
Abstract
DNA damage causes a local distortion of chromatin that triggers the sequential processes that participate in specific DNA repair mechanisms. This initiation of the repair response requires the involvement of a protein whose activity can be regulated by histones. Kinases are candidates to regulate and coordinate the connection between a locally altered chromatin and the response initiating signals that lead to identification of the type of lesion and the sequential steps required in specific DNA damage responses (DDR). This initiating kinase must be located in chromatin, and be activated independently of the type of DNA damage. We review the contribution of the Ser-Thr vaccinia-related kinase 1 (VRK1) chromatin kinase as a new player in the signaling of DNA damage responses, at chromatin and cellular levels, and its potential as a new therapeutic target in oncology. VRK1 is involved in the regulation of histone modifications, such as histone phosphorylation and acetylation, and in the formation of γH2AX, NBS1 and 53BP1 foci induced in DDR. Induction of DNA damage by chemotherapy or radiation is a mainstay of cancer treatment. Therefore, novel treatments can be targeted to proteins implicated in the regulation of DDR, rather than by directly causing DNA damage.
Similar content being viewed by others
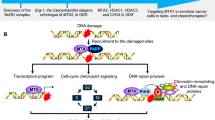


Introduction
Genome stability
Genome stability is essential for the maintenance of species, but, at the same time, genetic variation is necessary for their evolution. Therefore, in all species, there are several mechanisms aiming to protect DNA from genetic damage of endogenous or exogenous origin. Endogenous DNA damage is a consequence of the biological properties of cells, and includes oxidative stress, replication errors, transcriptional errors, or metabolism of DNA, to which cells are continuously exposed [1]. Alternatively, exogenous factors such as ultraviolet light, ionizing radiation, or chemicals also cause DNA damage to which exposure is frequently transient. The DNA damage has many different forms, single- or double-strand breaks, nucleotide, or base modification [1]. To cope with all of them, cells have developed several specific DNA repair mechanisms, which increase their complexity in higher organisms because of the chromatin organization. Double-strand breaks constitute the most serious form of DNA damage that has two alternative repair mechanisms depending of the situation of the cell cycle. During replication, DNA double-strand breaks (DSBs) are repaired by homologous recombination (HR) using as template the other chromatid. In non-dividing-cells or in G0/G1 phases, DSBs are repaired by non-homologous end-joining (NHEJ) [2].
Independently of the origin of DNA lesions, these lesions have to be detected rapidly and efficiently before cells divide, to avoid transmitting the damage to their progeny. Because nuclear kinases are capable of rapidly and reversibly responding to changes in the cell and its environment, and of integrating diverse stimuli, they are likely to be involved in sensing, triggering, regulating, and organizing the sequential steps that are needed for a correct and specific DNA damage response.
Cells are continuously exposed to DNA damage and it can occur at any time during the cellular lifetime. The number of normal cell division is limited to approximately 40 because of telomere shortening, which implies that, in the life of the organism, most cells are not dividing at the time of exposure to DNA damage [3, 4]. Furthermore, cells are most of their lifetime in the G0/G1 phases, in which homologous recombination is not functional [5], but are exposed to DNA damage. Furthermore, stem cells have an enhanced response to DNA damage mediated by the NHEJ pathway [6]. Therefore, most of the DNA lesions will occur and have to be repaired in the absence of replication. Very often, there is a large time interval between the moment in which DNA damage occurs and the time when an individual cell replicates, in which most cells are non-dividing, and are thus able to pass the mutation to their daughter cells. Consequently, each cell has to deal individually with this problem and to respond independently of its particular situation, which is very variable within a tissue. Cells are either resting or dividing, and their individual position within a tissue implies that cellular interactions are heterogeneous depending on its location. DNA repair mechanisms have to function in all these different cellular contexts. In the particular case of neurons, by their exposition to oxidative stress, the accumulation of DNA damage might be a pathogenic mechanism for deterioration of neurological functions associated with aging. Recent evidence indicates that a significant proportion of the DNA damage is of endogenous origin [7, 8]. Francis Crick predicted that several redundant mechanisms must exist to repair damaged DNA and maintain genome integrity [9]. Since then, several pathways have been identified [10,11,12,13]. Induction of DNA damage is a mainstay of cancer treatment, and the specific targeting of regulatory proteins implicated in DDR can lead to the development of new drugs.
Chromatin and DNA damage
The cellular response to DNA damage has to be initiated and triggered at the site of the DNA lesion, independent of its type. DNA damage causes a local distortion of the double helix, and of its associated nucleosomes, which is reflected in the local chromatin organization. Chromatin can function as a signaling platform that has effects not only on its remodeling, but can also send signals to other processes involved in nuclear dynamics [14]. When cells encounter a stress such as DNA damage, the activation of complex signaling networks triggers the detection and repair of the damage in a specific and sequential process, before returning to the homeostatic equilibrium. These networks integrate a wide variety of signals from inside the cell, transduced through protein kinases [10,11,12], to ultimately control cell cycle arrest or progression in the case of dividing cells [15]. Moreover, the chromatin-signaling platform regulates DDR, cell cycle checkpoints, cell death, and senescence, among others. All these processes are associated with the maintenance of genetic stability and the transmission of a mutation-free genome to daughter cells. The major pathological consequence of DNA damage is the potential transmission of mutations to their progeny [16], which are implicated in aging and cancer [17]. In addition to the role of DNA damage in cancer, alterations in DNA repair genes are also associated with neurodegenerative diseases [18], since neurons are not dividing in most of the individual lifetime and have to repair these DNA lesions. Most research into DNA damage responses has been studied in the context of replication and cell division [16].
In the highly organized eukaryotic chromatin, the most vulnerable DNA is the fraction that is transcriptionally active at the time of exposure to damaging agents, particularly in resting or non-dividing cells, such as stem cells or neurons. In these locations, DNA has to relax and open to allow the access of RNA polymerase and permit gene transcription. In these transcriptionally active regions, DNA is more exposed and vulnerable, particularly in non-dividing or cells in G0/G1. Therefore, in an individual resting cell, the response to DNA damage does not have to be linked to cell division, differentiation state, or the cell location and its interactions within a tissue. Even in dividing cells, the G1 phase last several hours before entering replication. DNA damage has to be detected, identified, and repaired immediately in all different types of situations.
Cellular response to DNA damage
The cellular reaction to DNA damage involves two major aims; one is to protect the DNA, and the other to protect cells and the organisms from the consequences of unrepaired DNA damage. The cellular protection against DNA damage is mediated by arresting cell cycle in proliferating cells, so that damage can be repaired before its transmission to daughter cells. However, if DNA damage is excessive and cannot be repaired, the alternative response is mediated by the induction of cell death, and in that way, there is no progeny of mutated cells. These two types of responses are associated with p53 and activated by different types of DNA damage.
DNA repair requires a sequential reorganization of chromatin to allow for the different and consecutive steps in each repair pathway, which includes protection of damaged DNA, recognition of the type of lesion, recruitment of specific repair mechanisms, ligation of DNA ends, and restoration to its normal chromatin organization. After DNA damage, in addition to the DNA lesion, the initial effect is a local distortion of chromatin, which is the initiating event to trigger the cascade of DNA repair processes. As organisms increased in their complexity, new regulatory elements are necessary not only to coordinate different functions in DDR, but also to adjust to their much more complex and dynamic structure of chromatin. Therefore, new regulatory mechanisms that integrate and coordinate basic processes are necessary. In this context, new regulatory elements have evolved from preexisting proteins. A candidate for this role must be a chromatin protein with a reversible enzymatic activity. Among the 518 kinase of the human kinome, vaccinia-related kinase-1 (VRK1) is a potential candidate for this role because of its association to chromatin and its targets, with the exception of chromosomes condensed in mitosis [19, 20].
VRK1 roles in chromatin
The VRK1 chromatin kinase
VRK1 is a Ser-Thr kinase that belongs to the VRK family that diverged early from branch of the human kinome that led the casein kinase family [21]. Bacteria and yeast have no VRK or p53 members, invertebrates such as D. melanogaster or C. elegans have one member, and mammals have three members in their respective families. The complexity of VRK family [22] parallels that of p53 [23] and the autophagic DRAM (death-related autophagic modulator) [24]. This increased complexity during evolution is likely to reflect the need for additional regulatory or coordinating roles as organisms and their functions became more complex. In mammals and C. elegans, it is known as Vrk-1 [25], and in D. melanogaster as nucleosomal histone kinase 1 (NHK-1) [26].
VRK1 is a Ser-Thr kinase in nuclei [19] that is located on chromatin in resting cells and in all phases of the cell cycle covering all DNA, except when chromosomes are already condensed in mitosis [27, 28], in which VRK1 is ejected from mitotic chromosomes. When chromosomes segregate, VRK1 returns to chromatin in daughter cells. VRK1 forms stable complexes with several different types of chromatin proteins, ranging from histones, transcription factors, and proteins involved in DNA repair processes (Fig. 1). The proteins more closely associated with DNA are histones [29], which are organized in nucleosomes and in direct contact with DNA, contributing to chromatin spatial organization. VRK1 is detected in the chromatin fraction forming a stable complex with histone H3 [29]. Moreover, VRK1 phosphorylates histones H3 [27, 29, 30], H2A [31, 32] and H2AX [29]. Therefore, it is very likely that nucleosome organization can be modified by covalent modifications because of histone phosphorylations by VRK1. This regulation of histone covalent modifications is essential for different functions, normal or pathological, requiring a dynamic chromatin reorganization [33, 34].

VRK1 relation with transcription factors and DNA damage response proteins in chromatin
An additional role of VRK1 as a chromatin kinase is its association with transcriptional complexes, where it interacts and phosphorylates several transcription factors that include p53 [28], CREB [35], ATF2 [36], c-Jun [37], Sox2 [38], and the farnesoid X receptor (FXR) [39].
VRK1 as a sensor of chromatin alterations
Chromatin in interphase has a very large size and DNA lesions can occur at any place, heterochromatin and euchromatin, which are likely to have a different sensitivity to DNA damage. Alterations of DNA by strand breaks or chemical modifications, such as oxidation, alkylation, or intercalation among others, will alter the chromatin organization by introducing a local distortion [40, 41], which is a likely initiating event for triggering the complex processes of DNA repair [42,43,44]. However, responding to DNA damage requires the coupling of chromatin distortion to a signal transduction system, probably mediated by a nuclear chromatin kinase.
A requirement for a sensor kinase is that its activation is independent of the type of DNA damage and, therefore, is not associated to any particular type of DNA damage. In this latter case, the kinase involved will participate in specific steps of a particular DNA damage, as is the case for ATM, ATR, or DNA-PK in the response to double-strand DNA breaks [45]. In the particular case of VRK1, its kinase activity increases tenfold after induction of DNA damage independently of its type, which includes pyrimidine dimers caused by ultraviolet light, single-strand DNA breaks caused by hydroxyurea treatment, or double-strand DNA breaks induced by either doxorubicin or ionizing radiation [46].
Early sensor mechanism of DNA damage must fulfill some basic requirements, be a nuclear enzyme that interacts with basic chromatin components in nucleosomes, and be a capable of an immediate signaling reaction that is also reversible. In this context, a kinase, such as VRK1, is a very suitable candidate for this role [29, 46, 47].
Other important early proteins at the site of specific types of DNA damage are Ku70/Ku80 (XRCC6/XRCC5), which have to re-localize and interact with free DNA ends at the breakpoints, mainly in double-strand breaks [48], a subtype of DNA damage, or in telomeres [48, 49]. It is unknown whether these proteins are targets of VRK1, but it is a real possibility. Ku70 and Artemis have multiple phosphorylation sites, but the kinases involved in their specific phosphorylation and their regulation are unknown. Telomeres are naturally occurring DNA ends in chromosomes and there is evidence for a role of VRK1 in their maintenance [50]. Moreover, VRK1 phosphorylates hnRNP A1 (heterogeneous nuclear ribonucleoprotein A1) and facilitates its binding to telomeric ssDNA and telomeric RNA [50].
VRK1, chromatin relaxation, and histone acetylation
DNA damage and local disruption of chromatin are associated with an increase in histone acetylation, which is mediated by KAT (lysine acetyl transferase) proteins. Histone acetylation extends over an area of several hundred kilobases flanking the damaged DNA site [51, 52], which requires the local activation of KATs by a not yet identified mechanism. Defects in histone acetylation are associated with an increase in cellular sensitivity to DNA damage as a consequence of a defective DNA repair [53, 54]. Furthermore, acetylation of histone H4 in Lys16 disrupts the interaction between H4 and H2A–H2B, and facilitates the relaxation of chromatin [55, 56]. Consistently, the inactivation of KAT5/Tip60 blocks the opening of chromatin at DSBs (double-strand breaks) that are required to facilitate the repair process [52]. Induction of DNA damage by UV light or radiation causes an increase in histone acetylation [52, 57]. Depletion of VRK1, a nucleosomal chromatin kinase, causes a loss of histones H3 and H4 acetylation, which are necessary for chromatin relaxation, either in basal conditions or after DNA damage, independently of ATM and p53 [29]. VRK1 knockdown also causes a loss of specific histone acetylations, including H4K16 acetylation (H4K16ac), induced by DNA damage [29]. ATM-null cells, such as the HT144 cell line, has a high endogenous level of H4K16ac that is also lost by depletion of VRK1 [29]. In ATM+/+ cells, this acetylation induced by IR does not occur in the absence of VRK1 [29]. These results indicate that VRK1 is a good candidate to regulate the enzymes involved in epigenetic modifications of chromatin. DNA damage causes a local distortion of chromatin that can affect its different covalent modifications. Consequently, the regulation and coordination of histone modifiers such as acetylases, deacetylases, methylases, and demethylases is very poorly understood. Moreover, VRK1 also directly phosphorylates histone H2A in T120 [32], which is next to K119 ubiquitinated, and both modifications are functional alternatives, being T120 phosphorylation an activator of chromatin. Thus, two histones in nucleosomes, H3 and H2A, are directly regulated by VRK1. Furthermore, histone H4 is not a phosphorylation target of VRK1, but its covalent modification by acetylation is sensitive to VRK1 in an ATM-independent manner, since it is detected in ATM-null cells [29].
It is important to remark that the sensor kinase activity has to be regulated by protein–protein interactions. In this context, the C-terminal region of VRK1 has a low complexity structure, which is very flexible and can adopt different conformations [58]. This C-terminal region can fold and block the active site of the kinase [58] and proteins interacting with this region can modulate the activity of VRK1. Two proteins that inhibit the VRK1 kinase activity have been identified, macrohistone H2A1.2 in interphase [59], and Ran-GDP, but not Ran-GTP [60], which have an asymmetric nuclear distribution [61].
VRK1 in DNA damage responses
VRK1 and histone H2AX
VRK1 directly and stably interacts with histones H2AX and H3 in basal conditions, and is able to phosphorylate them in vitro with purified proteins in Ser139 and Thr3, respectively [29]. The early response to DNA damage requires the phosphorylation of H2AX in Ser139 (γH2AX). γH2AX covers large areas of DNA surrounding the site of DNA damage [62] and protects DNA from exonuclease attack. This γH2AX organization can also function as a platform for the recruitment of proteins that participate in sequential DDR steps, such as NBS1, 53BP1, or BRCA1, among others [40, 63]. Phosphorylation of histone H2AX in Ser139 (γH2AX) is a mark of an early reaction to DNA damage that can be detected by formation of γH2AX foci [62, 64]. The phosphorylation of H2AX and the formation of γH2AX foci induced by ionizing radiation (IR) are lost by depletion of VRK1 and can be rescued by kinase-active, but not by kinase-dead, VRK1 [29]. This effect of VRK1 is also independent of ATM, suggesting that VRK1 is an upstream participant. VRK1 is also necessary for the activation of ATM and CHK2 in response to IR [46]. However, in the absence of ATM, the γH2AX foci induced by IR have a smaller size, which indicates that both kinases cooperate either in the formation or stabilization of the foci [29]. This latter possibility might be a consequence of the effect of VRK1 on the stability of NBS1 [47]. In this context, VRK1 is a novel chromatin component that reacts to its alterations and participates very early in DDR by itself and in cooperation with ATM [29].
VRK1 and specific DNA damage response proteins
Because of the physical association of VRK1 with chromatin, VRK1 has also been implicated in the regulation of DDR proteins. The VRK1 kinase has also been directly associated with different components in DDR pathways, which have been studied in the context of the response to DSBs, in both resting and cycling cells as well as in ATM-null and p53-null cells. VRK1 physically interacts and directly phosphorylates specific proteins participating at different sequential stages of DDR, which include H2AX [29], NBS1 [47], and 53BP1 [46, 65] in NHEJ [66, 67]; and all of these activating phosphorylations are lost by VRK1 depletion. Intermediate steps in DDR signal transmission are well known. The most common pathways in DNA damage response (DDR) implicate protein phosphorylation by different kinases such as ATM [10], ATR [11], and DNA-PK [12]; all members of the PI-3K family, which have been mostly studied in the context of cell division and cell cycle checkpoints [15]. In response to double-strand breaks induced by ionizing radiation (IR), the 53BP1 scaffold protein is recruited to IR-induced foci (IRIF), and is an important marker for monitoring cellular DDR by NHEJ. 53BP1 foci induced by ionizing radiation or doxorubicin are intermediate steps in DDR activation [68, 69] and are known to be regulated by ATM in response to DSBs [70], and by ATR in response to replication stress [71]. However, it is also known that DNA damage response can be ATM-independent [72]. The effect of VRK1 in DDR is insensitive to inhibitors of PI3KK proteins that target ATM and DNA-PK [46]. This suggests that there are alternative kinases participating in DDR induced by ionizing radiation. The complete molecular components that sequentially participate in DDR, particularly regulatory proteins, remain unknown. In this context, VRK1 knockdown also prevents the activating phosphorylations of ATM in Ser1981, CHK2 in Thr68, and DNA-PK in Ser2056, all induced in response to IR [46], suggesting that VRK1 is an early and upstream component in this DDR process.
VRK1 and NBS1 in early DDR
Cellular responses to DNA damage require the formation of protein complexes in a highly organized fashion. In resting cells, VRK1 plays an important role in the formation of ionizing radiation-induced foci formed by γH2AX, NBS1, and 53BP1 during DDR. The MRE11 complex holds together the two free ends of the broken DNA. This complex, formed NBS1–Mre11–Rad50, is highly dynamic and has a very complex organization [66]. Phosphorylation of NBS1/nibrin is necessary for the recruitment of ATM to damaged sites and for the stabilization of the repair complex [73]. VRK1 is activated by DNA double-strand breaks induced by ionizing radiation (IR) or doxorubicin, and specifically phosphorylates NBS1 in Ser343 [47] and 53BP1 in serum-starved cells and ATM-null and p53-null cells [46], indicating that they are independent of both p53 and ATM activation [47], and consistent with VRK1 role as an early step in the response to DNA damage. Depletion of VRK1 causes a loss of NBS1 stability that is prevented by treatment with the MG132 proteasome inhibitor [47]. This phosphorylation mediated by VRK1 protects the NBS1 protein of RNF8-mediated ubiquitination [47]. Therefore, it is likely that NBS1 phosphorylation by VRK1 contributes to the stabilization of foci, and facilitates the recruitment of additional participants in the specific DNA repair process, such as kinases of the PI3K family, ATM, ATR, or DNA-PK, for specific signaling steps or pathways in DDR [45].
VRK1 and 53BP1 in NHEJ
Double-strand breaks are the most serious form of DNA damage, particularly in cells that are resting or in the early phases of the cell cycle, which includes differentiated resting cells, as neurons, and stem cells. Under these conditions, these DSBs are repaired by non-homologous end-joining (NHEJ); and one of its main components is 53BP1, a scaffold protein that forms foci induced by DNA damage [74]. VRK1 stably interacts with 53BP1 in the region comprised between residues 955-1354, which is implicated in the interaction with H2AX, but its phosphorylation site is in Ser25/29 within the 53BP1N-terminal region and occurs even in the absence of ATM (null cells) [46]. VRK1 depletion causes a defective formation of 53BP1 foci induced by ionizing radiation or doxorubicin, both in number and size, which requires a kinase-active VRK1 protein for their rescue [46]. Moreover, this effect of VRK1 on 53BP1 foci is insensitive to ATM and DNA-PK inhibitors and is functional in p53-null and ATM-null cells. All these data indicate that the effect of VRK1 is independent of both ATM and p53 [46], and that VRK1 activation in response to DNA damage is a novel participant in the NHEJ mechanism of mammalian DNA damage responses [29, 46, 47].
Cellular protection mediated by VRK1 and its target p53
VRK1 forms a complex and phosphorylates p53
The p53-mediated responses induced by DNA damage have two major roles in the context of cellular protection (Fig. 2). The first one is preventing the transmission of damaged DNA to daughter cells during cell proliferation. This p53 action is mediated by the induction of a cell cycle arrest, and forms part of cell cycle checkpoints [75, 76]. The other role is the protection of the organism from the consequences of accumulating cells with damaged DNA, which is mediated by induction of cell death [77]. The p53 transcription factor mediates these two main protective responses that are regulated by p53 immediate phosphorylation in response to DNA damage. These cellular responses have a different temporal order because of the covalent modifications, which are immediate as the stabilization of p53, or require hours, such as the induction of specific gene expression.

Kinase activation induced by DNA damage and the regulation of p53 in G0/G1 cells. An enzyme (X) activated by VRK1, and that has not yet been identified, mediates the activation of ATM
The stabilization and activation of p53 is performed by several Ser-Thr kinases that target different residues within the p53N-terminal transactivation domain (Fig. 2), and have different sequential roles [78,79,80]. In the absence of stress or DNA damage, the basal intracellular level of the p53 protein is very low, but it is always present. This basal low level of p53 is necessary to initiate a fast response to cellular stress by its immediate phosphorylation. The p53 phosphorylation in several residues within its TAD1 region (residues 1–46) is the main determinants of the stress response [79]. To trigger an immediate reaction to DNA damage, the response will be greatly facilitated by the formation of a stable and inactive complex between p53 and one of its regulatory kinases that are activated by DNA damage. In non-damaged cells, the basal low p53 level is partially forming a stable complex with VRK1, which are detected by reciprocal immunoprecipitations, and are detected in resting and cycling cells [81]. This basal VRK1-p53 complex forms a basic early warning system for detection of cellular stress and its activation is induced by DNA damage caused by ultraviolet light, ionizing radiation, or doxorubicin treatments. All these types of DNA damage activate the kinase activity of VRK1, a previous step required for the specific phosphorylation of p53 at Thr18 [28, 81]. Therefore, the subpopulation of basal p53 that is forming a complex with VRK1 facilitates a readiness state of p53 to initiate an immediate activating response in different cellular stress situations [28, 81]. Furthermore, the p53 protein also indirectly plays an important role in epigenetic regulation of chromatin [82].
Phosphorylation of p53 by VRK1 prevents the interaction with MDM2 and regulates the switch between ubiquitination and transcription
Non-phosphorylated p53 binds to the human MDM2 (HDM2) ubiquitin ligase [83]. VRK1 uniquely and specifically phosphorylates p53 in Thr-18 [28, 84, 85]. This Thr18 residue is critical to maintain the folding of the p53 α-helix required for its binding to a hydrophobic pocket in MDM2 [86]. The phosphorylation of p53 in Thr18 alters the alignment of hydrophobic residues in this α-helix, and this altered conformation permits the p53 binding to transcriptional cofactors. Moreover, this phosphorylation of Thr18 determines the change in binding mode from ubiquitin ligases to transcription factors, and additional p53 phosphorylation in Ser15 or Ser20 [87] contributes to the selection of specific transcriptional cofactors [88]. The specific phosphorylation of p53 in Thr18 places VRK1 upstream of additional phosphorylation in Ser15 and Ser20 mediated by other kinases [79]. The activated ATM-CHK2, ATR-CHK1, or DNA-PK pathways mediate the phosphorylation p53 in Ser15 or Ser20 [78, 79], and all of them are necessary to achieve the full transcriptional activation of p53 [88]. These additional p53 phosphorylations, and their combination, select transcriptional cofactors and activate p53-dependent genes such as CDKN1A (p21) expression [89], which induces a cell cycle arrest and senescence [90], and BAX that facilitates apoptosis [91, 92], among others. The role of p53 in transcription in these processes has been extensively reviewed [93].
The kinase activity of all these p53 kinases, VRK1, ATM, ATR, and DNA-PK, are inducible by DNA damage, but their spatial organization, coordination, and sequential activation require further studies for its complete understanding. In this context, because of its interactions with histones, VRK1 is a new component that participates very early in the response mechanisms to DNA damage, as well as in specific steps of DDR.
Activated p53 induces the downregulation of VRK1
Once DNA damage has been repaired, the cell cycle arrest induced by activated p53 has to be reverted. Otherwise, p53 will maintain the cell cycle arrest or even induce apoptosis. This reversal requires the deactivation of p53, which is mediated by its dephosphorylation and subsequent interaction with MDM2. However, the phosphorylation of p53 in Thr18 by VRK1 blocks its interaction with MDM2 and other phosphorylations, in Ser15 and Ser20 further interfere with the interaction [88]. All these phosphorylations have to be removed, to revert the p53-mediated responses, such as a cell cycle arrest, in viable cells. This is accomplished by the regulation by p53 of different target genes that range from ubiquitin ligases, phosphatases, to autophagic proteins [94]. The reversion of activated p53 also requires downregulation of p53 activating kinases, including VRK1 and ATM, so that dephosphorylated p53 is not re-phosphorylated and becomes accessible to MDM2. In this context, p53 induces of the expression of DUSP6 and WIP1 phosphatases targeting ATM [95,96,97], and that of the DUSP4 phosphatase targeting VRK1 [98, 99]. However, downregulation of p53 activation is more complex and also requires additional deactivation of other kinases, which are mediated by phosphatases, and deacetylation of p53 [94].
The stabilization and accumulation of p53 by VRK1 in response to DNA damage is reverted by a novel p53-dependent activation of autophagy that removes its activating VRK1 [100], a p53 stabilizer, and thus permits p53 dephosphorylation and its downregulation by MDM2 [85, 100, 101]. Among the degradation processes regulated by p53 is autophagy. In normal cells, autophagy contributes to regulate basal levels of cytosolic and particulate proteins [102], a process that is further activated in response to several types of stress, including DNA damage. Autophagy is required for recycling of proteins implicated in negative cell cycle regulation, and can provide a survival strategy to tumor cells [103]. By this process, regulated by p53, cells remove and digest endogenous proteins, particularly those that are very stable, functioning as an important mechanism for tissue remodeling [103] and maintenance of cellular homeostasis [104], but it can also result in a form of cell death, thus having a dual role [105, 106].
The downregulation of VRK1 is a late response that is also mediated by the p53-dependent transcription of DRAM (death-related autophagic modulator) [107]. DRAM is a small hydrophobic protein located in the membrane of autophagosomes [108]. Expression of DRAM facilitates degradation of VRK1 in the lysosome, and the elimination of DRAM or Beclin1 prevents the downregulation of VRK1 by proteolytic degradation [85, 100] (Fig. 3). This degradation of VRK1 takes place in the cytosol and is sensitive to the inhibition of nuclear export with leptomycin B and to lysosomal inhibitors [100]. DRAM expression induced by p53 regulates the degradation of stable proteins. DRAM is a novel component of the cell autophagic response [107]. Autophagy is partly regulated by p53-induced DRAM expression [107], and p53-induced VRK1 degradation requires entry in the endosomal–lysosomal pathway [85]. In this way, DRAM downregulates VRK1 forming an autoregulatory loop [101] (Fig. 3). Moreover, this autophagic downregulation of VRK1 is altered in tumors with p53 mutations that affect its DNA-binding domain, including all the most frequent mutations detected in human cancer [85, 109], because they disrupt this autoregulatory loop. Consequently, tumors harboring p53 mutations also have very high levels of VRK1, as it has been shown in head and neck squamous cell carcinomas [110] and lung cancer [109], which can also facilitate cell proliferation.

Downregulation of VRK1 by DRAM1 in the autophagic pathway induced by p53 and deactivation of p53 by phosphatases and ubiquitin ligases the proteasome in DNA damage response. Solid black lines represent the activation route. Dashed lines represent the downregulatory routes and each color represents a different route. Kinases: VRK1 and ATM. Phosphatases: WIP1 (wild-type P53-induced phosphatase 1) and DUSP4 (dual specificity phosphatase 4). DRAM1: damage-regulated autophagy modulator 1
In conclusion, the main mechanism of downregulation of p53 is mediated MDM2, but for this to occur, it is necessary to previously dephosphorylate p53 and its activating kinases, all of which are regulated by p53 [94]. Once p53 is dephosphorylated, it becomes available for its ubiquitination by MDM2 and degradation in the proteasome, which has been extensively reviewed and has become a target for therapeutic intervention with drugs that interfere with the p53-MDM2 interaction, such as nutlins [111].
Implications of VRK1 in cancer biology
The functions of VRK1 suggest that it is likely to actively participate in tumor biology. Knockdown experiments indicate that VRK1 plays a major role in cell cycle progression and proliferation [27, 112, 113]. Moreover, VRK1 elimination by CRISPR/Cas9 identifies wild-type VRK1 as an overexpressed oncogenic driver gene [114], consistent with its role in lung adenocarcinomas [115]. In most cell types, the human VRK1 gene is expressed at different levels and is not mutated in cancer, and it is overexpressed in many cancer types of different origins correlating with a poorer prognosis in breast [116, 117], lung [115], liver [118], glioblastoma [119], head and neck [110], and esophageal cancer [120]. Some driver genes are oncogenic in situations in which they are overexpressed by different mechanisms, as it occurs with members of the MYC and EGFR families that promote cell proliferation. In the context of tumor growth, the human VRK1 protein has been implicated in the regulation of proliferation and cell cycle progression, where it plays several roles [121]. VRK1 is required for G0 exit, behaving like an early gene such as MYC and FOS, which facilitate the progression in G1 and passing the restriction point [113]. In this context, depletion of VRK1 prevents the expression of CCND1 (cyclin D1), since VRK1 directly binds to the human CCND1 promoter [32], and consequently, retinoblastoma cannot be phosphorylated [110]. Later, in cell cycle progression, VRK1 is also required for the phosphorylation of histone H3 that facilitates the initiation of chromatin compaction in G2/M [27] and cooperates with AURKB in the sequential remodeling of chromatin in the progression of mitosis [122].
However, based on the biological actions of VRK1, either in cell proliferation or DNA damage responses indicates that depending on the cellular context, VRK1 might function as an oncogene or a tumor suppressor or predisposition gene. VRK1 might behave an oncogene because of its roles in the promotion of cell cycle progression and proliferation. However, in other contexts, VRK1 might behave as a tumor suppressor or a tumor susceptibility gene represented by the effects mediated by p53 and those associated with genome stability. These properties, in the context of cancer, can contribute to a poorer prognosis of tumors overexpressing VRK1 because of its contribution to the promotion of cell proliferation and resistance to treatments based on DNA damage.
Due to the essential role played by VRK1, attempts to generate knock-out mice have been unsuccessful. However, the consequences of VRK1 deficiency in animal models have been studied in gene-trap mice with a fifteen percent residual level of VRK1 [123,124,125]. In this model, deficient animals were sterile, both male and female [123,124,125], preventing additional studies. The role that VRK1 plays in response to DNA damage in this model was not studied. In one of the studies, the problem was identified as lagging chromosomes during meiosis leading to sterility [124], an observation consistent with the role of VRK1 in dynamic chromatin reorganization. In addition, VRK1 regulates the attachment of chromatin to the nuclear envelop that is mediated by the phosphorylation of BANF1 [126]. The disruption of this process can also lead to alterations of chromatin reorganization in mitosis and affect cell viability [127].
VRK1 potential as therapeutic target in oncology
Protein kinases, because of their structural characteristics, are candidates for development of inhibitors. Knockdown screening is a useful approach to identify potential therapeutic targets. Knockdown of VRK1 sensitizes cells to other cancer treatments based on DNA damage such as ionizing radiation or doxorubicin by impairing the DNA damage response [46, 65]. Moreover, depletion of VRK1 inhibits cell proliferation [113, 128]. VRK1 has been identified as a potential target in a screening of synthetic lethal relationships in a massive siRNA screening [129]. These observations suggests that inhibitors of VRK1 can be of potential use in cancer treatments, by themselves or in combinations, by facilitating inhibition of proliferation and at the same time sensitizing cells to treatments based on DNA damage. In cancer treatment, many drugs are directed to the main driver as targets. However, cancer cells can be derailed if alternative pathways that impinge on basic processes of the tumor phenotype are targeted. These alternative targets will open a wide range of possibilities, as well as provide with alternatives to manage individual cases.
Kinases share a common structure in their catalytic kinase domain and are druggable proteins [130]. Therefore, the likelihood of cross inhibition with other kinases is very high and makes many kinase inhibitors promiscuous. In the human kinome, there are kinases that are isolated from other major branches, among which is VRK1. The VRK family has some structural differences, which makes its members susceptible of highly specific inhibition with no promiscuity as detected in kinase assays or by structural thermal shift upon binding to inhibitors [131, 132]. However, at this time, there is no specific inhibitor for VRK1. Testing kinases inhibitors that target the main kinome families did not detect any that has an effect in assays of VRK1 autophosphorylation and VRK1 phosphorylation of p53 or H3 [30]. This is due to the very high inhibitor concentrations needed because of their low affinity, which required doses in the micromolar range that have a very high risk of cross inhibition and of the high probability of high toxicity and side effects. The elimination of VRK1 causes a defective DDR that facilitates and increases the sensitivity to DNA damaging agents, such as ionizing radiation or doxorubicin [65]. Depletion of VRK1 sensitizes cells to these treatments because of defective DNA repair, and thus permits the use of lower doses of toxic drugs to achieve the same result. This is important, because this sensitization also occurs in non-dividing cells, and might be useful for targeting non-dividing cells within a tumor that later might cause a relapse. Moreover, the treatment with a lower dose of commonly used cancer drugs can contribute to a reduction of the toxicity associated with them.
Facilitating some degree of DNA damage in tumor cells can contribute to the generation of new antigens and facilitate the response to new therapies based on manipulation of the immune system, as supported by the evidence that tumors with an intrinsic higher genome instability are better responders to these new therapies [133].
In conclusion, the pharmacological targeting of VRK1 will impair p53-mediated responses, prevent cell cycle progression and proliferation, and sensitize cells to treatments based on DNA damage, such as ionizing radiation and some chemotherapeutic drugs. The consequence of therapeutically exploiting this target will be a better control of the tumor if the new drugs are selective regarding both its molecular target and the specific tumor cell.
Abbreviations
- VRK1:
-
Vaccinia-related kinase 1
- DSB:
-
DNA double-strand break
- DDR:
-
DNA damage response
- NBS1:
-
Nijmegen breakage syndrome 1 (nibrin)
- NHEJ:
-
Non-homologous end-joining
- 53BP1:
-
Tumor protein P53 binding protein 1
- ATM:
-
Ataxia-telangiectasia-mutated Ser/Thr kinase
References
Friedberg EC, McDaniel LD, Schultz RA (2004) The role of endogenous and exogenous DNA damage and mutagenesis. Curr Opin Genet Dev 14(1):5–10. https://doi.org/10.1016/j.gde.2003.11.001
Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40(2):179–204. https://doi.org/10.1016/j.molcel.2010.09.019
Flores I, Blasco MA (2010) The role of telomeres and telomerase in stem cell aging. FEBS Lett 584(17):3826–3830. https://doi.org/10.1016/j.febslet.2010.07.042
Henriques CM, Ferreira MG (2012) Consequences of telomere shortening during lifespan. Curr Opin Cell Biol 24(6):804–808. https://doi.org/10.1016/j.ceb.2012.09.007
Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A, Munro M, Pinder J, Salsman J, Dellaire G, Xia B, Peter M, Durocher D (2015) A mechanism for the suppression of homologous recombination in G1 cells. Nature 528(7582):422–426. https://doi.org/10.1038/nature16142
Soteriou D, Fuchs Y (2018) A matter of life and death: stem cell survival in tissue regeneration and tumour formation. Nat Rev Cancer 18(3):187–201. https://doi.org/10.1038/nrc.2017.122
Tomasetti C, Vogelstein B (2015) Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347(6217):78–81. https://doi.org/10.1126/science.1260825
Tomasetti C, Li L, Vogelstein B (2017) Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 355(6331):1330–1334. https://doi.org/10.1126/science.aaf9011
Crick F (1974) The double helix: a personal view. Nature 248(5451):766–769
Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali SR, Simmons A, Clines GA, Sartiel A, Gatti RA, Chessa L, Sanal O, Lavin MF, Jaspers NG, Taylor AM, Arlett CF, Miki T, Weissman SM, Lovett M, Collins FS, Shiloh Y (1995) A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268(5218):1749–1753. https://doi.org/10.1126/science.7792600
Lakin ND, Hann BC, Jackson SP (1999) The ataxia-telangiectasia related protein ATR mediates DNA-dependent phosphorylation of p53. Oncogene 18(27):3989–3995. https://doi.org/10.1038/sj.onc.1202973
Lee SE, Mitchell RA, Cheng A, Hendrickson EA (1997) Evidence for DNA-PK-dependent and -independent DNA double-strand break repair pathways in mammalian cells as a function of the cell cycle. Mol Cell Biol 17(3):1425–1433. https://doi.org/10.1128/MCB.17.3.1425
Soria G, Polo SE, Almouzni G (2012) Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell 46(6):722–734. https://doi.org/10.1016/j.molcel.2012.06.002
Bustin M, Misteli T (2016) Nongenetic functions of the genome. Science 352(6286):aad6933. https://doi.org/10.1126/science.aad6933
Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15(17):2177–2196. https://doi.org/10.1101/gad.914401
Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461(7267):1071–1078. https://doi.org/10.1038/nature08467
Hoeijmakers JH (2009) DNA damage, aging, and cancer. N Engl J Med 361(15):1475–1485. https://doi.org/10.1056/NEJMra0804615
Rass U, Ahel I, West SC (2007) Defective DNA repair and neurodegenerative disease. Cell 130(6):991–1004. https://doi.org/10.1016/j.cell.2007.08.043
Varjosalo M, Sacco R, Stukalov A, van Drogen A, Planyavsky M, Hauri S, Aebersold R, Bennett KL, Colinge J, Gstaiger M, Superti-Furga G (2013) Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized AP-MS. Nat Methods 10(4):307–314. https://doi.org/10.1038/nmeth.2400
Cantarero L, Moura DS, Salzano M, Monsalve DM, Campillo-Marcos I, Martín-Doncel E, Lazo PA (2017) VRK1 (vaccinia-related kinase 1). Encyclopedia of signaling molecules, 2nd edn. Springer Science. https://doi.org/10.1007/978-1-4614-6438-9_561-2
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298(5600):1912–1934. https://doi.org/10.1126/science.1075762
Nichols RJ, Traktman P (2004) Characterization of three paralogous members of the mammalian vaccinia related kinase family. J Biol Chem 279(9):7934–7946. https://doi.org/10.1074/jbc.M310813200
Belyi VA, Ak P, Markert E, Wang H, Hu W, Puzio-Kuter A, Levine AJ (2010) The origins and evolution of the p53 family of genes. Cold Spring Harb Perspect Biol 2(6):a001198. https://doi.org/10.1101/cshperspect.a001198
Mrschtik M, Ryan KM (2016) Another DRAM involved in autophagy and cell death. Autophagy 12(3):603–605. https://doi.org/10.1080/15548627.2015.1137412
Gorjanacz M, Klerkx EP, Galy V, Santarella R, Lopez-Iglesias C, Askjaer P, Mattaj IW (2008) Caenorhabditis elegans BAF-1 and its kinase VRK-1 participate directly in post-mitotic nuclear envelope assembly. EMBO J 26(1):132–143. https://doi.org/10.1038/sj.emboj.7601470
Cullen CF, Brittle AL, Ito T, Ohkura H (2005) The conserved kinase NHK-1 is essential for mitotic progression and unifying acentrosomal meiotic spindles in Drosophila melanogaster. J Cell Biol 171(4):593–602. https://doi.org/10.1083/jcb.200706067
Kang TH, Park DY, Choi YH, Kim KJ, Yoon HS, Kim KT (2007) Mitotic histone H3 phosphorylation by vaccinia-related kinase 1 in mammalian cells. Mol Cell Biol 27(24):8533–8546. https://doi.org/10.1128/MCB.00018-07
Vega FM, Sevilla A, Lazo PA (2004) p53 Stabilization and accumulation induced by human vaccinia-related kinase 1. Mol Cell Biol 24(23):10366–10380. https://doi.org/10.1128/MCB.24.23.10366-10380.2004
Salzano M, Sanz-Garcia M, Monsalve DM, Moura DS, Lazo PA (2015) VRK1 chromatin kinase phosphorylates H2AX and is required for foci formation induced by DNA damage. Epigenetics 10(5):373–383. https://doi.org/10.1080/15592294.2015.1028708
Vazquez-Cedeira M, Barcia-Sanjurjo I, Sanz-Garcia M, Barcia R, Lazo PA (2011) Differential inhibitor sensitivity between human kinases VRK1 and VRK2. PLoS One 6(8):e23235. https://doi.org/10.1371/journal.pone.0023235
Aihara H, Nakagawa T, Yasui K, Ohta T, Hirose S, Dhomae N, Takio K, Kaneko M, Takeshima Y, Muramatsu M, Ito T (2004) Nucleosomal histone kinase-1 phosphorylates H2A Thr 119 during mitosis in the early Drosophila embryo. Genes Dev 18(8):877–888. https://doi.org/10.1101/gad.1184604
Aihara H, Nakagawa T, Mizusaki H, Yoneda M, Kato M, Doiguchi M, Imamura Y, Higashi M, Ikura T, Hayashi T, Kodama Y, Oki M, Nakayama T, Cheung E, Aburatani H, Takayama KI, Koseki H, Inoue S, Takeshima Y, Ito T (2016) Histone H2A T120 phosphorylation promotes oncogenic transformation via upregulation of cyclin D1. Mol Cell 64(1):176–188. https://doi.org/10.1016/j.molcel.2016.09.012
Tessarz P, Kouzarides T (2014) Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol 15(11):703–708. https://doi.org/10.1038/nrm3890
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21(3):381–395. https://doi.org/10.1038/cr.2011.22
Kang TH, Park DY, Kim W, Kim KT (2008) VRK1 phosphorylates CREB and mediates CCND1 expression. J Cell Sci 121(Pt 18):3035–3041. https://doi.org/10.1242/jcs.026757
Sevilla A, Santos CR, Vega FM, Lazo PA (2004) Human vaccinia-related kinase 1 (VRK1) activates the ATF2 transcriptional activity by novel phosphorylation on Thr-73 and Ser-62 and cooperates with JNK. J Biol Chem 279(26):27458–27465. https://doi.org/10.1074/jbc.M401009200
Sevilla A, Santos CR, Barcia R, Vega FM, Lazo PA (2004) c-Jun phosphorylation by the human vaccinia-related kinase 1 (VRK1) and its cooperation with the N-terminal kinase of c-Jun (JNK). Oncogene 23(55):8950–8958. https://doi.org/10.1038/sj.onc.1208015
Moura DS, Fernandez IF, Marin-Royo G, Lopez-Sanchez I, Martin-Doncel E, Vega FM, Lazo PA (2016) Oncogenic Sox2 regulates and cooperates with VRK1 in cell cycle progression and differentiation. Sci Rep 6:28532. https://doi.org/10.1038/srep28532
Hashiguchi T, Arakawa S, Takahashi S, Gonzalez FJ, Sueyoshi T, Negishi M (2016) Phosphorylation of Farnesoid X receptor at serine 154 links ligand activation with degradation. Mol Endocrinol 30(10):1070–1080. https://doi.org/10.1210/me.2016-1105
Shi L, Oberdoerffer P (2012) Chromatin dynamics in DNA double-strand break repair. Biochim Biophys Acta 7:811–819. https://doi.org/10.1016/j.bbagrm.2012.01.002
Deem AK, Li X, Tyler JK (2012) Epigenetic regulation of genomic integrity. Chromosoma 121(2):131–151. https://doi.org/10.1007/s00412-011-0358-1
Raschella G, Melino G, Malewicz M (2017) New factors in mammalian DNA repair—the chromatin connection. Oncogene 36(33):4673–4681. https://doi.org/10.1038/onc.2017.60
Ceccaldi R, Rondinelli B, D’Andrea AD (2016) Repair pathway choices and consequences at the double-strand break. Trends Cell Biol 26(1):52–64. https://doi.org/10.1016/j.tcb.2015.07.009
Polo SE, Jackson SP (2011) Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 25(5):409–433. https://doi.org/10.1101/gad.2021311
Falck J, Coates J, Jackson SP (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434(7033):605–611. https://doi.org/10.1038/nature03442
Sanz-Garcia M, Monsalve DM, Sevilla A, Lazo PA (2012) Vaccinia-related Kinase 1 (VRK1) is an upstream nucleosomal kinase required for the assembly of 53BP1 foci in response to ionizing radiation-induced DNA damage. J Biol Chem 287(28):23757–23768. https://doi.org/10.1074/jbc.M112.353102
Monsalve DM, Campillo-Marcos I, Salzano M, Sanz-Garcia M, Cantarero L, Lazo PA (2016) VRK1 phosphorylates and protects NBS1 from ubiquitination and proteasomal degradation in response to DNA damage. BBA Mol Cell Res 4:760–769. https://doi.org/10.1016/j.bbamcr.2016.02.005
Fell VL, Schild-Poulter C (2015) The Ku heterodimer: function in DNA repair and beyond. Mutat Res Rev Mutat Res 763:15–29. https://doi.org/10.1016/j.mrrev.2014.06.002
Celli GB, Denchi EL, de Lange T (2006) Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat Cell Biol 8(8):885–890. https://doi.org/10.1038/ncb1444
Choi YH, Lim JK, Jeong MW, Kim KT (2012) HnRNP A1 phosphorylated by VRK1 stimulates telomerase and its binding to telomeric DNA sequence. Nucleic Acids Res 40(17):8499–8518. https://doi.org/10.1093/nar/gks634
Downs JA, Allard S, Jobin-Robitaille O, Javaheri A, Auger A, Bouchard N, Kron SJ, Jackson SP, Cote J (2004) Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell 16(6):979–990. https://doi.org/10.1016/j.molcel.2004.12.003
Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, Herceg Z (2006) Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol 8(1):91–99. https://doi.org/10.1038/ncb1343
Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC, Grant PA, Smith MM, Christman MF (2002) Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 419(6905):411–415. https://doi.org/10.1038/nature01035
Ikura T, Tashiro S, Kakino A, Shima H, Jacob N, Amunugama R, Yoder K, Izumi S, Kuraoka I, Tanaka K, Kimura H, Ikura M, Nishikubo S, Ito T, Muto A, Miyagawa K, Takeda S, Fishel R, Igarashi K, Kamiya K (2007) DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol Cell Biol 27(20):7028–7040. https://doi.org/10.1128/MCB.00579-07
Robinson PJ, An W, Routh A, Martino F, Chapman L, Roeder RG, Rhodes D (2008) 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J Mol Biol 381(4):816–825. https://doi.org/10.1016/j.jmb.2008.04.050
Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL (2006) Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311(5762):844–847. https://doi.org/10.1126/science.1124000
Li X, Corsa CA, Pan PW, Wu L, Ferguson D, Yu X, Min J, Dou Y (2010) MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Mol Cell Biol 30(22):5335–5347. https://doi.org/10.1128/MCB.00350-10
Shin J, Chakraborty G, Bharatham N, Kang C, Tochio N, Koshiba S, Kigawa T, Kim W, Kim KT, Yoon HS (2011) NMR solution structure of human vaccinia-related kinase 1 (VRK1) reveals the C-terminal tail essential for its structural stability and autocatalytic activity. J Biol Chem 286(25):22131–22138. https://doi.org/10.1074/jbc.M110.200162
Kim W, Chakraborty G, Kim S, Shin J, Park CH, Jeong MW, Bharatham N, Yoon HS, Kim KT (2012) Macro histone H2A1.2 (MacroH2A1) protein suppresses mitotic kinase VRK1 during interphase. J Biol Chem 287(8):5278–5289. https://doi.org/10.1074/jbc.M111.281709
Sanz-Garcia M, Lopez-Sanchez I, Lazo PA (2008) Proteomics identification of nuclear Ran GTPase as an inhibitor of human VRK1 and VRK2 (vaccinia-related kinase) activities. Mol Cell Proteom 7(11):2199–2214. https://doi.org/10.1074/mcp.M700586-MCP200
Kalab P, Pralle A, Isacoff EY, Heald R, Weis K (2006) Analysis of a RanGTP-regulated gradient in mitotic somatic cells. Nature 440(7084):697–701. https://doi.org/10.1038/nature04589
Rogakou EP, Boon C, Redon C, Bonner WM (1999) Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 146(5):905–916. https://doi.org/10.1083/jcb.146.5.905
Nakamura AJ, Rao VA, Pommier Y, Bonner WM (2010) The complexity of phosphorylated H2AX foci formation and DNA repair assembly at DNA double-strand breaks. Cell Cycle 9(2):389–397. https://doi.org/10.4161/cc.9.2.10475
Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 10(15):886–895. https://doi.org/10.1016/S0960-9822(00)00610-2
Salzano M, Vazquez-Cedeira M, Sanz-Garcia M, Valbuena A, Blanco S, Fernandez IF, Lazo PA (2014) Vaccinia-related kinase 1 (VRK1) confers resistance to DNA-damaging agents in human breast cancer by affecting DNA damage response. Oncotarget 5(N7):1770–1778. https://doi.org/10.18632/oncotarget.1678
Williams GJ, Lees-Miller SP, Tainer JA (2010) Mre11–Rad50–Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair (Amst) 9(12):1299–1306. https://doi.org/10.1016/j.dnarep.2010.10.001
Williams GJ, Hammel M, Radhakrishnan SK, Ramsden D, Lees-Miller SP, Tainer JA (2014) Structural insights into NHEJ: building up an integrated picture of the dynamic DSB repair super complex, one component and interaction at a time. DNA Repair (Amst) 17:110–120. https://doi.org/10.1016/j.dnarep.2014.02.009
Schultz LB, Chehab NH, Malikzay A, Halazonetis TD (2000) p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol 151(7):1381–1390. https://doi.org/10.1083/jcb.151.7.1381
Wang B, Matsuoka S, Carpenter PB, Elledge SJ (2002) 53BP1, a mediator of the DNA damage checkpoint. Science 298(5597):1435–1438. https://doi.org/10.1126/science.1076182
Mochan TA, Venere M, DiTullio RA Jr, Halazonetis TD (2003) 53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage. Cancer Res 63(24):8586–8591
Ward IM, Chen J (2001) Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem 276(51):47759–47762. https://doi.org/10.1074/jbc.C100569200
Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, Parker AR, Jackson SP, Gennery A, Jeggo PA, Lobrich M (2004) A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell 16(5):715–724. https://doi.org/10.1016/j.molcel.2004.10.029
Wen J, Cerosaletti K, Schultz KJ, Wright JA, Concannon P (2013) NBN phosphorylation regulates the accumulation of MRN and ATM at sites of DNA double-strand breaks. Oncogene 32(37):4448–4456. https://doi.org/10.1038/onc.2012.443
Panier S, Boulton SJ (2014) Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 15(1):7–18. https://doi.org/10.1038/nrm3719
Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88(3):323–331
Selivanova G, Wiman KG (1995) p53: a cell cycle regulator activated by DNA damage. Adv Cancer Res 66:143–180
Oren M (2003) Decision making by p53: life, death and cancer. Cell Death Differ 10(4):431–442. https://doi.org/10.1038/sj.cdd.4401183
Toledo F, Wahl GM (2006) Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer 6(12):909–923. https://doi.org/10.1038/nrc2012
Meek DW, Anderson CW (2009) Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol 1:a000950. https://doi.org/10.1101/cshperspect.a000950
Meek DW (2009) Tumour suppression by p53: a role for the DNA damage response? Nat Rev Cancer 9(10):714–723. https://doi.org/10.1038/nrc2716
Lopez-Sanchez I, Valbuena A, Vazquez-Cedeira M, Khadake J, Sanz-Garcia M, Carrillo-Jimenez A, Lazo PA (2014) VRK1 interacts with p53 forming a basal complex that is activated by UV-induced DNA damage. FEBS Lett 588(5):692–700. https://doi.org/10.1016/j.febslet.2014.01.040
Levine AJ (2017) The p53 protein plays a central role in the mechanism of action of epigenetic drugs that alter the methylation of cytosine residues in DNA. Oncotarget 8(5):7228–7230. https://doi.org/10.18632/oncotarget.14805
Moll UM, Petrenko O (2003) The MDM2–p53 interaction. Mol Cancer Res 1(14):1001–1008
Lopez-Borges S, Lazo PA (2000) The human vaccinia-related kinase 1 (VRK1) phosphorylates threonine-18 within the mdm-2 binding site of the p53 tumour suppressor protein. Oncogene 19(32):3656–3664. https://doi.org/10.1038/sj.onc.1203709
Valbuena A, Vega FM, Blanco S, Lazo PA (2006) p53 downregulates its activating vaccinia-related kinase 1, forming a new autoregulatory loop. Mol Cell Biol 26(13):4782–4793. https://doi.org/10.1128/MCB.00069-06
Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP (1996) Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274(5289):948–953. https://doi.org/10.1126/science.274.5289.948
Teufel DP, Freund SM, Bycroft M, Fersht AR (2007) Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proc Natl Acad Sci USA 104(17):7009–7014. https://doi.org/10.1073/pnas.0702010104
Teufel DP, Bycroft M, Fersht AR (2009) Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 28(20):2112–2118. https://doi.org/10.1038/onc.2009.71
Del Sal G, Murphy M, Ruaro E, Lazarevic D, Levine AJ, Schneider C (1996) Cyclin D1 and p21/waf1 are both involved in p53 growth suppression. Oncogene 12(1):177–185
Ferbeyre G, de Stanchina E, Lin AW, Querido E, McCurrach ME, Hannon GJ, Lowe SW (2002) Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol 22(10):3497–3508. https://doi.org/10.1128/MCB.22.10.3497-3508.2002
Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80(2):293–299. https://doi.org/10.1016/0092-8674(95)90412-3
Miyashita T, Krajewski S, Krajewski M, Wang HG, Lin HK, Lieberman DA, Hoffman B, Reed JC (1994) Tumor suppressor p53 is a regulator of Bcl-2 and Bax gene expression in vitro and in vivo. Oncogene 9:1799–1805
Menendez D, Inga A, Resnick MA (2009) The expanding universe of p53 targets. Nat Rev Cancer 9(10):724–737. https://doi.org/10.1038/nrc2730
Lazo PA (2017) Reverting p53 activation after recovery of cellular stress to resume with cell cycle progression. Cell Signal 33:49–58. https://doi.org/10.1016/j.cellsig.2017.02.005
Piya S, Kim JY, Bae J, Seol DW, Moon AR, Kim TH (2012) DUSP6 is a novel transcriptional target of p53 and regulates p53-mediated apoptosis by modulating expression levels of Bcl-2 family proteins. FEBS Lett 586(23):4233–4240. https://doi.org/10.1016/j.febslet.2012.10.031
Yamaguchi H, Durell SR, Chatterjee DK, Anderson CW, Appella E (2007) The Wip1 phosphatase PPM1D dephosphorylates SQ/TQ motifs in checkpoint substrates phosphorylated by PI3K-like kinases. Biochemistry 46(44):12594–12603. https://doi.org/10.1021/bi701096s
Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, Timofeev ON, Dudgeon C, Fornace AJ, Anderson CW, Minami Y, Appella E, Bulavin DV (2006) Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell 23(5):757–764. https://doi.org/10.1016/j.molcel.2006.07.010
Shen WH, Wang J, Wu J, Zhurkin VB, Yin Y (2006) Mitogen-activated protein kinase phosphatase 2: a novel transcription target of p53 in apoptosis. Cancer Res 66(12):6033–6039. https://doi.org/10.1158/0008-5472.CAN-05-3878
Jeong MW, Kang TH, Kim W, Choi YH, Kim KT (2013) Mitogen-activated protein kinase phosphatase 2 regulates histone H3 phosphorylation via interaction with vaccinia-related kinase 1. Mol Biol Cell 24(3):373–384. https://doi.org/10.1091/mbc.E12-06-0456
Valbuena A, Castro-Obregon S, Lazo PA (2011) Downregulation of VRK1 by p53 in response to DNA damage is mediated by the autophagic pathway. PLoS One 6(2):e17320. https://doi.org/10.1371/journal.pone.0017320
Valbuena A, Blanco S, Vega FM, Lazo PA (2008) The C/H3 domain of p300 is required to protect VRK1 and VRK2 from their downregulation induced by p53. PLoS One 3(7):e2649. https://doi.org/10.1371/journal.pone.0002649
Baehrecke EH (2005) Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol 6(6):505–510. https://doi.org/10.1038/nrm1666
Cecconi F, Levine B (2008) The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell 15(3):344–357. https://doi.org/10.1016/j.devcel.2008.08.012
Mizushima N (2007) Autophagy: process and function. Genes Dev 21(22):2861–2873. https://doi.org/10.1101/gad.1599207
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8(9):741–752. https://doi.org/10.1038/nrm2239
Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D’Amelio M, Djavaheri-Mergny M, Cecconi F, Tavernarakis N, Kroemer G (2008) A dual role of p53 in the control of autophagy. Autophagy 4(6):810–814. https://doi.org/10.4161/auto.6486
Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM (2006) DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126(1):121–134. https://doi.org/10.1016/j.cell.2006.05.034
Mah LY, O’Prey J, Baudot AD, Hoekstra A, Ryan KM (2012) DRAM-1 encodes multiple isoforms that regulate autophagy. Autophagy 8(1):18–28. https://doi.org/10.4161/auto.8.1.18077
Valbuena A, Suarez-Gauthier A, Lopez-Rios F, Lopez-Encuentra A, Blanco S, Fernandez PL, Sanchez-Cespedes M, Lazo PA (2007) Alteration of the VRK1-p53 autoregulatory loop in human lung carcinomas. Lung Cancer 58(3):303–309. https://doi.org/10.1016/j.lungcan.2007.06.023
Santos CR, Rodriguez-Pinilla M, Vega FM, Rodriguez-Peralto JL, Blanco S, Sevilla A, Valbuena A, Hernandez T, van Wijnen AJ, Li F, de Alava E, Sanchez-Cespedes M, Lazo PA (2006) VRK1 signaling pathway in the context of the proliferation phenotype in head and neck squamous cell carcinoma. Mol Cancer Res 4(3):177–185. https://doi.org/10.1158/1541-7786.MCR-05-0212
Wang S, Zhao Y, Aguilar A, Bernard D, Yang CY (2017) Targeting the MDM2-p53 protein–protein interaction for new cancer therapy: progress and challenges. Cold Spring Harb Perspect Med 7(5):a026245. https://doi.org/10.1101/cshperspect.a026245
Vega FM, Gonzalo P, Gaspar ML, Lazo PA (2003) Expression of the VRK (vaccinia-related kinase) gene family of p53 regulators in murine hematopoietic development. FEBS Lett 544(1–3):176–180. https://doi.org/10.1016/S0014-5793(03)00501-5
Valbuena A, Lopez-Sanchez I, Lazo PA (2008) Human VRK1 is an early response gene and its loss causes a block in cell cycle progression. PLoS One 3(2):e1642. https://doi.org/10.1371/journal.pone.0001642
Kiessling MK, Schuierer S, Stertz S, Beibel M, Bergling S, Knehr J, Carbone W, de Valliere C, Tchinda J, Bouwmeester T, Seuwen K, Rogler G, Roma G (2016) Identification of oncogenic driver mutations by genome-wide CRISPR-Cas9 dropout screening. BMC Genom 17(1):723. https://doi.org/10.1186/s12864-016-3042-2
Kim IJ, Quigley D, To MD, Pham P, Lin K, Jo B, Jen KY, Raz D, Kim J, Mao JH, Jablons D, Balmain A (2013) Rewiring of human lung cell lineage and mitotic networks in lung adenocarcinomas. Nat Commun 4:1701. https://doi.org/10.1038/ncomms2660
Fournier MV, Martin KJ, Kenny PA, Xhaja K, Bosch I, Yaswen P, Bissell MJ (2006) Gene expression signature in organized and growth-arrested mammary acini predicts good outcome in breast cancer. Cancer Res 66(14):7095–7102. https://doi.org/10.1158/0008-5472.CAN-06-0515
Martin KJ, Patrick DR, Bissell MJ, Fournier MV (2008) Prognostic breast cancer signature identified from 3D culture model accurately predicts clinical outcome across independent datasets. PLoS One 3(8):e2994. https://doi.org/10.1371/journal.pone.0002994
Huang W, Cui X, Chen Y, Shao M, Shao X, Shen Y, Liu Q, Wu M, Liu J, Ni W, Lu C, Wan C (2016) High VRK1 expression contributes to cell proliferation and survival in hepatocellular carcinoma. Pathol Res Pract 212(3):171–178. https://doi.org/10.1016/j.prp.2015.11.015
Varghese RT, Liang Y, Guan T, Franck CT, Kelly DF, Sheng Z (2016) Survival kinase genes present prognostic significance in glioblastoma. Oncotarget 7:20140–20151. https://doi.org/10.18632/oncotarget.7917
Li J, Wang T, Pei L, Jing J, Hu W, Sun T, Liu H (2017) Expression of VRK1 and the downstream gene BANF1 in esophageal cancer. Biomed Pharmacother 89:1086–1091. https://doi.org/10.1016/j.biopha.2017.02.095
Valbuena A, Sanz-Garcia M, Lopez-Sanchez I, Vega FM, Lazo PA (2011) Roles of VRK1 as a new player in the control of biological processes required for cell division. Cell Signal 23(8):1267–1272. https://doi.org/10.1016/j.cellsig.2011.04.002
Moura DS, Campillo-Marcos I, Vazquez-Cedeira M, Lazo PA (2018) VRK1 and AURKB form a complex that cross inhibit their kinase activity and the phosphorylation of histone H3 in the progression of mitosis. Cell Mol Life Sci. https://doi.org/10.1007/s00018-018-2746-7
Wiebe MS, Nichols RJ, Molitor TP, Lindgren JK, Traktman P (2010) Mice deficient in the serine/threonine protein kinase VRK1 are infertile due to a progressive loss of spermatogonia. Biol Reprod 82(1):182–193. https://doi.org/10.1095/biolreprod.109.079095
Schober CS, Aydiner F, Booth CJ, Seli E, Reinke V (2011) The kinase VRK1 is required for normal meiotic progression in mammalian oogenesis. Mech Dev 128(3–4):178–190. https://doi.org/10.1016/j.mod.2011.01.004
Kim J, Choi YH, Chang S, Kim KT, Je JH (2012) Defective folliculogenesis in female mice lacking vaccinia-related kinase 1. Sci Rep 2:468. https://doi.org/10.1038/srep00468
Nichols RJ, Wiebe MS, Traktman P (2006) The vaccinia-related kinases phosphorylate the N’ terminus of BAF, regulating its interaction with DNA and its retention in the nucleus. Mol Biol Cell 17(5):2451–2464. https://doi.org/10.1091/mbc.E05-12-1179
Jamin A, Wiebe MS (2015) Barrier to Autointegration Factor (BANF1): interwoven roles in nuclear structure, genome integrity, innate immunity, stress responses and progeria. Curr Opin Cell Biol 34:61–68. https://doi.org/10.1016/j.ceb.2015.05.006
Molitor TP, Traktman P (2013) Molecular genetic analysis of VRK1 in mammary epithelial cells: depletion slows proliferation in vitro and tumor growth and metastasis in vivo. Oncogenesis 2:e48. https://doi.org/10.1038/oncsis.2013.11
McDonald ER, 3rd, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E, Gampa K, Golji J, Kao I, Li L, Megel P, Perkins TA, Ramadan N, Ruddy DA, Silver SJ, Sovath S, Stump M, Weber O, Widmer R, Yu J, Yu K, Yue Y, Abramowski D, Ackley E, Barrett R, Berger J, Bernard JL, Billig R, Brachmann SM, Buxton F, Caothien R, Caushi JX, Chung FS, Cortes-Cros M, deBeaumont RS, Delaunay C, Desplat A, Duong W, Dwoske DA, Eldridge RS, Farsidjani A, Feng F, Feng J, Flemming D, Forrester W, Galli GG, Gao Z, Gauter F, Gibaja V, Haas K, Hattenberger M, Hood T, Hurov KE, Jagani Z, Jenal M, Johnson JA, Jones MD, Kapoor A, Korn J, Liu J, Liu Q, Liu S, Liu Y, Loo AT, Macchi KJ, Martin T, McAllister G, Meyer A, Molle S, Pagliarini RA, Phadke T, Repko B, Schouwey T, Shanahan F, Shen Q, Stamm C, Stephan C, Stucke VM, Tiedt R, Varadarajan M, Venkatesan K, Vitari AC, Wallroth M, Weiler J, Zhang J, Mickanin C, Myer VE, Porter JA, Lai A, Bitter H, Lees E, Keen N, Kauffmann A, Stegmeier F, Hofmann F, Schmelzle T, Sellers WR (2017) Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell 170(3):577–592 e510. https://doi.org/10.1016/j.cell.2017.07.005
Knight ZA, Lin H, Shokat KM (2010) Targeting the cancer kinome through polypharmacology. Nat Rev Cancer 10(2):130–137. https://doi.org/10.1038/nrc2787
Fedorov O, Marsden B, Pogacic V, Rellos P, Muller S, Bullock AN, Schwaller J, Sundstrom M, Knapp S (2007) A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc Natl Acad Sci USA 104(51):20523–20528. https://doi.org/10.1073/pnas.0708800104
Fedorov O, Sundstrom M, Marsden B, Knapp S (2007) Insights for the development of specific kinase inhibitors by targeted structural genomics. Drug Discov Today 12(9–10):365–372. https://doi.org/10.1016/j.drudis.2007.03.006
Nebot-Bral L, Brandao D, Verlingue L, Rouleau E, Caron O, Despras E, El-Dakdouki Y, Champiat S, Aoufouchi S, Leary A, Marabelle A, Malka D, Chaput N, Kannouche PL (2017) Hypermutated tumours in the era of immunotherapy: the paradigm of personalised medicine. Eur J Cancer 84:290–303. https://doi.org/10.1016/j.ejca.2017.07.026
Acknowledgements
I.C-M was supported by FPI-MINECO-Fondo Social Europeo predoctoral contract (BES-2014-06772). The laboratory was supported by grants from Agencia Estatal de Investigación-MINECO (SAF2016-75744-R) and Consejería de Educación de la Junta de Castilla y León (CSI001U16, UIC-017) to P.A.L.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interests.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Campillo-Marcos, I., Lazo, P.A. Implication of the VRK1 chromatin kinase in the signaling responses to DNA damage: a therapeutic target?. Cell. Mol. Life Sci. 75, 2375–2388 (2018). https://doi.org/10.1007/s00018-018-2811-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-018-2811-2