Model 13:1173–1213
Dang LX, Kollman PA (1990) Molecular dynamics simulations study of the free energy of association of 9-methyladenine and 1-methylthymine bases in water. J Am Chem Soc 112:503–507
Danilov VI, Dailidonis VV, van Mourik T, Früchtl HA (2011) A study of nucleic acid base-stacking by the Monte Carlo method: extended cluster approach. Centr Eur J Chem 9:720–727
Danilov VI, Dailidonis VV, van Mourik T, Früchtl HA (2011) The study of the nucleic acid base-stacking by Monte Carlo method: extended cluster approach. J Biomol Struct Dyn 28:1140–1141 (Book of Abstracts: Albany 2011, Conversation 17, June 14–18 2011). http://www.jbsdonline.com/The-Study-of-the-Nucleic-Acid-Base-Stacking-by-Monte-Carlo-Method-Extended-Cluster-Approach-p18003.html
Cieplak P, Kollman PA (1988) Calculation of the free-energy of association of nucleic-acid bases in vacuo and water solution. J Am Chem Soc 110:3734–3739
Danilov VI, Tolokh IS (1984) Nature of the stacking of nucleotide bases in water—a Monte Carlo simulation. J Biomol Struct Dyn 2:119–130
Danilov VI, Tolokh IS (1984) On the role of hydrophobic groups in nucleotide base stacking—a Monte-Carlo study of hydration of thymine dimers. FEBS Lett 173:347–350
Danilov VI, Tolokh IS, Poltev VI (1984) A Monte Carlo study of the hydration of thymine molecule associates. FEBS Lett 171:325–328
Danilov VI, Tolokh IS, Poltev VI, Malenkov GG (1984) Nature of the stacking interaction of nucleotide bases in water—a Monte-Carlo study of the hydration of uracil molecule associates. FEBS Lett 167:245–248
Pohorille A, Burt SK, MacElroy RD (1984) Monte Carlo simulation of the influence of solvent on nucleic acid base associations. J Am Chem Soc 106:402–409
Pohorille A, Pratt LR, Burt SK, MacElroy RD (1984) Solution influence on biomolecular equilibria—nucleic-acid base associations. J Biomol Struct Dyn 1:1257–1280
Danilov VI, Tolokh IS (1985) Nature of the stacking of nucleic acid bases in water: a Monte Carlo study. J Mol Struct (THEOCHEM) 123:109–119
Danilov VI (1986) Application of the Monte Carlo method for studying the hydration of molecules: base stacking. J Mol Struct (THEOCHEM) 138:239–242
Danilov VI (1986) In: Trinajstic N (ed) Mathematics and computational concepts in chemistry. Elis Horwood Lmd, Chichester, p 48
Lee JK, Barker JA, Abraham FF (1973) Theory and Monte Carlo simulation of physical clusters in the imperfect vapor. J Chem Phys 58:3166–3180
Abraham FF (1974) Monte Carlo simulation of physical clusters of water molecules. J Chem Phys 61:1221–1225
Abraham FF, Mruzik MR, Pound GM (1976) The thermodynamics and structure of hydrated halide and alkali ions. Faraday Discuss Chem Soci 61:34–47
Mruzik MR, Abraham FF, Schreiber DE, Pound GM (1976) A Monte Carlo study of ion-water clusters. J Chem Phys 64:481–491
Mruzik MR (1977) A Monte Carlo study of water clusters. Chem Phys Lett 48:171–175
Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E (1953) J Chem Phys 21:1087–1092
Poltev VI, Grokhlina TI, Malenkov GG (1984) Hydration of nucleic-acid bases studied using novel atom–atom potential functions. J Biomol Struct Dyn 2:413–429
González-Jiménez E, Castro-Valdez I, López-Apresa E, Filippov SV, Teplukhin AV, Poltev VI (1999) Computer study of the role of hydration in the accuracy of nucleic acid biosynthesis. J Mol Struct (Theochem) 493:301–308
González E, Cedeño FI, Teplukhin AV, Malenkov GG, Poltev VI (2000) Refinement of methodology for simulation of nucleic acid hydration. Rev Mex Fis 46:142–147
Poltev VI, Deryabina AS, González E, Grokhlina TI (2002) Interactions between nucleic acid bases. New parameters of potential functions and new energy minima. Biofizika 47:996–1004
Zhurkin VB, Poltev VI, Florentev VL (1980) Atom-atom potential functions for conformational calculations of nucleic-acids. Mol Biol 14:882–895
Poltev VI, Shulyupina NV (1986) Simulation of interactions between nucleic-acid bases by refined atom–atom potential functions. J Biomol Struct Dyn 3:739–765
Poltev VI, Malenkov GG, Gonzalez EJ, Teplukhin A, Rein R, Shibata M, Miller JH (1996) Modeling DNA hydration: comparison of calculated and experimental hydration properties of nucleic acid bases. J Biomol Struct Dyn 13:717–725
Grimme S, Antony J, Ehrlich S, Krieg H (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 132:154104
TURBOMOLE V6.0 2009, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007. Available from http://www.turbomole.com
Dailidonis VV, Danilov VI (2010) The extended cluster approach in the Metropolis-Monte Carlo algorithm, see http://www.biophys.in.ua/
El Hassan MA, Calladine CR (1997) Conformational characteristics of DNA: empirical classifications and a hypothesis for the conformational behaviour of dinucleotide steps. Phil Trans Roy Soc Lond A 355:43–100
Danilov VI, Stewart JPP, van Mourik T (2007) A PM6 study of the “hydration shell” of nucleic acid bases in small water clusters. J Biomol Struct Dyn 24:652–653 (Book of Abstracts: Albany 2007, Conversation 15, June 19–23 2007). http://www.jbsdonline.com/A-PM6-Study-of-the-Hydration-Shell-of-Nucleic-Acid-Bases-in-Small-Water-Clusters-p15619.html
Gusarov S, Malmqvist P-Å, Lindh R, Roos BO (2004) Correlation potentials for a multiconfigurational-based density functional theory with exact exchange. Theor Chem Acc 112:84–94
Gräfenstein J, Cremer D (2005) Development of a CAS-DFT method covering non-dynamical and dynamical electron correlation in a balanced way. Mol Phys 103:279–308
Weimer M, Sala FD, Görling A (2008) Multiconfiguration optimized effective potential method for a density-functional treatment of static correlation. J Chem Phys 128:144109
Acknowledgments
We are grateful to Professor Stefan Grimme for providing us with BLYP-D3 energies for the AT, AU and GC stacks. TvM and HAF thank EaStCHEM for support via the EaStCHEM Research Computing Facility. TvM and VID acknowledge financial support from the Royal Society through an International Joint Project grant.
Author information
Authors and Affiliations
Corresponding author
Additional information
Dedicated to Professor Akira Imamura on the occasion of his 77th birthday and published as part of the Imamura Festschrift Issue.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 2 (MPG 1,522 kb)
Supplementary material 3 (MPG 1,326 kb)
Supplementary material 4 (MPG 1,484 kb)
Supplementary material 5 (MPG 1,436 kb)
Rights and permissions
About this article
Cite this article
Dailidonis, V.V., Danilov, V.I., Früchtl, H.A. et al. The nature of base stacking: a Monte Carlo study. Theor Chem Acc 130, 859–870 (2011). https://doi.org/10.1007/s00214-011-1046-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00214-011-1046-1