
Abstract
Rationale
Disturbances in impulse control are key features of substance abuse disorders, and conversely, many drugs of abuse are known to elicit impulsive behavior both clinically and preclinically. To date, little is known with respect to the involvement of the opioid system in impulsive behavior, although recent findings have demonstrated its involvement in delay discounting processes. The aim of the present study was to further investigate the role of the opioid system in varieties of impulsivity.
Materials and methods
To this end, groups of rats were trained in the five-choice serial reaction time task (5-CSRTT) and stop-signal task (SST), operant paradigms that provide measures of inhibitory control and response inhibition, respectively. In addition, another group of rats was trained in the delayed reward paradigm, which measures the sensitivity towards delay of gratification and as such assesses impulsive choice.
Results and discussion
Results demonstrated that morphine, a selective µ-opioid receptor agonist, primarily impaired inhibitory control in the 5-CSRTT by increasing premature responding. In addition, in keeping with previous data, morphine decreased the preference for the large over small reward in the delayed reward paradigm. The effects of morphine on measures of impulsivity in both the 5-CSRTT and delayed reward paradigm were blocked by naloxone, a µ-opioid receptor antagonist. Naloxone by itself did not alter impulsive behavior, suggesting limited involvement of an endogenous opioid tone in impulsivity. Response inhibition measured in the SST was neither altered by morphine nor naloxone, although some baseline-dependent effects of morphine on response inhibition were observed.
Conclusion
In conclusion, the present data demonstrate that acute challenges with morphine modulate distinct forms of impulsive behavior, thereby suggesting a role for the opioid system in impulsivity.
Similar content being viewed by others

Introduction
A commonly observed effect of many substances of abuse and in particular psychostimulants such as amphetamine, cocaine, and nicotine is that they impair behavioral inhibition and increase impulsivity in humans (Fillmore et al. 2002; Mitchell 2004) as well as laboratory animals (Cole and Robbins 1987, 1989; Harrison et al. 1997; Paine and Olmstead 2004; Van Gaalen et al. 2006a). Furthermore, a wealth of findings have demonstrated that chronic psychostimulant abuse is associated with a variety of cognitive disturbances including elevated levels of impulsivity (e.g., Clark et al. 2006; Coffey et al. 2003; Ersche et al. 2008; Kirby and Petry 2004; Monterosso et al. 2005; Verdejo-Garcia et al. 2007; for review, see Perry and Carroll 2008). However, somewhat contradictory findings have been reported in laboratory animals depending on forced (Jentsch et al. 2002; Paine et al. 2003; Simon et al. 2007) or volitional administration of psychostimulant drugs (Dalley et al. 2005a, b, 2007; but see, Winstanley et al. 2009).
To date, the effects of opioid drugs on impulsivity have been less well documented, although recent evidence points towards a role for the opioid system in impulsivity. For instance, abstinent alcoholics have been shown to make impulsive decisions in a delay discounting task (Mitchell et al. 2005), operationalized as a preference for smaller and sooner over larger and later hypothetical monetary reward. Interestingly, in these subjects, the opioid receptor antagonist naltrexone reduced impulsive decisions in this task depending on the subjects' locus of control personality trait (Mitchell et al. 2007). Also, in control subjects, naltrexone improved performance on a measure of inhibitory motor control in this task. Collectively, these findings suggest that opioid activity might modulate aspects of impulsive behavior. In support of this, in rats, acute challenges with the μ-opioid receptor agonist morphine induce impulsive decisions in an adjusting amount procedure (Kieres et al. 2004) and seem to induce delay aversion in a delay discounting task (Pitts and McKinney 2005). Thus, these observations confirm a role for µ-opioid receptors in impulsive decision making.
In addition to the aforementioned acute opioid drug challenges on measures of impulsivity, several studies have reported increased risky decision making (Odum et al. 2000), as well as impulsive decision making in opiate users (Clark et al. 2006; Madden et al. 1997; Petry et al. 1998; Kirby and Petry 2004). To date, there is little evidence of chronic opiate use on perturbations in measures of inhibitory response control (Verdejo-Garcia and Perez-Garcia 2007; Verdejo-Garcia et al. 2007; Passetti et al. 2008). In contrast to acute opioid effects on impulsivity, elevated impulsivity in opiate addicts might also have resulted from neuroadaptations following prolonged opiate use. In this regard, alterations in brain opioid and α2 adrenergic receptor densities have been reported in opiate addicts (Gabilondo et al. 1994; Kling et al. 2000). Studying the acute effects of opioids on impulsivity might help to interpret the aforementioned observations in opiate addicts.
There is broad consensus that impulsivity is multifactorial with distinct behavioral expressions that might be under control of partly separate underlying neuroanatomical circuits and neurotransmitter systems (Evenden 1999; Pattij and Vanderschuren 2008; Winstanley et al. 2006). Poor inhibitory response control (impulsive action) and impulsive decision making (impulsive choice) are prominent behavioral phenomena of impulsivity, the latter often operationalized as delay aversion in delay discounting tasks. The aforementioned observations primarily suggest a role for the opioid system in impulsive decision making. Therefore, the purpose of the present experiments was to further examine the role of μ-opioid receptors in distinct forms of impulsive behavior. To this extent, we investigated the acute effects of morphine and naloxone in three translational rodent paradigms: (1) the five-choice serial reaction time task (5-CSRTT; Bari et al. 2008) to measure aspects of inhibitory response control, reflected by the ability to withhold inappropriate responses; (2) the stop-signal task (Eagle et al. 2008) to measure response inhibition, i.e., the ability to inhibit a response that already has been initiated; and (3) the delayed reward paradigm (Cardinal 2006) to measure delay aversion.
Materials and methods
Subjects
Male Wistar rats were obtained from Harlan CPB (Horst, The Netherlands). At the start of the experiments, animals were 12 weeks old, weighed approximately 250 g, and were housed in pairs in macrolon cages (42.5 × 26.6 × 18.5 cm; l × w × h) under a reversed 12-h light/dark cycle (lights on at 7:00 p.m.) at controlled room temperature (21 ± 2°C) and relative humidity of 60 ± 15%. Animals were maintained at approximately 90% of their free-feeding weight, starting 1 week prior to the beginning of the experiments by restricting the amount of standard rodent food pellets (Harlan Teklad Global Diet, Blackthorn, UK). Water was available ad libitum throughout the entire experiment. All experiments were conducted with the approval of the animal ethical committee of the Vrije Universiteit, Amsterdam, The Netherlands.
Apparatus
Experiments were conducted in identical rat five-hole nose poke operant chambers with stainless steel grid floors (MED-NPW-5L, Med Associates, St. Albans, VT, USA) housed in sound-insulating and ventilated cubicles. Set in the curved wall of each box was an array of five holes. Each nose poke unit was equipped with an infrared detector and a yellow light emitting diode stimulus light. Rodent food pellets (45 mg, Formula P, Bioserv, Frenchtown, USA) could be delivered at the opposite wall via a dispenser. In addition, a white house light could illuminate the chamber and a 4,500-Hz tone could be delivered through a speaker. A computer equipped with MED-PC version 1.17 (Med Associates) controlled experimental sessions and recorded data. Animals were tested once daily from Monday until Friday, during the dark phase of the light/dark cycle.
Behavioral procedures
Separate groups of n = 16 animals were trained for all paradigms for which similar habituation and magazine training protocols were followed. This protocol consisted of a habituation exposure to the operant chambers for 20 min with the houselight on and the food cup containing three food pellets for two consecutive sessions. Subsequently, in the next two sessions, pellets (100 per session) were delivered with an average delay of 15 s to allow the animals to associate the sound of pellet delivery with reward.
Five-choice serial reaction time task experiments
A detailed description of the 5-CSRTT behavioral procedure in our laboratory has been provided previously (Van Gaalen et al. 2006a). In short, rats were trained to detect and respond to a brief visual stimulus in one of five nose poke units in order to obtain a food reward. Each session terminated after 100 trials or 30 min, whichever occurred first. Initially, the duration of this stimulus was 32 s and was gradually decreased to 1 s over sessions until animals reached stable baseline performance (accuracy >80% correct choice and <20% errors of omission). Responding during stimulus presentation or within the limited hold (LH) period of 2 s was counted as a correct response. Incorrect, premature responses during the fixed 5-s intertrial interval and errors of omission (no responses or a response after the LH) did not lead to the delivery of a food reward and resulted in a 5-s timeout period during which the houselight was extinguished, whereas perseverative responses, i.e., repeated responding during the presentation of the stimulus, were measured but did not have any programmed consequences. Two different measures of inhibitory control were measured, namely, (1) the number of premature responses before the onset of the visual stimulus, reflecting aspects of loss of inhibitory control and (2) the number of perseverative responses into the stimulus unit after correct choice, a measure of compulsive behavior. In addition, the following other behavioral parameters were measured that reflect task performance, namely, (3) accurate choice, i.e., percentage correct responses calculated as [number correct trials/(correct + incorrect trials)] × 100; (4) latency to make a correct choice, i.e., the mean time between stimulus onset and nose poke in the illuminated unit; (5) omission errors, i.e., the number of omitted trials during a session; and (6) feeder latency, i.e., the latency to collect a pellet following correct choice.
Stop-signal task experiments
Training
A detailed description of training in the stop-signal task in our laboratory has been provided previously with some minor modifications in the current behavioral procedure (Pattij et al. 2007a). In the current study, the outer nose poke units were used instead of those immediately adjacent to the middle nose poke unit as described previously. In terms of behavioral performance, this modification primarily resulted in higher mean reaction times, and therefore, stop-signal delays (SSD) were also adjusted accordingly as described below. In brief, rats were trained in the same boxes used in the 5-CSRTT experiments and a typical stop-signal task session consisted of 200 trials; 25% of these trials were stop trials and the remainder go trials. To start a go trial, rats were trained to respond into the middle illuminated nose poke unit stimulus light (start stimulus) under a variable ratio 2 schedule which subsequently switched off this stimulus light and resulted into illumination of the outer left (or right, counterbalanced across rats) nose poke unit stimulus light (go stimulus). Rats were then required to respond as quickly as possible during a LH period to this go stimulus in order to obtain a food reward. Initially, the LH period was set at 5 s and in subsequent sessions was individually titrated to meet go-trial accuracy criterion of approximately 80% successful hits. A stop trial did not differ from a go trial; however, contingent with the illumination of the go stimulus or following a SSD, a stop signal (duration 50 ms; frequency 4500 Hz; intensity 80 dB) was presented and successfully refraining from responding during the LH period of a stop trial resulted in delivery of a food reward. In the final stages of training, the LH periods during both go and stop trials were equal for each individual rat. The SSDs including zero delays were presented in a pseudorandom order within a session. To compensate for differences between rats, SSDs were based on each individual rat's mean reaction time on go trials (mGoRT) in the preceding drug-free training session. Thus, SSDs were calculated as follows: mGoRT minus either 25, 75, 150, 300, or 600 ms. During all stop trials, responding during the onset of the stop signal or during the LH immediately extinguished the go stimulus, houselight, and, if applicable, turned off the stop signal and was followed by a 5-s timeout period.
Estimation stop-signal reaction time
Calculations to estimate the stop-signal reaction times (SSRT) and a correction for omission errors were adapted from Logan (1994) and Solanto et al. (2001). For estimating the SSRT, data of three SSDs (mRT minus 300, 150, and 75 ms) were used, as the probability of correct inhibition on these SSDs was within the range of 0.2 < p < 0.8 and thus most informative for estimation of SSRT (Band et al. 2003). For each of the three SSDs, the probability of responding was calculated including a correction for non-responses based on the number of omissions during the go trials, the latter since omissions cannot be distinguished from successful inhibitions during stop trials. The following formula, adapted from Solanto et al. (2001), was used for these calculations:
where x is the number of stop-signal trials at each delay interval; correct inhibitions are the number of correctly inhibited trials and y is the probability of omissions during the go trials within the entire session. To calculate SSRTs, reaction times on all go trials were rank-ordered. From this list with RTs, the “nth” RT was taken, where “n” was obtained by multiplying the total number of go trials by the probability of responding for a particular SSD. This RT value approximates the latency between onset of the go stimulus and completion of the stopping process. The SSRT for each interval is then obtained by subtracting the SSD interval from this RT. The average estimated SSRT that is used for the analyses in the present study is calculated by taking the mean of each SSRT at the aforementioned three SSDs.
Delayed reward paradigm experiments
The delayed reward paradigm used in our laboratory has been described previously (Van Gaalen et al. 2006b). Briefly, in the final stages of training and during drug testing, a session was divided into five blocks of 12 trials, each block started with two forced choice trials. Each rat received a left forced and a right forced trial. The order of these was counterbalanced between subjects. In the next ten trials, the animals had a free choice and both the left and right unit were illuminated. Poking into one position resulted in the immediate delivery of a small reinforcer (one food pellet), whereas a nose poke into the other position resulted in the delivery of a large, but delayed, reinforcer (four food pellets). If an animal did not make a response during this choice phase within 10 s, an intertrial interval was initiated and the trial was counted as an omission. The position associated with the small and large reinforcer was always the same for each individual and counterbalanced for the group of rats. Delays for the large reinforcer progressively increased within a session per block of 12 trials as follows: 0, 5, 10, 20, and 40 s. Responding into non-illuminated units during the test was recorded, but had no further programmed consequences. The behavioral measure to assess task performance, i.e., the percentage preference for the large reinforcer as a function of delay, was calculated as the number of choices for the large reinforcer/(number choices large + small reinforcers) × 100. Furthermore, we calculated the total number of omitted choice trials per block of ten trials within a session. In addition to the percentage preference data, hyperbolic curves were fitted by employing the least-squares criterion on individual data obtained in the drug studies using GraphPad Prism version 5 (GraphPad Software, La Jola, CA, USA). The following equation from Mazur (1987) was used: \( Y = {A \mathord{\left/ {\vphantom {A {1 + kD}}} \right. } {1 + kD}} \), where Y is the mean percentage preference for the large reinforcer, D is the delay to obtain the large reinforcer, A is a free parameter related to the amount of reinforcement, and k is free parameter indicating the steepness of the discounting curve.
Drugs
Morphine hydrochloride (OPG, Utrecht, The Netherlands) and naloxone hydrochloride (Sigma, St. Louis, MO, USA) were dissolved in sterile saline. Drug doses were based on a previous study investigating the effects of morphine and naloxone on an operant progressive ratio schedule (Solinas and Goldberg 2005). In all experiments, drugs were injected 30 min before testing, whereas in drug combination studies, naloxone was injected 30 min and morphine was injected 20 min prior to testing. Drugs were freshly prepared each day before testing and intraperitoneally injected in a volume of 1-ml/kg bodyweight according to a Latin square within-subjects design for both the dose–response studies as well as the drug combination studies on Tuesdays and Fridays with baseline training sessions on the other weekdays.
Statistical analyses
Data were subjected to repeated measures analysis of variance with drug dose (all paradigms) and delay to large reinforcer (delayed reward paradigm only) as within-subjects variables using the Statistical Package for the Social Sciences version 14 (SPSS, Chicago, IL, USA). The homogeneity of variance across groups was determined using Mauchly's tests for equal variances, and in case of violation of homogeneity, Huynh–Feldt epsilon (ε) adjusted degrees of freedom were applied and the resulting more conservative probability values depicted and used for subsequent analyses. In case of statistically significant main effects, further post hoc comparisons were conducted using Newman–Keuls multiple comparison tests. The level of probability for statistically significant effects was set at 0.05. In the delayed reward experiments, the highest dose of morphine (6.0 mg/kg) increased choice for the small reward at 0-s delays. Therefore, the delay discounting curves in the morphine/ naloxone combination study were also analyzed as percentage change in preference from the 0-s delay in order to control for these baseline differences. In the stop-signal task, Pearson's correlation coefficients were also calculated in order to explore the relationship between the magnitude of drug effects, i.e., change in SSRT from vehicle to drug (morphine, naloxone) and baseline SSRT under vehicle condition.
Results
Effects of morphine on impulsive action in the five-choice serial reaction time task
As depicted in Fig. 1a, c and Table 1, under stable baseline responding, morphine significantly increased the number of premature responses and decreased the number of perseverative responses after correct choice [F(4,60) = 4.14, ε = 0.46, p = 0.030 and F(4,60) = 4.15, ε = 0.67, p = 0.015, respectively]. Further comparisons indicated that 3.0 and 6.0 mg/kg morphine significantly increased the number of premature responses, whereas only 6.0 mg/kg morphine decreased the number of perseverative responses. In addition to increasing impulsive action, the highest dose of morphine also significantly increased the number of omissions [F(4,60) = 14.32, ε = 0.65, p < 0.001] and increased correct response latencies [F(4,60) = 4.29, ε = 0.60, p = 0.016]. Attentional performance measured by the percentage accurate choice was not affected by morphine [F(4,60) = 1.09, p = 0.37 and F(4,60) = 2.17, ε = 0.42, p = 0.10]. Likewise, the latency to collect food reward after correct choice was not significantly changed by morphine [F(4,60) = 2.67, ε = 0.40, p = 0.10].

Effects of morphine (a, b), naloxone (c, d), and their combination (e, f) on different measures of inhibitory control in the 5-CSRTT. In total, n = 16 (a–d) and n = 15 (e, f) animals were included in the analyses and data depict mean (±SEM) numbers of premature responses (a, c, e) and perseverative responses after correct choice (b, d, f). *p < 0.05 versus vehicle and vehicle–vehicle (0–0) injections and + p < 0.05, ++ p < 0.005 versus vehicle/3.0 mg-kg morphine injections
The effects of naloxone on performance in the 5-CSRTT are summarized in Fig. 1b, d and Table 1. Naloxone increased correct response latencies [F(3,45) = 4.59, p = 0.007], and further comparisons revealed that only the highest dose significantly increased latencies by approximately 20 ms. In addition, the lowest dose of 0.3 mg/kg naloxone significantly reduced the number of perseverative responses after correct choice [F(3,45) = 2.96, p = 0.042]. Other performance measures in the 5-CSRTT reflecting impulsive action, attentional performance, and motivation were not affected by naloxone [accurate choice: F(3,45) = 0.60, p = 0.62; premature responses: F(3,45) = 2.46, p = 0.075; omissions: F(3,45) = 0.26, ε = 0.81, p = 0.81; feeder latency: F(3,45) = 1.28, ε = 0.43, p = 0.29].
In the morphine/naloxone combination study (morphine 3.0 mg/kg and naloxone 0.3 and 1.0 mg/kg), one animal was excluded from the analyses due to high number of omissions displayed by this animal on two different test days (68 and 72 omissions, respectively). The effects of 3.0 mg/kg morphine on impulsive action in the 5-CSRTT were fully antagonized by both doses of naloxone (Fig. 1e) [F(3,42) = 6.98, ε = 0.66, p = 0.004]. Likewise, 0.3 and 1.0 mg/kg naloxone blocked the small but significant increase in the number of omissions by 3.0 mg/kg morphine [F(3,42) = 3.44, ε = 0.77, p = 0.038]. In contrast to the morphine alone study, in the combination study, 3.0 mg/kg morphine slightly but significantly decreased the number of perseverative responses after correct choice from approximately seven to five responses, which was antagonized by both doses naloxone (Fig. 1f) [F(3,42) = 5.78, p = 0.002]. In addition, in the combined morphine/ naloxone experiments, morphine significantly speeded the latency to collect food reward after correct choice, and these effects of morphine were antagonized by both doses of naloxone [F(3,42) = 18.34, p < 0.001]. Attentional parameters in the 5-CSRTT were not altered by the combination of morphine and naloxone [accurate choice: F(3,42) = 2.26, p = 0.095; correct response latency: F(3,42) = 0.49, p = 0.69].
Effects of morphine on response inhibition in the stop-signal task
In the stop-signal task experiments, one animal was excluded from all analyses because it did not acquire stable performance. Moreover, two additional animals were excluded from all analyses because of their low go trial accuracy and high omission rate of around 50% of all go trials during training and test sessions.
Morphine did not alter the estimated SSRT, the primary measure of response inhibition, and, in addition, mGoRTs were also not changed by morphine (Fig. 2 a, c) [F(3,36) = 0.76, p = 0.52 and F(3,36) = 2.54, ε = 0.63, p = 0.10, respectively]. Only the high dose of 3.0 mg/kg morphine did, however, significantly affect behavior during the go trials and decreased go trial accuracy from 77% under vehicle conditions to 57% [F(3,36) = 6.17, ε = 0.55, p = 0.012]. Further inspection of the data revealed that this decrease was primarily caused by two rats displaying a go trial accuracy of 17% and 21%, respectively, only at the high (3.0 mg/kg) dose morphine. In order to explore whether morphine had differentially affected response inhibition depending on baseline SSRT under vehicle condition, a correlation analysis was performed. This analysis revealed that the effect size of morphine on SSRT (averaged over the three doses 0.3, 1.0, and 3.0 mg/kg) correlated negatively with baseline SSRT as shown in Fig. 2e (Pearson’s r = −0.83, p < 0.001). The effect size of morphine on mGoRT did, however, not correlate with baseline SSRT (data not shown; Pearson’s r = −0.32, p = 0.23).

Effects of morphine and naloxone on response inhibition as measured in the stop-signal paradigm. In total, n = 13 animals were included in the morphine and naloxone analyses and data are depicted as mean (±SEM) estimated stop-signal reaction times (a, b) and go reaction times (c, d). In addition, Pearson’s r indicated a negative correlation between the magnitude morphine’s effects on SSRT averaged over all three doses and baseline SSRT under vehicle condition (e). *p < 0.05 versus vehicle
Comparable to the effects of morphine, naloxone did neither affect estimated SSRTs nor mGoRTs (Fig. 2b, d) [F(3,36) = 0.69, p = 0.56 and F(3,36) = 2.73, p = 0.058, respectively]. Furthermore, naloxone had no effects on go trial accuracy [F(3,36) = 0.39, p = 0.76]. In contrast to the effects of morphine, there was no relationship between the effects of naloxone on response inhibition and baseline SSRT (Pearson’s r = −0.36, p = 0.089).
Effects of morphine on impulsive choice in the delayed reward paradigm
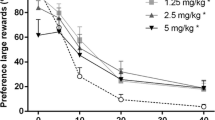
The percentage choice for the larger delayed reward significantly declined depending on the delay that was imposed on the animals (Fig. 3a) [delay: F(4,60) = 156.15, p < 0.001]. Morphine significantly reduced the preference for the large delayed reward [dose: F(4,60) = 3.35, ε = 0.64, p = 0.035; dose × delay: F(16,240) = 1.58, p = 0.082], and further, post hoc comparisons revealed that this effect was significant in the highest dose morphine. However, analyses of the steepness (k) of the discounting curves with morphine revealed no significant differences between doses, thereby supporting the lack of a dose × delay interaction effect [F(4,60) = 1.40, ε = 0.25, p = 0.26]. The number of omissions to start a trial (range, 1.03 ± 0.23 to 1.67 ± 0.35 omissions/block) was not significantly affected by morphine [dose: F(4,60) = 1.71, p = 0.16; dose × delay: F(16,240) = 1.22, p = 0.25]. It should be noted that in two individuals, 6.0 mg/kg morphine did bias choice towards one response option irrespective of the delay. One animal preferably chose the response alternative associated with the immediate small reward, whereas the other mainly preferred the delayed large reward. All other rats displayed a delay-dependent decrease in preference for the large reward at this dose of morphine, although the majority did show a modest reduction in choice for the large reward at the 0-s delay with 6.0 mg/kg morphine. Excluding these two individuals with biased choice from the analyses did not alter the main findings with morphine [dose: F(4,52) = 2.72, p = 0.039; dose × delay: F(16,208) = 1.05, p = 0.40].

Effects of morphine (a), naloxone (b), and their combination (c) on the mean percentage preference for the large reinforcer and change in preference from the 0-s delay (d) in the delayed reward paradigm. In total, n = 16 animals were included in the analyses. SED standard error of differences between means. *p < 0.05 versus vehicle/vehicle and + p < 0.05 versus vehicle/6.0-mg/kg morphine injections
Naloxone slightly but significantly shifted the preference for the larger delayed reward (Fig. 3b) [dose: F(3,45) = 0.46, p = 0.71; dose × delay: F(12,180) = 2.04, ε = 0.66, p = 0.048]. However, further comparisons revealed no significant dose × delay interaction effects of naloxone at any dose compared with vehicle. This suggests that the overall dose × delay interaction effect lacks biological significance. Further analyses of the steepness of the discounting curves supported this notion [F(3,45) = 1.19, ε = 0.44, p = 0.31]. Similarly, naloxone did not affect the number of omissions to start a trial [dose: F(3,45) = 2.28, ε = 0.65, p = 0.12; dose × delay: F(12,180) = 1.07, p = 0.39].
In the combination study (Fig. 3c), the effects of 6.0 mg/kg morphine were antagonized by naloxone [dose: F(3,45) = 4.63, ε = 0.74, p = 0.014; dose × delay: F(12,180) = 2.62, ε = 0.93, p = 0.004], and further comparisons showed that this was the case for 1.0 mg/kg naloxone. This significant dose × delay interaction effect was further substantiated by the slope analysis of the discounting curves [F(3,45) = 2.91, ε = 0.51, p = 0.045] which revealed significant differences in steepness of the slopes between vehicle–vehicle versus vehicle–morphine and 0.3 mg/kg naloxone–morphine. Similar to the morphine dose–response experiment, one individual always preferred the response alternative associated with the immediate small reward. Excluding this individual from the analyses did not change the main findings [dose: F(3,42) = 3.77, ε = 0.71, p = 0.032; dose × delay: F(12,168) = 2.46, ε = 0.89, p = 0.008].
To control for the decrease in preference for the large reward that occurred with 6.0 mg/kg morphine at the 0-s delay, further analyses were performed. To this end, all data from the morphine/naloxone combination study were also calculated as percentage change in preference from the 0-s delay (Fig. 3d). Analyses on these data revealed that morphine significantly shifted the preference for the large reward depending on the delay [dose: F(3,45) = 1.79, ε = 0.71, p = 0.18; dose × delay: F(12,180) = 2.40, ε = 0.81, p = 0.012]. These effects of morphine on the preference for the large reward depending on the delay were reversed by 1.0 mg/kg naloxone.
Discussion
The main findings of the present study are that acute challenges with morphine modulate impulsive behavior, as expressed in impaired inhibitory response control and—to a somewhat lesser extent—impulsive decisions. The number of premature responses in the 5-CSRTT was dose-dependently increased following morphine, and in addition, morphine also biased the preference from larger more beneficial delayed rewards towards small and immediate rewards. The observation that the effects of morphine were abolished by naloxone strongly suggests μ-opioid receptor involvement in different forms of impulsive behavior. In contrast, morphine did not change response inhibition as measured in the stop-signal task, although in a correlation analysis, baseline-dependent effects of morphine on response inhibition were detected and morphine seemed to improve response inhibition in rats with “slow” stopping abilities. In the absence of an effect of naloxone on the different forms of impulsive behavior, our data do not provide clear evidence for a role of tonically activated μ-opioid receptors by endogenous opioids in measures of impulsivity. In as much as these observations can be extrapolated to clinical data, they do not support the recently reported beneficial effects of a single dose of naltrexone in healthy volunteers on a measure of inhibitory response control (Mitchell et al. 2007). However, in that study, the effects of naltrexone (50 mg) on inhibitory response control were derived from a calculated measure in a delay discounting task and not from results obtained in a separate task measuring inhibitory response control, which may have accounted for this discrepancy in findings.
The observation that morphine robustly increased premature responding in the 5-CSRTT extends previous reported acute effects of psychostimulants such as amphetamine, cocaine, and nicotine on impulsive action (Cole and Robbins 1987, 1989; Evenden 1998; Harrison et al. 1997; Paine and Olmstead 2004; Pattij et al. 2007b; Van Gaalen et al. 2006a). Strikingly, the high dose of morphine (6.0 mg/kg) was found to improve a different measure of inhibitory response control in this paradigm by reducing the number of perseverative responses following correct choice and before food reward collection (see for review, Robbins 2002). This finding may suggest beneficial effects of morphine on aspects of compulsive behavior. However, this dose of morphine also strongly increased the number of omissions and lengthened response latencies, and therefore, these effects on perseverative responding are likely due to non-specific drug effects on motor behavior. In contrast to the effects of morphine on measures of inhibitory control, other parameters in the 5-CSRTT reflecting attentional performance, such as accurate choice, were not affected by morphine. These latter findings are supported by recent observations showing that morphine in a similar dose range did not influence measures of sustained attention in a modified 5-CSRTT with a stimulus duration of 5 s and a test session consisting of 250 trials (Boyette-Davis et al. 2008).
To date, clinical evidence demonstrating inhibitory response control deficits in opiate abusers is only scarcely available. While the majority of these studies do not find clear behavioral evidence of inhibitory response control disturbances in opiate addicts (Passetti et al. 2008; Verdejo-Garcia and Perez-Garcia 2007; Verdejo-Garcia et al. 2007), some changes in brain activation patterns following a Go/NoGo task have been detected in opiate users (Forman et al. 2004). Furthermore, a recent study found evidence for reflection impulsivity in opiate users (Clark et al. 2006), i.e., the tendency to act before all necessary information is evaluated. Although reflection impulsivity may conceptually be regarded as a separate form of impulsive behavior (see for discussion, Evenden 1999), it might contain aspects of both deficient inhibitory response control and impulsive decision making.
Consistent with the aforementioned paucity of evidence demonstrating inhibitory control deficits in opiate users, morphine had no clear effects on response inhibition as measured in the stop-signal task. Thus, our stop-signal task data suggest minor μ-opioid receptor involvement in the ability to inhibit ongoing behavior and contrast the findings in the 5-CSRTT. The latter task measures the ability to withhold inappropriate (premature) responses, and clearly, the opioid system modulates these inhibitory response control processes. These observations add to the growing literature demonstrating that the 5-CSRTT and stop-signal task measure different cognitive processes that can be dissociated neuroanatomically and pharmacologically (Eagle et al. 2008; Winstanley et al. 2006). Further in-depth analyses on the stop-signal data revealed that the effect size of morphine on estimated SSRT, the primary parameter reflecting response inhibition, correlated negatively with SSRTs under vehicle condition. Thus, in individuals with a low baseline SSRT and better stopping abilities, morphine seemed to “impair” response inhibition by increasing the SSRT. In contrast, in “slow” stopping rats with higher baseline SSRTs, morphine seemed to “improve” response inhibition by decreasing the SSRT. These findings suggest baseline-dependent effects of morphine on response inhibition that could not be detected when the data were calculated over the entire population of rats. This conclusion should be drawn with caution, as we only explored such a relationship by correlation analyses and did not preselect groups of individuals based on response inhibition capacities. Notwithstanding this limitation, various clinical and preclinical studies have indeed supported this notion and shown that the behavioral effects of, e.g., amphetamine, in healthy volunteers (De Wit et al. 2000, 2002) as well as amphetamine and methylphenidate in rats on response inhibition largely seem to depend on baseline performance (Feola et al. 2000; Eagle et al. 2007).
The observation that morphine shifted more self-controlled decisions by decreasing preference for the large over small reward in our delayed reward paradigm is in line with previous observations in rats (Kieres et al. 2004) and numerous findings of delay discounting in opiate users (Clark et al. 2006; Madden et al. 1997; Petry et al. 1998; Kirby and Petry 2004). With regard to these observations in human opiate users, it is unclear whether increased delay discounting in opiate users results from premorbid levels of impulsive decision making or is the consequence of chronic opiate abuse. Our findings and those of Kieres et al. (2004) at least suggest acute effects of opiates on impulsive decision making. While opiate addicts generally discount the value of delayed monetary rewards or the drug itself, in the present study, rats were food-restricted and highly palatable food pellets served as food reinforcement. Given the involvement of the central opioid system in food reward, and in particular its modulatory role on the hedonic properties of appetitive foods (e.g., Solinas and Goldberg 2005; for review, see Cota et al. 2006), it is possible that morphine might have enhanced the hedonic property of the highly palatable food pellets to the extent that rats shifted their preference from larger delayed food reward to immediate reward. Nonetheless, the finding that the number of omissions to initiate a trial, a parameter that reflects aspects of food-motivated behavior in the delayed reward paradigm (Cardinal et al. 2000), did neither decrease following morphine nor increase following naloxone argues against such an explanation. Similarly, in the 5-CSRTT experiments, neither morphine nor naloxone altered the primary indices of food-motivated behavior, namely, the feeder latency and number of omissions at doses that increased impulsivity (Table 1).
It is important to emphasize that the highest morphine dose modestly reduced the preference for the large reward at the 0-s delay by approximately 15% compared to vehicle. This observation may confound a solid interpretation of morphine’s effects on impulsive choice. It is not entirely clear what caused these effects, and this observation is not unique to the present study. In previous experiments, pharmacological challenges with, e.g., ethanol, the 5-HT1A and 5-HT2 receptor agonists 8-OH-DPAT and DOI, and the dopamine D1 receptor antagonist SCH23390 have also been found to reduce the preference for the large reward at the 0-s delay (Evenden and Ryan 1999; Van Gaalen et al. 2006b; Winstanley et al. 2005). As discussed above, effects on food-motivated behavior seem an unlikely explanation, although positive reinforcing drugs including morphine are known to possess aversive stimulus properties and elicit conditioned taste aversions (Hunt and Amit 1987). The observation that following 6.0 mg/kg morphine rats still earned and consumed a similar amount of pellets compared to baseline training and vehicle conditions (data not shown) argues against morphine-induced emetic effects. Alternatively, it is possible that morphine caused difficulties for the animals to distinguish between two behavioral outcomes or induced a positional bias. However, if the former is the case, the question remains why—on average—this only applied to two out of ten choice trials at the 0-s delay and did not result in chance performance throughout the entire session. The finding that similar doses of morphine did not induce a positional bias in the 5-CSRTT (data not shown) does not support such an explanation. Also, if morphine did bias the responses of the animals towards one side of the operant chamber, one would expect animals to always choose either the immediate small reward or delayed large reward irrespective of the delay. Although few individuals (n = 2 in the morphine dose–response experiment and n = 1 in the morphine–naloxone experiment) did indeed display biased responding with 6.0 mg/kg morphine, excluding these individuals from the analyses did not change the overall effects of morphine on delay discounting. Importantly, all other animals highly preferred the large reward at short delays and shifted their preference with longer delays, although in the large majority of animals, the preference for the large reward at the 0-s delay was modestly reduced when treated with 6.0 mg/kg morphine. Analyses on the steepness of the delay discounting curves suggest that the effects of morphine became stronger after repeated exposure. Whereas in the dose–response study with morphine the slope of the delay discounting curve with vehicle did not differ from 6.0 mg/kg, in the morphine–naloxone study, the steepness of these curves did differ significantly. Although speculative, perhaps tolerance to putative behavioral “disruptive” effects of morphine at the 0-s delay explains these observations. Indeed, in a parallel study, we found that repeated exposure to 6 mg/kg morphine appeared to induce tolerance to this “disruptive” effect of the opiate at the 0-s delay, whereas at larger delays, morphine still significantly reduced preference for the larger reward. Notwithstanding these possible confounds, the fact that naloxone blocked morphine’s effects in the delayed reward indicates a µ-receptor-dependent mechanism.
A possible mechanism of action by which morphine might elevate impulsive behavior is through its modulatory action on mesolimbic dopamine transmission. Opiates such as heroin and morphine enhance dopamine transmission in the nucleus accumbens (e.g., Di Chiara and Imperato 1986, 1988; Rada et al. 1991; Shoaib et al. 1995; Wise et al. 1995). These effects are primarily mediated through µ-opioid-mediated disinhibition of dopamine neuron activity in the ventral tegmental area (e.g., Kalivas et al. 1990; Spanagel et al. 1992). We have previously shown that the detrimental effects of psychostimulants on impulsivity in the 5-CSRTT largely depend on dopamine receptor activation (Van Gaalen et al. 2006a). More specifically, previous studies have pinpointed the nucleus accumbens (Cole and Robbins 1989; Murphy et al. 2008) and in particular dopamine within this brain region as critical (Cole and Robbins 1987; Pattij et al. 2007b). Thus, increments in accumbal dopamine transmission might explain the effects of morphine on inhibitory control in the 5-CSRTT. Alternatively, altered dopamine function in the prefrontal cortex has been related to impulse action in the 5-CSRTT (Dalley et al. 2002). Nonetheless, doses of morphine that elevate extracellular dopamine in the nucleus accumbens were ineffective in altering dopamine in the prefrontal cortex (Bassareo et al. 1996). Furthermore, other basal ganglia and connected nuclei, such as the subthalamic nucleus (Baunez and Robbins 1997) and pedunculopontine tegmental nucleus (Inglis et al. 2001), have been implicated in impulsivity in the 5-CSRTT. To our knowledge, no studies have directly examined (1) dopaminergic modulation of impulsive action or (2) opioid modulation of dopamine transmission within these nuclei. Taken together, future work should elucidate whether altered dopamine transmission and which specific brain regions are critically involved in morphine-induced impulsivity.
Whether opioid modulation of mesolimbic dopamine transmission is responsible for the decrease in preference for the large over small reward induced by morphine also remains a topic for further experiments. Given previous data, this seems an unlikely explanation. In contrast to the aforementioned detrimental effects of elevating dopamine transmission on impulsive action, in general, elevating dopamine transmission results in more self-controlled decisions in rodent delayed reward paradigms (Cardinal et al. 2000; Isles et al. 2003; Van Gaalen et al. 2006b; Wade et al. 2000; Winstanley et al. 2005; for review, Pattij and Vanderschuren 2008) and in healthy volunteers (De Wit et al. 2002).
Alternatively, acute challenges with morphine at doses of 5 mg/kg have been demonstrated to reduce extracellular levels of noradrenaline in the prefrontal cortex (Devoto et al. 2002; Rossetti et al. 1993), presumably mediated via presynaptic µ-opioid receptors on noradrenergic nerve terminals in the cortex (Mulder et al. 1987). Yet, to our knowledge, no evidence is available unequivocally demonstrating a role for cortical noradrenaline in impulsive choice. However, the α2 adrenoceptor agonist clonidine induces impulsive choice in the delayed reward paradigm (Van Gaalen et al. 2006b). Presumably, this effect is mediated through activation of presynaptic α2 receptors, which in turn strongly inhibits noradrenergic neurotransmission (Starke 2001). Furthermore, recent findings demonstrate that increasing noradrenaline transmission by the selective noradrenaline reuptake inhibitor atomoxetine reduces impulsivity in the delayed reward paradigm (Robinson et al. 2008), a finding that is possibly explained by the effects of atomoxetine on cortical noradrenaline transmission (Bymaster et al. 2002; Swanson et al. 2006).
Taken together, two conclusions can be drawn from the present data. First, our findings provide evidence that acute challenges with morphine impair measures of inhibitory control. Second, in support of previous data demonstrating acute morphine effects on impulsive decision making (Kieres et al. 2004), our findings also suggest acute opioid effects on this measure of impulsivity. Accumulating evidence has now implicated impulsivity in the vulnerability towards and maintenance of drug addiction (see for reviews, Perry and Carroll 2008; Verdejo-Garcia et al. 2008). In view of this, an implication of our findings might be that the acute effects of opiates on impulsivity partly contribute to the persistence of opiate use.
References
Band GP, van der Molen MW, Logan GD (2003) Horse-race model simulations of the stop-signal procedure. Acta Psychol 112:105–142
Bari A, Dalley JW, Robbins TW (2008) The application of the 5-choice serial reaction time task for the assessment of visual attentional processes and impulse control in rats. Nat Protoc 3:759–767
Bassareo V, Tanda G, Petromilli P, Giua C, Di CG (1996) Non-psychostimulant drugs of abuse and anxiogenic drugs activate with differential selectivity dopamine transmission in the nucleus accumbens and in the medial prefrontal cortex of the rat. Psychopharmacology (Berl) 124:293–299
Baunez C, Robbins TW (1997) Bilateral lesions of the subthalamic nucleus induce multiple deficits in an attentional task in rats. Eur J Neurosci 9:2086–2099
Boyette-Davis JA, Thompson CD, Fuchs PN (2008) Alterations in attentional mechanisms in response to acute inflammatory pain and morphine administration. Neuroscience 151:558–563
Bymaster FP, Katner JS, Nelson DL, Hemrick-Luecke SK, Threlkeld PG, Heiligenstein JH, Morin SM, Gehlert DR, Perry KW (2002) Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology 27:699–711
Cardinal RN (2006) Neural systems implicated in delayed and probabilistic reinforcement. Neural Netw 19:1277–1301
Cardinal RN, Robbins TW, Everitt BJ (2000) The effects of d-amphetamine, chlordiazepoxide, alpha-flupenthixol and behavioural manipulations on choice of signalled and unsignalled delayed reinforcement in rats. Psychopharmacology 152:362–375
Clark L, Robbins TW, Ersche KD, Sahakian BJ (2006) Reflection impulsivity in current and former substance users. Biol Psychiatry 60:515–522
Coffey SF, Gudleski GD, Saladin ME, Brady KT (2003) Impulsivity and rapid discounting of delayed hypothetical rewards in cocaine-dependent individuals. Exp Clin Psychopharmacol 11:18–25
Cole BJ, Robbins TW (1987) Amphetamine impairs the discriminative performance of rats with dorsal noradrenergic bundle lesions on a 5-choice serial reaction time task: new evidence for central dopaminergic–noradrenergic interactions. Psychopharmacology (Berl) 91:458–466
Cole BJ, Robbins TW (1989) Effects of 6-hydroxydopamine lesions of the nucleus accumbens septi on performance of a 5-choice serial reaction time task in rats: implications for theories of selective attention and arousal. Behav Brain Res 33:165–179
Cota D, Tschop MH, Horvath TL, Levine AS (2006) Cannabinoids, opioids and eating behavior: the molecular face of hedonism? Brain Res Rev 51:85–107
Dalley JW, Theobald DE, Eagle DM, Passetti F, Robbins TW (2002) Deficits in impulse control associated with tonically-elevated serotonergic function in rat prefrontal cortex. Neuropsychopharmacology 26:716–728
Dalley JW, Theobald DE, Berry D, Milstein JA, Laane K, Everitt BJ, Robbins TW (2005a) Cognitive sequelae of intravenous amphetamine self-administration in rats: evidence for selective effects on attentional performance. Neuropsychopharmacology 30:525–537
Dalley JW, Laane K, Pena Y, Theobald DE, Everitt BJ, Robbins TW (2005b) Attentional and motivational deficits in rats withdrawn from intravenous self-administration of cocaine or heroin. Psychopharmacology (Berl) 182:579–587
Dalley JW, Laane K, Theobald DE, Pena Y, Bruce CC, Huszar AC, Wojcieszek M, Everitt BJ, Robbins TW (2007) Enduring deficits in sustained visual attention during withdrawal of intravenous methylenedioxymethamphetamine self-administration in rats: results from a comparative study with d-amphetamine and methamphetamine. Neuropsychopharmacology 32:1195–1206
De Wit H, Crean J, Richards JB (2000) Effects of d-amphetamine and ethanol on a measure of behavioral inhibition in humans. Behav Neurosci 114:830–837
De Wit H, Enggasser JL, Richards JB (2002) Acute administration of d-amphetamine decreases impulsivity in healthy volunteers. Neuropsychopharmacology 27:813–825
Devoto P, Flore G, Pira L, Diana M, Gessa GL (2002) Co-release of noradrenaline and dopamine in the prefrontal cortex after acute morphine and during morphine withdrawal. Psychopharmacology (Berl) 160:220–224
Di Chiara G, Imperato A (1986) Preferential stimulation of dopamine release in the nucleus accumbens by opiates, alcohol, and barbiturates: studies with transcerebral dialysis in freely moving rats. Ann NY Acad Sci 473:367–381
Di Chiara G, Imperato A (1988) Opposite effects of mu and kappa opiate agonists on dopamine release in the nucleus accumbens and in the dorsal caudate of freely moving rats. J Pharmacol Exp Ther 244:1067–1080
Eagle DM, Tufft MR, Goodchild HL, Robbins TW (2007) Differential effects of modafinil and methylphenidate on stop-signal reaction time task performance in the rat, and interactions with the dopamine receptor antagonist cis-flupenthixol. Psychopharmacology (Berl) 192:193–206
Eagle DM, Bari A, Robbins TW (2008) The neuropsychopharmacology of action inhibition: cross-species translation of the stop-signal and go/no-go tasks. Psychopharmacology (Berl) 199:439–456
Ersche KD, Roiser JP, Robbins TW, Sahakian BJ (2008) Chronic cocaine but not chronic amphetamine use is associated with perseverative responding in humans. Psychopharmacology (Berl) 197:421–431
Evenden JL (1998) The pharmacology of impulsive behaviour in rats II: the effects of amphetamine, haloperidol, imipramine, chlordiazepoxide and other drugs on fixed consecutive number schedules (FCN 8 and FCN 32). Psychopharmacology (Berl) 138:283–294
Evenden JL (1999) Varieties of impulsivity. Psychopharmacology (Berl) 146:348–361
Evenden JL, Ryan CN (1999) The pharmacology of impulsive behaviour in rats VI: the effects of ethanol and selective serotonergic drugs on response choice with varying delays of reinforcement. Psychopharmacology (Berl) 146:413–421
Feola TW, de Wit H, Richards JB (2000) Effects of d-amphetamine and alcohol on a measure of behavioral inhibition in rats. Behav Neurosci 114:838–848
Fillmore MT, Rush CR, Hays L (2002) Acute effects of oral cocaine on inhibitory control of behavior in humans. Drug Alcohol Depend 67:157–167
Forman SD, Dougherty GG, Casey BJ, Siegle GJ, Braver TS, Barch DM, Stenger VA, Wick-Hull C, Pisarov LA, Lorensen E (2004) Opiate addicts lack error-dependent activation of rostral anterior cingulate. Biol Psychiatry 55:531–537
Gabilondo AM, Meana JJ, Barturen F, Sastre M, Garcia-Sevilla JA (1994) mu-Opioid receptor and alpha 2-adrenoceptor agonist binding sites in the postmortem brain of heroin addicts. Psychopharmacology (Berl) 115:135–140
Harrison AA, Everitt BJ, Robbins TW (1997) Central 5-HT depletion enhances impulsive responding without affecting the accuracy of attentional performance: interactions with dopaminergic mechanisms. Psychopharmacology (Berl) 133:329–342
Hunt T, Amit Z (1987) Conditioned taste aversion induced by self-administered drugs: paradox revisited. Neurosci Biobehav Rev 11:107–130
Inglis WL, Olmstead MC, Robbins TW (2001) Selective deficits in attentional performance on the 5-choice serial reaction time task following pedunculopontine tegmental nucleus lesions. Behav Brain Res 123:117–131
Isles AR, Humby T, Wilkinson LS (2003) Measuring impulsivity in mice using a novel operant delayed reinforcement task: effects of behavioural manipulations and d-amphetamine. Psychopharmacology 170:376–382
Jentsch JD, Olausson P, De La Garza R Jr, Taylor JR (2002) Impairments of reversal learning and response perseveration after repeated, intermittent cocaine administrations to monkeys. Neuropsychopharmacology 26:183–190
Kalivas PW, Duffy P, Eberhardt H (1990) Modulation of A10 dopamine neurons by gamma-aminobutyric acid agonists. J Pharmacol Exp Ther 253:858–866
Kieres AK, Hausknecht KA, Farrar AM, Acheson A, de Wit H, Richards JB (2004) Effects of morphine and naltrexone on impulsive decision making in rats. Psychopharmacology (Berl) 173:167–174
Kirby KN, Petry NM (2004) Heroin and cocaine abusers have higher discount rates for delayed rewards than alcoholics or non-drug-using controls. Addiction 99:461–471
Kling MA, Carson RE, Borg L, Zametkin A, Matochik JA, Schluger J, Herscovitch P, Rice KC, Ho A, Eckelman WC, Kreek MJ (2000) Opioid receptor imaging with positron emission tomography and [(18) F]cyclofoxy in long-term, methadone-treated former heroin addicts. J Pharmacol Exp Ther 295:1070–1076
Logan GD (1994) On the ability to inhibit thought and action. In: Dagenbach D, Carr TH (eds) Inhibitory processes in attention, memory, and language. Academic, San Diego, pp 189–239
Madden GJ, Petry NM, Badger GJ, Bickel WK (1997) Impulsive and self-control choices in opioid-dependent patients and non-drug-using control participants: drug and monetary rewards. Exp Clin Psychopharmacol 5:256–262
Mazur JE (1987) An adjusting procedure for studying delayed reinforcement. In: Commons ML, Mazur JE, Nevin JA, Rachlin H (eds) The effect of delay and of intervening events on reinforcement value. Erlbaum, Hillsdale, NJ, pp 55–73
Mitchell SH (2004) Measuring impulsivity and modeling its association with cigarette smoking. Behav Cogn Neurosci Rev 3:261–275
Mitchell JM, Fields HL, D’Esposito M, Boettiger CA (2005) Impulsive responding in alcoholics. Alcohol Clin Exp Res 29:2158–2169
Mitchell JM, Tavares VC, Fields HL, D'Esposito M, Boettiger CA (2007) Endogenous opioid blockade and impulsive responding in alcoholics and healthy controls. Neuropsychopharmacology 32:439–449
Monterosso JR, Aron AR, Cordova X, Xu J, London ED (2005) Deficits in response inhibition associated with chronic methamphetamine abuse. Drug Alcohol Depend 79:273–277
Mulder AH, Hogenboom F, Wardeh G, Schoffelmeer AN (1987) Morphine and enkephalins potently inhibit [3H]noradrenaline release from rat brain cortex synaptosomes: further evidence for a presynaptic localization of mu-opioid receptors. J Neurochem 48:1043–1047
Murphy ER, Robinson ES, Theobald DE, Dalley JW, Robbins TW (2008) Contrasting effects of selective lesions of nucleus accumbens core or shell on inhibitory control and amphetamine-induced impulsive behaviour. Eur J Neurosci 28:353–363
Odum AL, Madden GJ, Badger GJ, Bickel WK (2000) Needle sharing in opioid-dependent outpatients: psychological processes underlying risk. Drug Alcohol Depend 60:259–266
Paine TA, Olmstead MC (2004) Cocaine disrupts both behavioural inhibition and conditional discrimination in rats. Psychopharmacology (Berl) 175:443–450
Paine TA, Dringenberg HC, Olmstead MC (2003) Effects of chronic cocaine on impulsivity: relation to cortical serotonin mechanisms. Behav Brain Res 147:135–147
Passetti F, Clark L, Mehta MA, Joyce E, King M (2008) Neuropsychological predictors of clinical outcome in opiate addiction. Drug Alcohol Depend 94:82–91
Pattij T, Vanderschuren LJ (2008) The neuropharmacology of impulsive behaviour. Trends Pharmacol Sci 29:192–199
Pattij T, Janssen MC, Schepers I, Gonzalez-Cuevas G, De Vries TJ, Schoffelmeer AN (2007a) Effects of the cannabinoid CB(1) receptor antagonist rimonabant on distinct measures of impulsive behavior in rats. Psychopharmacology (Berl) 193:85–96
Pattij T, Janssen MC, Vanderschuren LJ, Schoffelmeer AN, van Gaalen MM (2007b) Involvement of dopamine D(1) and D(2) receptors in the nucleus accumbens core and shell in inhibitory response control. Psychopharmacology (Berl) 191:587–598
Perry JL, Carroll ME (2008) The role of impulsive behavior in drug abuse. Psychopharmacology (Berl) 200:1–16
Petry NM, Bickel WK, Arnett M (1998) Shortened time horizons and insensitivity to future consequences in heroin addicts. Addiction 93:729–738
Pitts RC, McKinney AP (2005) Effects of methylphenidate and morphine on delay-discount functions obtained within sessions. J Exp Anal Behav 83:297–314
Rada P, Mark GP, Pothos E, Hoebel BG (1991) Systemic morphine simultaneously decreases extracellular acetylcholine and increases dopamine in the nucleus accumbens of freely moving rats. Neuropharmacology 30:1133–1136
Robbins TW (2002) The 5-choice serial reaction time task: behavioural pharmacology and functional neurochemistry. Psychopharmacology (Berl) 163:362–380
Robinson ES, Eagle DM, Mar AC, Bari A, Banerjee G, Jiang X, Dalley JW, Robbins TW (2008) Similar effects of the selective noradrenaline reuptake inhibitor atomoxetine on three distinct forms of impulsivity in the rat. Neuropsychopharmacology 33:1028–1037
Rossetti ZL, Longu G, Mercuro G, Gessa GL (1993) Extraneuronal noradrenaline in the prefrontal cortex of morphine-dependent rats: tolerance and withdrawal mechanisms. Brain Res 609:316–320
Shoaib M, Spanagel R, Stohr T, Shippenberg TS (1995) Strain differences in the rewarding and dopamine-releasing effects of morphine in rats. Psychopharmacology (Berl) 117:240–247
Simon NW, Mendez IA, Setlow B (2007) Cocaine exposure causes long-term increases in impulsive choice. Behav Neurosci 121:543–549
Solanto MV, Abikoff H, Sonuga-Barke E, Schachar R, Logan GD, Wigal T, Hechtman L, Hinshaw S, Turkel E (2001) The ecological validity of delay aversion and response inhibition as measures of impulsivity in AD/HD: a supplement to the NIMH multimodal treatment study of AD/HD. J Abnorm Child Psychol 29:215–228
Solinas M, Goldberg SR (2005) Motivational effects of cannabinoids and opioids on food reinforcement depend on simultaneous activation of cannabinoid and opioid systems. Neuropsychopharmacology 30:2035–2045
Spanagel R, Herz A, Shippenberg TS (1992) Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci U S A 89:2046–2050
Starke K (2001) Presynaptic autoreceptors in the third decade: focus on alpha2-adrenoceptors. J Neurochem 78:685–693
Swanson CJ, Perry KW, Koch-Krueger S, Katner J, Svensson KA, Bymaster FP (2006) Effect of the attention deficit/hyperactivity disorder drug atomoxetine on extracellular concentrations of norepinephrine and dopamine in several brain regions of the rat. Neuropharmacology 50:755–760
Van Gaalen MM, Brueggeman RJ, Bronius PFC, Schoffelmeer AN, Vanderschuren LJ (2006a) Behavioral disinhibition requires dopamine receptor activation. Psychopharmacology 187:73–85
Van Gaalen MM, van Koten R, Schoffelmeer AN, Vanderschuren LJ (2006b) Critical involvement of dopaminergic neurotransmission in impulsive decision making. Biol Psychiatry 60:66–73
Verdejo-Garcia A, Perez-Garcia M (2007) Profile of executive deficits in cocaine and heroin polysubstance users: common and differential effects on separate executive components. Psychopharmacology (Berl) 190:517–530
Verdejo-Garcia AJ, Perales JC, Perez-Garcia M (2007) Cognitive impulsivity in cocaine and heroin polysubstance abusers. Addict Behav 32:950–966
Verdejo-Garcia A, Lawrence AJ, Clark L (2008) Impulsivity as a vulnerability marker for substance-use disorders: review of findings from high-risk research, problem gamblers and genetic association studies. Neurosci Biobehav Rev 32:777–810
Wade TR, De Wit H, Richards JB (2000) Effects of dopaminergic drugs on delayed reward as a measure of impulsive behavior in rats. Psychopharmacology (Berl) 150:90–101
Winstanley CA, Theobald DE, Dalley JW, Robbins TW (2005) Interactions between serotonin and dopamine in the control of impulsive choice in rats: therapeutic implications for impulse control disorders. Neuropsychopharmacology 30:669–682
Winstanley CA, Eagle DM, Robbins TW (2006) Behavioral models of impulsivity in relation to ADHD: translation between clinical and preclinical studies. Clin Psychol Rev 26:379–395
Winstanley CA, Bachtell RK, Theobald DE, Laali S, Green TA, Kumar A, Chakravarty S, Self DW, Nestler EJ (2009) Increased impulsivity during withdrawal from cocaine self-administration: role for deltaFosB in the orbitofrontal cortex. Cereb Cortex 19:435–444
Wise RA, Leone P, Rivest R, Leeb K (1995) Elevations of nucleus accumbens dopamine and DOPAC levels during intravenous heroin self-administration. Synapse 21:140–148
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Pattij, T., Schetters, D., Janssen, M.C.W. et al. Acute effects of morphine on distinct forms of impulsive behavior in rats. Psychopharmacology 205, 489–502 (2009). https://doi.org/10.1007/s00213-009-1558-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-009-1558-8