
Abstract
Sepsis is a significant cause of death worldwide. Although the prevailing theory of the sepsis syndrome has been that of a condition of uncontrolled inflammation in response to infection, sepsis is increasingly being recognized as an immunosuppressive state. The immune modulations of sepsis result in altered innate and adaptive immune responses, thereby rendering the septic host susceptible to secondary infections. In this review, we present an overview of the clinical and experimental evidence for sepsis-induced immunosuppression and outline the mechanisms that underlie this phenotype. With an improved understanding of how host immune states may be altered during sepsis, better immunomodulatory therapies may be developed to address the immune derangements observed in patients with sepsis.
Similar content being viewed by others


Introduction
Sepsis remains a major cause of morbidity and mortality in the United States and worldwide, despite advances in supportive care [1, 2]. Historically, sepsis was defined as the presence of pathogenic microorganisms or their toxins in the bloodstream and the term was used interchangeably with bacteremia [3]. More recently, the prevailing theory of sepsis has been that of a condition of uncontrolled inflammation in response to infection. In 1972, Lewis Thomas described sepsis in the following way: “It is our response to [the microorganism’s] presence that makes the disease. Our arsenals for fighting off bacteria are so powerful...that we are more in danger from them than the invaders.” [4]. In 1992, the American College of Chest Physicians and Society of Critical Care Medicine consensus conference officially defined sepsis as the systemic inflammatory response syndrome (SIRS) occurring as a result of infection [5]. While certain cases of sepsis, such as meningococcemia, are accurately characterized in this way [6], subsequent clinical and immunologic discoveries have challenged this view. In clinical trials, attempts at neutralizing inflammation and inflammatory mediators during sepsis, using agents such as high-dose corticosteroids, tumor necrosis factor (TNF) antagonists, and interleukin-1 (IL-1) pathway inhibitors, have been largely unsuccessful in terms of improving survival or other clinical outcomes [7–9].
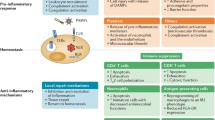
With a better understanding of the complexity of sepsis pathophysiology, it is now appreciated that anti-inflammatory immunologic events develop concurrently or subsequently during the time-course of sepsis (Fig. 1). Although anti-inflammatory responses likely are essential to the restoration of immune homeostasis following an inflammatory stimulus such as infection, this anti-inflammatory state can result in immunosuppression and subsequent death because of the inability to fight secondary infections in the post-septic period [10]. This has been demonstrated in animal models of sepsis using animals rendered septic by cecal ligation and puncture (CLP)-induced peritonitis. As early as 24 h following CLP, septic animals had marked impairment in their ability to clear secondary intrapulmonary challenge with Pseudomonas aeruginosa, as compared to non-septic controls. Even 2 weeks after CLP, enhanced susceptibility to Aspergillus infection was observed in mice with severe sepsis [11]. The immunosuppressive properties of the septic immune response are becoming increasingly relevant with continued improvements in critical care, as many deaths due to sepsis do not occur acutely but rather occur after a protracted hospital course [12, 13]. Therefore, therapies aimed at blocking pro-inflammatory mediators may be detrimental to septic hosts who are in a relatively immunosuppressed state.

Dynamics of the septic inflammatory response. This graphic illustrates the general framework for the immunologic response to sepsis over time. Upon a significant physiologic insult, such as overwhelming infection or traumatic injury, inflammatory components of the immune system are activated. At the same time, the body initiates a counteractive anti-inflammatory response presumably to restore immunologic homeostasis. After the initial septic period, however, anti-inflammatory components appear to predominate, resulting in a state of relative immunosuppression
Terms used to describe the anti-inflammatory events occurring during sepsis include “sepsis-induced immunosuppression,” “immunoparalysis,” and the “compensatory anti-inflammatory response syndrome” (CARS) [10, 13]. These terms reflect the presence of anergy, monocyte deactivation, and a potential increased risk of subsequent secondary infections among patients with sepsis [14]. Several main molecular and cellular mechanisms underlying the development of sepsis-induced immunosuppression (Figs. 1 and 2) have emerged from studies of septic patients and experimental animals, which will be discussed in detail in this review.

Sepsis-induced alterations in immune cell function. In septic patients, multiple aspects of leukocyte function are disrupted, leading to susceptibility to secondary infections among survivors of sepsis
Mechanisms of sepsis-induced immunosuppression
Anti-inflammatory cytokines
Many studies have examined cytokine levels during sepsis both in patients and experimental animals. Indeed, much of our understanding about sepsis pathophysiology is derived from analyzing patterns of cytokine expression, particularly over time. Experimental animal models of sepsis using systemic endotoxemia or CLP-induced septic peritonitis have demonstrated markedly increased levels of inflammatory cytokines such as IL-1, IL-6, and TNF-α [15, 16]. In contrast to the systemic endotoxemia model where animals develop a rapid spike and fall in inflammatory cytokine levels, mice that have undergone CLP initially develop concurrent elevations in circulating pro- and anti-inflammatory cytokines, but later on, acquire a predominantly anti-inflammatory profile [17, 18]. Similarly, in human patients with sepsis, a systemic elevation in pro-inflammatory cytokines is neither consistently nor persistently observed. On the other hand, anti-inflammatory cytokines, including IL-4, IL-10, IL-13, IL-1 receptor antagonist (IL-1ra), and transforming growth factor-beta (TGF-β) [14] are more consistently detected and typically elevated to a higher degree and for a longer duration of time [19].
These anti-inflammatory cytokines have the ability to inhibit the synthesis of IL-1, TNF-α, and other major pro-inflammatory cytokines such as IL-12 [20]. Of the anti-inflammatory cytokines, IL-10 has been the most studied and acts as a potent inhibitor of pro-inflammatory cytokine production by mononuclear cells. It also exerts a wide range of anti-inflammatory effects including inhibition of cell surface expression of class II major histocompatibility complex (MHC) expression by monocytes, downregulation of TNF receptors, inhibition of macrophage bactericidal activity, and inhibition of the crucial pro-inflammatory transcription factor, nuclear factor κB (NF-κB), after endotoxin stimulation [19, 20]. IL-10 has been found in multiple studies to be elevated in sepsis, with the degree of elevation correlating with fatal outcomes [21–23]. Levels of IL-10 remain higher in non-survivors of sepsis until 15 days after the onset of septic shock [22] and the ratio of IL-10 to TNF-α is significantly higher in patients who do not survive sepsis [21–23]. Furthermore, experimental animals rendered septic by CLP were significantly less susceptible to secondary pulmonary infection with P. aeruginosa when treated with IL-10 neutralizing antibodies [24].
Further support for the detrimental role of an anti-inflammatory response in sepsis has come from studies examining the role of caspases, a family of cysteinyl aspartate proteases involved in apoptosis and inflammatory cytokine processing. Several important recent studies performed by Saleh et al. have focused on the role of the anti-inflammatory caspase, caspase-12. They found that individuals who synthesized the long-form of caspase-12 were hyporesponsive to endotoxin and had a higher frequency of sepsis [25]. Further mechanistic basis for this observation was provided by a subsequent study showing that the targeted disruption of the caspase-12 gene in mice rendered the animals resistant to peritonitis and septic shock [26]. Splenocytes from caspase-12−/− animals elaborated higher levels of inflammatory cytokines including IL-1β and interferon-gamma (IFN-γ) following exposure to microbial ligands. Administration of neutralizing antibodies to IFN-γ receptors ablated the survival advantage that occurred in the caspase-12 deficient mice [26]. Collectively, these studies highlight how an imbalance towards an anti-inflammatory state is detrimental in host defense against sepsis.
Monocyte deactivation
During infection, monocytes are one of the primary effectors of innate immunity. Monocytes and macrophages ingest microbes and are a source of inflammatory mediators, which in turn, activate and recruit other innate immune cells. They can also exert direct antimicrobial effects via bactericidal activity and activate adaptive immune responses by serving as antigen-presenting cells [27].
Monocytes from patients with sepsis lose their ability to mount an inflammatory response after stimulation by bacterial products and instead increase their production of anti-inflammatory mediators such as IL-10 and IL-1ra [13, 28, 29]. In animal models of experimental sepsis, alveolar macrophages isolated as early as 24 h following CLP had decreased expression of TNF, IL-12, and other inflammatory cytokines in response to ex vivo stimulation by lipopolysaccharide (LPS). Peritoneal macrophages isolated from mice post-CLP were similarly hyporesponsive [30]. The hyporesponsiveness of macrophages from septic hosts to ex vivo stimulation with LPS has drawn many analogies to endotoxin tolerance, which will be discussed later in this review.
Another major characteristic of monocytes from sepsis is the decreased surface expression of human leukocyte antigen-DR (HLA-DR) [31, 32]. Low levels of HLA-DR expression are correlated with loss of monocyte functions such as the ability to produce pro-inflammatory cytokines and induce antigen-specific T cell responses [13]. This decrease in surface expression of MHC II molecules among septic and other critically ill patients has been well described in the literature. In the majority of these studies, low HLA-DR expression has been associated with poor outcomes [33]. In a prospective study of 93 patients with septic shock, persistently low monocyte HLA-DR values, defined as <30%, at days 3–4 after ICU admission were independently associated with mortality and served as a better predictor of mortality than organ dysfunction scores such as the Sequential Organ Failure Assessment (SOFA) score [34]. Similarly, persistently low HLA-DR expression in patients 4–10 days after severe burn injury predicted the development of secondary septic shock and death [35]. In sepsis survivors, a progressive elevation of HLA-DR expression has been shown in the first 2 weeks following shock, suggesting ongoing recovery of immunologic functions [22].
The clinical utility of identifying patients with decreased HLA-DR expression is limited by several unresolved issues. First, it remains uncertain whether decreased MHC class II expression actually leads to or merely is associated with sepsis-induced immunosuppression. Second, the benefit of treating septic patients with inflammatory cytokines—thereby enhancing HLA-DR levels—in large randomized clinical trials has not been demonstrated. Nonetheless, persistently low HLA-DR values have emerged as a potentially useful marker in identifying those patients with persistent immunoparalysis and may provide prognostic information. Ongoing studies are necessary to determine whether HLA-DR levels can be measured accurately and reproducibly in different cohorts of septic and critically ill patients or healthy controls.
Attempts have been made to restore macrophage function by treating with inflammatory cytokines. In one study, the transient transgenic expression of TNF-α in the lungs of septic animals restored multiple aspects of alveolar macrophage function, including phagocytic function, which resulted in an enhanced ability to clear secondary bacterial lung infection [36]. Two small studies have evaluated the use of IFN-γ in patients with sepsis and low monocytic HLA-DR expression [37, 38]. Treatment with IFN-γ increased monocytic HLA-DR expression and recovered their capacity to produce TNF-α and IL-6 [37, 38]. Eight out of nine patients in one study subsequently recovered from sepsis and also significantly improved their multiple organ dysfunction scores [37]. Finally, granulocyte macrophage-colony stimulating factor (GM-CSF) is a cytokine that increases HLA-DR expression on monocytes leading to increased pro-inflammatory cytokine production [39]. Down-modulation of the GM-CSF receptor on septic monocytes has recently been demonstrated and could partially explain the monocyte deactivation linked to sepsis [40]. In a study of nine patients with severe sepsis and associated low HLA-DR expression, administering three days of GM-CSF significantly increased HLA-DR expression as well as the ex vivo production of TNF-α after endotoxin stimulation [41]. It has also been shown to improve gas exchange in severe sepsis with respiratory failure [42] and improve mortality in the setting of neutropenic neonatal sepsis without any untoward side effects [41].
The loss and dysfunction of dendritic cells
Dendritic cells (DCs) play an essential role in adaptive immunity and immune activation as antigen-presenting cells. In addition, they are an important source of cytokines upon microbial challenge, thereby polarizing the subsequent T cell response to type 1, type 2, or regulatory phenotype. Human autopsy studies and studies of experimental animals undergoing CLP have shown a profound loss of splenic DCs, which leads to significant compromise of B and T cell function [43–45]. In animal studies, a similar loss of DCs has also been shown in the lymph nodes and lungs [46, 47]. There is evidence of increased apoptosis as the mechanism for this loss, but a detrimental influence on DC development may also play a role [46, 47].
In addition to depletion of DCs, sepsis also impairs the ability of DCs to initiate type 1 cytokine responses. Splenic and lung DCs isolated from mice subjected to CLP are skewed toward a TH2-type cytokine profile with enhanced IL-10 synthesis and reduced IL-12 synthesis following ex vivo challenge with microbial components [47–49]. In the lung, this phenotype persists even after restoration of the DC cell population in the post-septic period. In mice undergoing CLP, intrapulmonary instillation of DCs from non-septic mice was able to restore the antifungal host response in the lung and prevent fatal Aspergillus infection [48]. Similar impairment of DC function has been described following other forms of significant physiologic insults, such as trauma/hemorrhagic shock [50].
Impairment of neutrophils
Neutrophils are a critical cellular component of innate immunity against a wide variety of pathogens, including bacteria and fungi. Multiple aspects of neutrophil function are dysregulated during sepsis. While sepsis can cause elevated, normal, or reduced neutrophil counts, neutropenia has been linked to poorer outcomes in sepsis. Circulating neutrophils in patients with sepsis have been found to express increased levels of activation markers, including CD11b, ICAM-1, MPO, and CD66b on their cell surfaces [51]. Despite the increased expression of adhesion molecules, neutrophilic adherence and migration is impaired in sepsis [51–54]. Furthermore, expression of neutrophil activation markers is significantly lower in patients who do not survive sepsis [51]. In the lung, significant downregulation of the CXCR2 chemokine receptor also occurs on septic neutrophils leading to decreased neutrophil recruitment and impairment of bacterial clearance from the lung [55–57]. During sepsis, however, the beneficial effects of neutrophils (i.e., antimicrobial functions) must be weighed against the destructive potential of activated neutrophils, which have been shown to mediate lung injury and multiple organ failure [58]. A trial investigating the use of recombinant G-CSF, which augments neutrophil function and number, in septic patients was largely unsuccessful in terms of improving overall clinical outcomes [59].
Apoptosis and lymphocyte dysfunction
Multiple studies by Hotchkiss and Karl have demonstrated that apoptosis plays an important role in mediating sepsis-induced immunosuppression. In addition to contributing to the loss of DCs, apoptosis is the primary mechanism of lymphocyte cell death in the setting of sepsis [14]. In autopsy studies, the spleen and colon were the two organs exhibiting the greatest degree of cell death with apoptosis as the major mechanism of lymphocyte loss [60, 61]. Specifically, there was a profound depletion of B cells and CD4+ T cells in the spleens of septic patients [62] which was not observed in critically ill non-septic patients [60]. Because lymphocytes produce pro-inflammatory cytokines, activate macrophages, and produce antibodies, the loss of lymphocytes in sepsis may contribute to sepsis-induced immunosuppression [60, 61]. Intensive care unit patients who develop a decreased lymphocyte count for >3 days are at a greatly increased risk of nosocomial sepsis [63]. In experimental animals undergoing CLP, inhibition of apoptosis by using caspase inhibitors or performing studies in caspase-3 knockout mice resulted in improved survival [61, 64]. Apoptotic T lymphocyte loss in sepsis is believed to contribute to the development of anergy, which is defined as a lack of response to skin testing with antigens derived from microbes to which previous exposure would be expected. This state of impaired delayed hypersensitivity is commonly present in sepsis and reflects monocyte defects in antigen processing and defective T cell cytokine secretion in response to specific antigens [14, 65]. Anergy has been identified as a marker of sepsis and mortality in surgical patients. An early study by Meakins et al. reported a mortality of 5% in surgical patients who improved their response to skin testing compared to 74% mortality in patients whose skin tests failed to improve [66].
There is also abundant evidence that apoptotic cells themselves modulate the inflammatory response in sepsis. The presence of apoptotic cells during monocyte activation has been shown to increase their secretion of IL-10 and TGF-β while decreasing the secretion of TNF-α, IL-1, and IL-12 [67, 68]. This shift of pro-inflammatory to anti-inflammatory cytokines in response to endotoxin further impairs the host response to pathogens. In adoptive transfer experiments, transfer of apoptotic splenocytes to animals undergoing CLP resulted in decreased survival compared to untreated animals. Interestingly, in the same studies, transfer of necrotic cells was associated with higher levels of IFN-γ production by splenocytes and improved survival following CLP [69].
Toll-like receptors and endotoxin tolerance
Toll-like receptors (TLRs) are a critical family of pattern recognition receptors that recognize a variety of microbial components, referred to as pathogen-associated molecular patterns (PAMPs). PAMPs include LPS, double-stranded RNA, flagellin, and microbial DNA. Toll-like receptors are expressed on a variety of cell types, including leukocytes, endothelial cells, and fibroblasts. Upon TLR ligation, intracellular signaling kinases are activated, ultimately leading to activation of multiple transcription factors, particularly NF-κB, that lead to inflammatory gene expression.
Polymorphisms in the TLRs and signaling intermediates have been linked to increased risks of infection. Mutations in TLR4 and CD14, which form the main receptor complex for LPS, have been examined regarding their role in mediating endotoxin responsiveness. In mice, a TLR4 mutation confers resistance to endotoxin, but also leads to increased susceptibility to gram-negative infections [70]. In humans, single nucleotide polymorphisms (SNPs) identified in TLR4 and CD14 have been linked to an endotoxin hyporesponsive phenotype [71]. Specifically, the Asp299Gly and the Thr399Ile cosegregating SNPs occurring in the extracellular portion of TLR4 have been identified as occurring at a higher frequency among individuals displaying decreased airway responsiveness to inhaled LPS [71]. These SNPs are present in approximately 10% of white individuals and this population may be more susceptible to a systemic inflammatory response initiated or exacerbated by endotoxin [72]. It is interesting to note that carriers of these SNPs appear to have a higher incidence of gram-negative infections (post-surgical patients) [73] and higher rates of gram-negative septic shock (medical ICU patients) [74] Similarly a polymorphism has been identified for TLR2, the receptor for many gram-positive organisms and fungi, and this polymorphism may be associated with staphylococcal infections [75]. Thus, genetic factors may influence a host’s immune state during sepsis and these factors are discussed further below.
Endotoxin tolerance is the phenomenon whereby a cell develops reduced endotoxin responsiveness following repeated exposure to LPS [76]. Pro-inflammatory cytokine secretion, especially TNF-α, is markedly diminished in endotoxin-tolerant animals and humans [76]. The phenomenon of tolerance has also been described with other TLR ligands, including lipoteichoic acid and flagellin [77–79]. Mechanisms underlying endotoxin tolerance include downregulation of TLR-4 (LPS receptor) expression, inhibition of downstream TLR signaling intermediates such as Interleukin-1 Receptor Kinase (IRAK)-1, and alterations in the NF-κB subunits [80–85]. These mechanisms may serve as an important means whereby the host limits the inflammatory response to an ongoing immune stimulus, thereby protecting itself from further injury.
Striking similarities have been observed between sepsis-induced macrophage dysfunction and endotoxin-tolerized macrophages. Both endotoxin-tolerant cells and monocytes isolated from septic patients have a predominance of p50 homodimers, which is the functionally inactive form of NF-κB [86, 87]. Interleukin-1 receptor-associated kinase-M (IRAK-M) has recently been identified as an inhibitor of TLR signaling and is implicated in mediating both peptidoglycan (TLR2 ligand) and LPS tolerance [80, 82]. It is interesting to note that in macrophages and monocytes isolated from patients and experimental animals with sepsis, IRAK-M expression is upregulated upon ex vivo LPS stimulation [88, 89]. Furthermore, IRAK-M knockout mice are relatively resistant to the development of sepsis-induced immunosuppression in terms of their enhanced ability to clear a secondary lung bacterial challenge following CLP [88]. These studies suggest that common molecular mechanisms may underlie the hyporesponsiveness to LPS exhibited by monocytes from patients with sepsis and endotoxin-tolerant cells.
The clinical significance of hyporesponsiveness to LPS in sepsis has been examined. Lower levels of endotoxin-stimulated TNF-α production are associated with poorer outcomes in ICU patients [90, 91]. Monocytes isolated from patients who went on to survive their septic episode were found to regain LPS responsiveness whereas normal reactivity was never restored in non-survivors [29]. In surgical intensive care units, low levels of TNF-α and IL-6 following LPS stimulation correlated with longer ICU length of stay, more ventilator days, higher incidence of infection, and a higher white blood cell count [91]. Thus, the phenomenon of endotoxin/TLR tolerance is thought to play an important role in the susceptibility to reinfection in patients with severe sepsis [92].
Endotoxin tolerance can be prevented by the administration of IFN-γ or GM-CSF [93, 94]. Interferon-γ augments the mRNA and surface expression of TLR4 and counteracts the LPS-induced downregulation of TLR4. Human monocytes primed with IFN-γ show increased responsiveness to LPS by increasing NF-κB binding activity as well as the secretion of TNF-α [94]. When LPS-tolerized human monocytes are pretreated with GM-CSF or IFN-γ, they do not exhibit endotoxin tolerance and are able to activate NF-κB and secrete TNF-α [93]. This phenomenon appears to be independent of the modulation of TLR2 or TLR4 expression.
Other genetic factors
In addition to the TLR polymorphisms discussed above, a variety of known and unknown genetic factors play an important role in any individual patient’s susceptibility to sepsis. Given the immense literature in this field, a comprehensive examination of this topic is beyond the scope of this review, and we direct the reader towards two excellent recent reviews [95, 96]. Here, we will present a brief overview and some observations. Of all the cytokines, polymorphisms in TNF, IL-6, and IL-10 have been most extensively studied as they relate to sepsis risk. An inherited risk for death from meningococcemia has been shown in identical twins and families with a phenotype of decreased pro-inflammatory (TNF) or increased anti-inflammatory (IL-10) response [97]. The TNF2 allele, which correlates with enhanced TNF production, is more common in patients with septic shock than healthy controls and, in those with septic shock, it is more common in non-survivors [98]. Among coagulation pathway-related genes, polymorphisms in the protein C gene have affected survival and organ dysfunction scores in a study of Caucasian patients [98]. Following burn injury, polymorphisms within TNF-α, TLR4, IL-6, and CD14 have been associated with an increased risk for severe sepsis [99]. Gender itself appears to affect the response to infection as shown by a study of experimental human endotoxemia in which healthy females showed a more pro-inflammatory response compared to healthy male subjects [100].
Hyporesponsiveness to endotoxin per se, however, does not necessarily translate into a predisposition to sepsis or septic shock. For example, a polymorphism occurring in the CD14 promoter region (C-159T) has been studied extensively in various clinical populations with sepsis. Monocytes isolated from TT homozygous patients have been shown to produce more sCD14, IL-6, and TNF-α than C/T heterozygotes or CC homozygotes, thereby conferring a pro-inflammatory state systemically [101]. Studies of septic patient populations, however, have demonstrated inconsistent results in terms of whether possessing a particular allele (C or T) or genotype (CC vs. C/T vs. TT) is associated with increased rates of sepsis or poorer outcomes following onset of sepsis. Two German studies found no association between genotype and the development of sepsis, whereas one American study of burn victims demonstrated that the C allele was a risk factor for the development of sepsis [99, 102]. Conversely, in a French study of 90 Caucasian patients with septic shock, the TT genotype was found at higher frequency in patients with septic shock and non-survivors of septic shock, as compared to healthy controls or survivors of sepsis [103]. Given our current understanding that sepsis is clearly not a homogenous immune state, these studies highlight the inherent complexity of establishing causal associations between a host’s genetic predisposition and the development/outcomes of sepsis.
Furthermore, given the multi-faceted aspects of sepsis-induced immunosuppression, other factors may overwhelm the phenotype of a particular genetic polymorphism. For example, the TLR4 Asp299Gly/Thr399Ile SNP associated with reduced responsiveness to inhaled endotoxin has been examined in post-surgical patients. Carriers of this mutation had similar rates of hyporesponsiveness following whole blood stimulation by LPS as wildtype post-operative patients [104]. This suggests that certain inflammatory stimuli (in this case, major surgery) lead to a generalized modulation of the endotoxin response, which ultimately determines the host’s immune state more so than specific genotype. It is interesting to note that patients predisposed to produce a balanced anti-, pro-inflammatory response appear to have the best chance for survival in sepsis [105].
Clinical significance
Currently, a widely held model of sepsis is that immunologically, sepsis evolves from initial hyperinflammation (SIRS), towards a period of relative homeostasis (mixed anti-inflammatory response syndrome, or MARS), and, finally, a state of immunosuppression (CARS). Unfortunately, this orderly model is poorly supported by clinical or in vivo evidence [17]. Studies using experimental animal models of sepsis have suggested that the mechanisms of death occurring early after the onset of sepsis differ from those occurring at later timepoints. In one study, mice that died early (i.e., within 4 days) following CLP-induced septic peritonitis had elevated plasma IL-6, whereas animals that died later had variable IL-6 levels and hyporesponsive macrophages to inflammatory stimuli ex vivo [18]. In another study examining late sepsis mortality (days 6–28) in a murine model, the late prelethal inflammatory response varied considerably from a virtually non-existent response to the presence of nearly all measured pro- and anti-inflammatory cytokines [106]. This suggests that the SIRS-to-CARS transition is not linear but rather constantly fluctuating between hyper-responsiveness and hyporesponsiveness in different hosts. In the clinical setting, the immunologic state of a septic patient is further modulated by a variety of internal and external factors, including genetic makeup, comorbidities, medications with immunomodulatory properties (e.g. steroids), and therapeutic interventions (e.g. surgery, blood product transfusions, etc.; Fig. 3) Therefore, similar to the murine model, rather than undergoing a one-way transition from SIRS to CARS, a septic patient may very well fluctuate back and forth along this spectrum multiple times during their hospital course.

Temporal development of sepsis-induced fluctuations on immune response. Experimental and clinical evidence demonstrate that an orderly progression from pro-inflammatory/SIRS state to an anti-inflammatory/CARS state does not occur following sepsis. Particularly in the clinical setting, multiple other factors, including those listed in the box, will tilt the balance towards one end of the spectrum or the other. (SIRS, systemic inflammatory response syndrome; MARS, mixed anti-inflammatory response syndrome; CARS, compensatory anti-inflammatory response syndrome)
Nonetheless, it has been recognized for many years that survivors of sepsis have an increased risk of death for up to 5 years following the event, even when their underlying medical comorbidities are accounted for [107]. This increased risk of death correlates with the severity of the initial sepsis [107]. Survivors of sepsis also have a high rehospitalization rate of approximately 50% in the first year after their septic episode [108]. It could be hypothesized that this subsequent elevated risk of death and rehospitalization is secondary to a persistent state of immunosuppression and associated recurrent infections. Indeed, we have observed in an ongoing study that patients who survive sepsis have higher rates of infections during the first year as compared to survivors of other forms of critical illness (T. Wang, unpublished data). In the surgical literature, it has been shown that the majority of life-threatening complications that occur in surgical patients are secondary to the failure to control infections, and the phenomenon of immune paralysis has been well documented in this population after major surgery or trauma [109–111]. Thus, developing therapies to enhance host resistance against secondary infections will be an important therapeutic strategy in patients with sepsis.
Potential therapeutic applications
From previous failed clinical trials targeting inflammation, the therapeutic strategy for designing immunomodulatory treatments has changed. For example, high-dose corticosteroids were initially thought to be beneficial in the treatment of sepsis. Experimental data obtained from endotoxin shock models supported the use of high-dose steroids [112], before large clinical trials confirmed the lack of benefit as well as the increased risk of secondary infections [113, 114]. In retrospect, given our current understanding that many patients with sepsis develop a relative state of immunosuppression, high-dose glucocorticoids may serve to exacerbate the host’s susceptibility to secondary infections. Therefore, anti-inflammatory therapies that are beneficial in markedly inflammatory states such as that induced by experimental endotoxemia may not translate into clinical practice, where the immune status during sepsis is considerably more heterogeneous. Subsequently, corticosteroids have been shown to be beneficial only when used in replacement doses for those patients with relative adrenal insufficiency in sepsis [115].
It has become evident that patients have different immunologic profiles in sepsis and multiple factors, including genetic polymorphisms, the virulence of the organism, and timing, affect this profile. Thus, successful treatment of sepsis requires a better understanding of each individual patient’s immunologic status. An example of this was a large placebo-controlled trial of the monoclonal anti-TNF antibody afelimomab in which the investigators attempted to stratify patients based on their IL-6 levels. In this study, there was a small but significant reduction in 28-day all-cause mortality, which was more marked in the population of patients with elevated IL-6 levels [116]. As mentioned above, in small studies, IFN-γ and GM-CSF have shown some promise in increasing monocyte HLA-DR expression, preventing endotoxin tolerance and helping patients clear sepsis. Another cytokine that may be targeted in future therapeutic trials is IL-12, which has been examined in preclinical models of burn injury-induced immunosuppression. In these studies, administration of IL-12 to burn-injured animals has been shown to improve survival and enhance resistance against secondary sepsis [117, 118].
In general, however, cytokine targets should be used with caution given the complexity and ever-changing nature of each patient’s immunologic profile. Simply stated, if we are trying to replace something that is missing or block something that is deleterious, constant monitoring will have to be performed to assure that we do not shift the anti-inflammatory cascade back into an inflammatory one or vice-versa. It is also likely that “deleterious” cytokines have beneficial effects that may not be realized until they are neutralized.
Volk et al. utilized plasmapheresis in patients with persistently low HLA-DR expression with the hopes of eliminating anti-inflammatory cytokines and other inhibitory factors. The 28-day survival rate of the plasmapheresis group was significantly higher than the control group (48% vs. 20%). None of the patients who failed to improve their HLA-DR expression with plasmapheresis survived while only one patient out of 18 died in the group that normalized their HLA-DR expression with plasmapheresis [13]. A large placebo-controlled multi-center trial is needed to verify these promising results. There is also much hope and potential with the elucidation of TLR signaling pathways as they represent a novel target for therapeutic agents in sepsis. Depending on the phase of sepsis that a patient is in, agonists or antagonists of TLR signaling pathways could be utilized to boost or depress innate immunity respectively.
Recent interest has emerged in the use of statins prior to or during sepsis. In a large observational population based study, the use of statins in patients with atherosclerosis was associated with a reduced risk of subsequent sepsis [119]. A subsequent observational study did not confirm this finding for community-acquired pneumonia suggesting the possible presence of confounding factors and the need for formal clinical trials [120]. Since statins do not target individual inflammatory mediators but rather reduce the overall magnitude of the systemic response, this class of medications could provide a unique benefit in sepsis [121]. It remains to be determined whether statins will be beneficial as pre-treatment or treatment for sepsis in the clinical setting.
Conclusion
Sepsis can no longer be characterized as the systemic inflammatory response syndrome associated with infection. Rather, patients with sepsis acquire heterogeneous immune phenotypes with immunologic disequilibrium that vary not only from individual to individual, but over time within a given individual. An individual’s immune response to sepsis can be modulated by a variety of factors—the nature of the infectious stimulus itself, the host genetic makeup (i.e., predisposition to inflammation), comorbidities, and exogenous factors (e.g., medications, blood transfusions, etc.). Thus, septic patients are not a homogenous group and therein lies the difficulty that physicians and scientists have faced in prior attempts to design effective therapies. Some patients may need suppression of their inflammatory response but other patients, especially those who survive their initial bout of sepsis, may need therapies that enhance their immune system and restore their ability to mount an inflammatory response. As we identify more reliable markers of sepsis-induced immunosuppression, such as HLA-DR expression, and find effective ways of targeting these markers, we move closer to being able to make a significant impact on the high initial and subsequent mortality in patients suffering from sepsis.
References
Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR (2001) Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 29:1303–1310
Martin GS, Mannino DM, Eaton S, Moss M (2003) The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med 348:1546–1554
Bone RC (1991) Gram-negative sepsis. Background, clinical features, and intervention. Chest 100:802–808
Thomas L (1972) Germs. N Engl J Med 287:553–555
Bone RC, Balk RA, Cerra FB et al (1992) Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 101:1644–1655
Girardin E, Grau GE, Dayer JM, Roux-Lombard P, Lambert PH (1988) Tumor necrosis factor and interleukin-1 in the serum of children with severe infectious purpura. N Engl J Med 319:397–400
Cronin L, Cook DJ, Carlet J et al (1995) Corticosteroid treatment for sepsis: a critical appraisal and meta-analysis of the literature. Crit Care Med 23:1430–1439
Fisher CJ Jr., Agosti JM, Opal SM et al (1996) Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N Engl J Med 334:1697–1702
Remick DG (2003) Cytokine therapeutics for the treatment of sepsis: why has nothing worked? Curr Pharm Des 9:75–82
Bone RC (1996) Immunologic dissonance: a continuing evolution in our understanding of the systemic inflammatory response syndrome (SIRS) and the multiple organ dysfunction syndrome (MODS). Ann Intern Med 125:680–687
Benjamim CF, Hogaboam CM, Lukacs NW, Kunkel SL (2003) Septic mice are susceptible to pulmonary aspergillosis. Am J Pathol 163:2605–2617
Fink MP, Heard SO (1990) Laboratory models of sepsis and septic shock. J Surg Res 49:186–196
Volk HD, Reinke P, Krausch D et al (1996) Monocyte deactivation—rationale for a new therapeutic strategy in sepsis. Intensive Care Med 22(Suppl 4):S474–481
Hotchkiss RS, Karl IE (2003) The pathophysiology and treatment of sepsis. N Engl J Med 348:138–150
Evans GF, Snyder YM, Butler LD, Zuckerman SH (1989) Differential expression of interleukin-1 and tumor necrosis factor in murine septic shock models. Circ Shock 29:279–290
Villa P, Sartor G, Angelini M et al (1995) Pattern of cytokines and pharmacomodulation in sepsis induced by cecal ligation and puncture compared with that induced by endotoxin. Clin Diagn Lab Immunol 2:549–553
Osuchowski MF, Welch K, Siddiqui J, Remick DG (2006) Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J Immunol 177:1967–1974
Xiao H, Siddiqui J, Remick DG (2006) Mechanisms of mortality in early and late sepsis. Infect Immun 74:5227–5235
van der Poll T, van Deventer SJ (1999) Cytokines and anticytokines in the pathogenesis of sepsis. Infect Dis Clin North Am 13:413–426 ix
Opal SM, DePalo VA (2000) Anti-inflammatory cytokines. Chest 117:1162–1172
Gogos CA, Drosou E, Bassaris HP, Skoutelis A (2000) Pro- versus anti-inflammatory cytokine profile in patients with severe sepsis: a marker for prognosis and future therapeutic options. J Infect Dis 181:176–180
Monneret G, Finck ME, Venet F et al (2004) The anti-inflammatory response dominates after septic shock: association of low monocyte HLA-DR expression and high interleukin-10 concentration. Immunol Lett 95:193–198
van Dissel JT, van Langevelde P, Westendorp RG, Kwappenberg K, Frolich M (1998) Anti-inflammatory cytokine profile and mortality in febrile patients. Lancet 351:950–953
Steinhauser ML, Hogaboam CM, Kunkel SL, Lukacs NW, Strieter RM, Standiford TJ (1999) IL-10 is a major mediator of sepsis-induced impairment in lung antibacterial host defense. J Immunol 162:392–399
Saleh M, Vaillancourt JP, Graham RK et al (2004) Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature 429:75–79
Saleh M, Mathison JC, Wolinski MK et al (2006) Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature 440:1064–1068
Cavaillon JM, Adib-Conquy M (2005) Monocytes/macrophages and sepsis. Crit Care Med 33:S506–509
Astiz M, Saha D, Lustbader D, Lin R, Rackow E (1996) Monocyte response to bacterial toxins, expression of cell surface receptors, and release of anti-inflammatory cytokines during sepsis. J Lab Clin Med 128:594–600
Munoz C, Carlet J, Fitting C, Misset B, Bleriot JP, Cavaillon JM (1991) Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest 88:1747–1754
Ayala A, Kisala JM, Felt JA, Perrin MM, Chaudry IH (1992) Does endotoxin tolerance prevent the release of inflammatory monokines (interleukin 1, interleukin 6, or tumor necrosis factor) during sepsis? Arch Surg 127:191–196 discussion 196–197
Fumeaux T, Pugin J (2002) Role of interleukin-10 in the intracellular sequestration of human leukocyte antigen-DR in monocytes during septic shock. Am J Respir Crit Care Med 166:1475–1482
Lin RY, Astiz ME, Saxon JC, Rackow EC (1993) Altered leukocyte immunophenotypes in septic shock. Studies of HLA-DR, CD11b, CD14, and IL-2R expression. Chest 104:847–853
Fumeaux T, Pugin J (2006) Is the measurement of monocytes HLA-DR expression useful in patients with sepsis? Intensive Care Med 32:1106–1108
Monneret G, Lepape A, Voirin N et al (2006) Persisting low monocyte human leukocyte antigen-DR expression predicts mortality in septic shock. Intensive Care Med 32:1175–1183
Venet F, Tissot S, Debard AL et al (2007) Decreased monocyte human leukocyte antigen-DR expression after severe burn injury: correlation with severity and secondary septic shock. Crit Care Med 35:1910–1917
Chen GH, Reddy RC, Newstead MW, Tateda K, Kyasapura BL, Standiford TJ (2000) Intrapulmonary TNF gene therapy reverses sepsis-induced suppression of lung antibacterial host defense. J Immunol 165:6496–6503
Docke WD, Randow F, Syrbe U et al (1997) Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med 3:678–681
Kox WJ, Bone RC, Krausch D et al (1997) Interferon gamma-1b in the treatment of compensatory anti-inflammatory response syndrome. A new approach: proof of principle. Arch Intern Med 157:389–393
Hornell TM, Beresford GW, Bushey A, Boss JM, Mellins ED (2003) Regulation of the class II MHC pathway in primary human monocytes by granulocyte-macrophage colony-stimulating factor. J Immunol 171:2374–2383
Pangault C, Le Tulzo Y, Tattevin P, Guilloux V, Bescher N, Drenou B (2006) Down-modulation of granulocyte macrophage-colony stimulating factor receptor on monocytes during human septic shock. Crit Care Med 34:1193–1201
Nierhaus A, Montag B, Timmler N et al (2003) Reversal of immunoparalysis by recombinant human granulocyte-macrophage colony-stimulating factor in patients with severe sepsis. Intensive Care Med 29:646–651
Presneill JJ, Harris T, Stewart AG, Cade JF, Wilson JW (2002) A randomized phase II trial of granulocyte-macrophage colony-stimulating factor therapy in severe sepsis with respiratory dysfunction. Am J Respir Crit Care Med 166:138–143
Ding Y, Chung CS, Newton S et al (2004) Polymicrobial sepsis induces divergent effects on splenic and peritoneal dendritic cell function in mice. Shock 22:137–144
Hotchkiss RS, Tinsley KW, Swanson PE et al (2002) Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol 168:2493–2500
Tinsley KW, Grayson MH, Swanson PE et al (2003) Sepsis induces apoptosis and profound depletion of splenic interdigitating and follicular dendritic cells. J Immunol 171:909–914
Efron PA, Martins A, Minnich D et al (2004) Characterization of the systemic loss of dendritic cells in murine lymph nodes during polymicrobial sepsis. J Immunol 173:3035–3043
Wen H, Hogaboam CM, Gauldie J, Kunkel SL (2006) Severe sepsis exacerbates cell-mediated immunity in the lung due to an altered dendritic cell cytokine profile. Am J Pathol 168:1940–1950
Benjamim CF, Lundy SK, Lukacs NW, Hogaboam CM, Kunkel SL (2005) Reversal of long-term sepsis-induced immunosuppression by dendritic cells. Blood 105:3588–3595
Flohe SB, Agrawal H, Schmitz D, Gertz M, Flohe S, Schade FU (2006) Dendritic cells during polymicrobial sepsis rapidly mature but fail to initiate a protective Th1-type immune response. J Leukoc Biol 79:473–481
Kawasaki T, Hubbard WJ, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH (2006) Trauma-hemorrhage induces depressed splenic dendritic cell functions in mice. J Immunol 177:4514–4520
Muller Kobold AC, Tulleken JE, Zijlstra JG et al (2000) Leukocyte activation in sepsis; correlations with disease state and mortality. Intensive Care Med 26:883–892
Benjamim CF, Ferreira SH, Cunha FQ (2000) Role of nitric oxide in the failure of neutrophil migration in sepsis. J Infect Dis 182:214–223
Tavares-Murta BM, Zaparoli M, Ferreira RB et al (2002) Failure of neutrophil chemotactic function in septic patients. Crit Care Med 30:1056–1061
Terregino CA, Lubkin CL, Thom SR (1997) Impaired neutrophil adherence as an early marker of systemic inflammatory response syndrome and severe sepsis. Ann Emerg Med 29:400–403
Goodman R, Cummings C, Frevert C, Quan J, Martin T (1999) Functional significance of CXCR2 downregulation on neutrophils from patients with severe sepsis. Chest 116:111S–112S
Reddy RC, Chen GH, Tekchandani PK, Standiford TJ (2001) Sepsis-induced immunosuppression: from bad to worse. Immunol Res 24:273–287
Tsai WC, Strieter RM, Mehrad B, Newstead MW, Zeng X, Standiford TJ (2000) CXC chemokine receptor CXCR2 is essential for protective innate host response in murine Pseudomonas aeruginosa pneumonia. Infect Immun 68:4289–4296
Hoesel LM, Neff TA, Neff SB et al (2005) Harmful and protective roles of neutrophils in sepsis. Shock 24:40–47
Root RK, Lodato RF, Patrick W et al (2003) Multicenter, double-blind, placebo-controlled study of the use of filgrastim in patients hospitalized with pneumonia and severe sepsis. Crit Care Med 31:367–373
Hotchkiss RS, Swanson PE, Freeman BD et al (1999) Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med 27:1230–1251
Hotchkiss RS, Tinsley KW, Swanson PE et al (1999) Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci U S A 96:14541–14546
Hotchkiss RS, Tinsley KW, Swanson PE et al (2001) Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol 166:6952–6963
Rajan G, Sleigh JW (1997) Lymphocyte counts and the development of nosocomial sepsis. Intensive Care Med 23:1187
Hotchkiss RS, Chang KC, Swanson PE et al (2000) Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat Immunol 1:496–501
Pinsky MR (2004) Dysregulation of the immune response in severe sepsis. Am J Med Sci 328:220–229
Meakins JL, Pietsch JB, Bubenick O et al (1977) Delayed hypersensitivity: indicator of acquired failure of host defenses in sepsis and trauma. Ann Surg 186:241–250
Barker RN, Erwig L, Pearce WP, Devine A, Rees AJ (1999) Differential effects of necrotic or apoptotic cell uptake on antigen presentation by macrophages. Pathobiology 67:302–305
Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I (1997) Immunosuppressive effects of apoptotic cells. Nature 390:350–351
Hotchkiss RS, Chang KC, Grayson MH et al (2003) Adoptive transfer of apoptotic splenocytes worsens survival, whereas adoptive transfer of necrotic splenocytes improves survival in sepsis. Proc Natl Acad Sci U S A 100:6724–6729
Poltorak A, He X, Smirnova I et al (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–2088
Arbour NC, Lorenz E, Schutte BC et al (2000) TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet 25:187–191
Schroder NW, Schumann RR (2005) Single nucleotide polymorphisms of Toll-like receptors and susceptibility to infectious disease. Lancet Infect Dis 5:156–164
Agnese DM, Calvano JE, Hahm SJ et al (2002) Human toll-like receptor 4 mutations but not CD14 polymorphisms are associated with an increased risk of gram-negative infections. J Infect Dis 186:1522–1525
Lorenz E, Mira JP, Frees KL, Schwartz DA (2002) Relevance of mutations in the TLR4 receptor in patients with gram-negative septic shock. Arch Intern Med 162:1028–1032
Lorenz E, Mira JP, Cornish KL, Arbour NC, Schwartz DA (2000) A novel polymorphism in the toll-like receptor 2 gene and its potential association with staphylococcal infection. Infect Immun 68:6398–6401
Cavaillon JM, Adib-Conquy M (2006) Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit Care 10:233
Lehner MD, Morath S, Michelsen KS, Schumann RR, Hartung T (2001) Induction of cross-tolerance by lipopolysaccharide and highly purified lipoteichoic acid via different Toll-like receptors independent of paracrine mediators. J Immunol 166:5161–5167
Mizel SB, Snipes JA (2002) Gram-negative flagellin-induced self-tolerance is associated with a block in interleukin-1 receptor-associated kinase release from toll-like receptor 5. J Biol Chem 277:22414–22420
Sato S, Nomura F, Kawai T et al (2000) Synergy and cross-tolerance between toll-like receptor (TLR) 2- and TLR4-mediated signaling pathways. J Immunol 165:7096–7101
Kobayashi K, Hernandez LD, Galan JE, Janeway CA Jr., Medzhitov R, Flavell RA (2002) IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110:191–202
Sato S, Takeuchi O, Fujita T, Tomizawa H, Takeda K, Akira S (2002) A variety of microbial components induce tolerance to lipopolysaccharide by differentially affecting MyD88-dependent and -independent pathways. Int Immunol 14:783–791
Nakayama K, Okugawa S, Yanagimoto S et al (2004) Involvement of IRAK-M in peptidoglycan-induced tolerance in macrophages. J Biol Chem 279:6629–6634
Wang JH, Doyle M, Manning BJ, Di Wu Q, Blankson S, Redmond HP (2002) Induction of bacterial lipoprotein tolerance is associated with suppression of toll-like receptor 2 expression. J Biol Chem 277:36068–36075
Li L, Cousart S, Hu J, McCall CE (2000) Characterization of interleukin-1 receptor-associated kinase in normal and endotoxin-tolerant cells. J Biol Chem 275:23340–23345
Medvedev AE, Lentschat A, Wahl LM, Golenbock DT, Vogel SN (2002) Dysregulation of LPS-induced Toll-like receptor 4-MyD88 complex formation and IL-1 receptor-associated kinase 1 activation in endotoxin-tolerant cells. J Immunol 169:5209–5216
Adib-Conquy M, Adrie C, Moine P et al (2000) NF-kappaB expression in mononuclear cells of patients with sepsis resembles that observed in lipopolysaccharide tolerance. Am J Respir Crit Care Med 162:1877–1883
Ziegler-Heitbrock HW, Wedel A, Schraut W et al (1994) Tolerance to lipopolysaccharide involves mobilization of nuclear factor kappa B with predominance of p50 homodimers. J Biol Chem 269:17001–17004
Deng JC, Cheng G, Newstead MW et al (2006) Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J Clin Invest 116:2532–2542
Escoll P, del Fresno C, Garcia L et al (2003) Rapid up-regulation of IRAK-M expression following a second endotoxin challenge in human monocytes and in monocytes isolated from septic patients. Biochem Biophys Res Commun 311:465–472
Appoloni O, Vincent JL, Duchateau J (2002) Response of tumour necrosis factor-alpha to delayed in vitro monocyte stimulation in patients with septic shock is related to outcome. Clin Sci (Lond) 102:315–320
Heagy W, Nieman K, Hansen C, Cohen M, Danielson D, West MA (2003) Lower levels of whole blood LPS-stimulated cytokine release are associated with poorer clinical outcomes in surgical ICU patients. Surg Infect (Larchmt) 4:171–180
Broad A, Jones DE, Kirby JA (2006) Toll-like receptor (TLR) response tolerance: a key physiological “damage limitation” effect and an important potential opportunity for therapy. Curr Med Chem 13:2487–2502
Adib-Conquy M, Cavaillon JM (2002) Gamma interferon and granulocyte/monocyte colony-stimulating factor prevent endotoxin tolerance in human monocytes by promoting interleukin-1 receptor-associated kinase expression and its association to MyD88 and not by modulating TLR4 expression. J Biol Chem 277:27927–27934
Bosisio D, Polentarutti N, Sironi M et al (2002) Stimulation of toll-like receptor 4 expression in human mononuclear phagocytes by interferon-gamma: a molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood 99:3427–3431
Stuber F, Klaschik S, Lehmann LE, Schewe JC, Weber S, Book M (2005) Cytokine promoter polymorphisms in severe sepsis. Clin Infect Dis 41(Suppl 7):S416–420
Arcaroli J, Fessler MB, Abraham E (2005) Genetic polymorphisms and sepsis. Shock 24:300–312
Westendorp RG, Langermans JA, Huizinga TW et al (1997) Genetic influence on cytokine production and fatal meningococcal disease. Lancet 349:170–173
Pinsky MR (2007) Genetics of individualizing patient care. Crit Care Med 35:287–280
Barber RC, Chang LY, Arnoldo BD et al (2006) Innate immunity SNPs are associated with risk for severe sepsis after burn injury. Clin Med Res 4:250–255
van Eijk LT, Dorresteijn MJ, Smits P, van der Hoeven JG, Netea MG, Pickkers P (2007) Gender differences in the innate immune response and vascular reactivity following the administration of endotoxin to human volunteers. Crit Care Med 35:1464–1469
Lin J, Yao YM, Yu Y et al (2007) Effects of CD14-159 C/T polymorphism on CD14 expression and the balance between proinflammatory and anti-inflammatory cytokines in whole blood culture. Shock 28:148–153
Heesen M, Bloemeke B, Schade U, Obertacke U, Majetschak M (2002) The −260 C→T promoter polymorphism of the lipopolysaccharide receptor CD14 and severe sepsis in trauma patients. Intensive Care Med 28:1161–1163
Gibot S, Cariou A, Drouet L, Rossignol M, Ripoll L (2002) Association between a genomic polymorphism within the CD14 locus and septic shock susceptibility and mortality rate. Crit Care Med 30:969–973
Kumpf O, Hamann L, Schlag PM, Schumann RR (2006) Pre- and postoperative cytokine release after in vitro whole blood lipopolysaccharide stimulation and frequent toll-like receptor 4 polymorphisms. Shock 25:123–128
Papathanassoglou ED, Giannakopoulou MD, Bozas E (2006) Genomic variations and susceptibility to sepsis. AACN Adv Crit Care 17:394–422
Osuchowski MF, Welch K, Yang H, Siddiqui J, Remick DG (2007) Chronic sepsis mortality characterized by an individualized inflammatory response. J Immunol 179:623–630
Quartin AA, Schein RM, Kett DH, Peduzzi PN (1997) Magnitude and duration of the effect of sepsis on survival. Department of Veterans Affairs Systemic Sepsis Cooperative Studies Group. JAMA 277:1058–1063
Braun L, Riedel AA, Cooper LM (2004) Severe sepsis in managed care: analysis of incidence, one-year mortality, and associated costs of care. J Manag Care Pharm 10:521–530
Angele MK, Chaudry IH (2005) Surgical trauma and immunosuppression: pathophysiology and potential immunomodulatory approaches. Langenbecks Arch Surg 390:333–341
Angele MK, Faist E (2002) Clinical review: immunodepression in the surgical patient and increased susceptibility to infection. Crit Care 6:298–305
Munford RS, Pugin J (2001) Normal responses to injury prevent systemic inflammation and can be immunosuppressive. Am J Respir Crit Care Med 163:316–321
Hinshaw LB, Solomon LA, Freeny PC, Reins DA (1967) Endotoxin shock. Hemodynamic and survival effects of methylprednisolone. Arch Surg 94:61–66
Bone RC, Fisher CJ Jr., Clemmer TP, Slotman GJ, Metz CA, Balk RA (1987) A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med 317:653–658
Group TVASSCS (1987) Effect of high-dose glucocorticoid therapy on mortality in patients with clinical signs of systemic sepsis. The Veterans Administration Systemic Sepsis Cooperative Study Group. N Engl J Med 317:659–665
Annane D, Sebille V, Charpentier C et al (2002) Effect of treatment with low doses of hydrocortisone and fluorocortisone on mortality in patients with septic shock. JAMA 288:862–871
Panacek EA, Marshall JC, Albertson TE et al (2004) Efficacy and safety of the monoclonal anti-tumor necrosis factor antibody F(ab’)2 fragment afelimomab in patients with severe sepsis and elevated interleukin-6 levels. Crit Care Med 32:2173–2182
Goebel A, Kavanagh E, Lyons A et al (2000) Injury induces deficient interleukin-12 production, but interleukin-12 therapy after injury restores resistance to infection. Ann Surg 231:253–261
O'Suilleabhain C, O'Sullivan ST, Kelly JL, Lederer J, Mannick JA, Rodrick ML (1996) Interleukin-12 treatment restores normal resistance to bacterial challenge after burn injury. Surgery 120:290–296
Hackam DG, Mamdani M, Li P, Redelmeier DA (2006) Statins and sepsis in patients with cardiovascular disease: a population-based cohort analysis. Lancet 367:413–418
Majumdar SR, McAlister FA, Eurich DT, Padwal RS, Marrie TJ (2006) Statins and outcomes in patients admitted to hospital with community acquired pneumonia: population based prospective cohort study. Br Med J 333:999
Terblanche M, Almog Y, Rosenson RS, Smith TS, Hackam DG (2007) Statins and sepsis: multiple modifications at multiple levels. Lancet Infect Dis 7:358–368
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, T.S., Deng, J.C. Molecular and cellular aspects of sepsis-induced immunosuppression. J Mol Med 86, 495–506 (2008). https://doi.org/10.1007/s00109-007-0300-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-007-0300-4