
Abstract
Bone morphogenetic proteins (BMPs) are important extracellular cytokines that play critical roles in embryogenesis and tissue homeostasis. BMPs signal via transmembrane type I and type II serine/threonine kinase receptors and intracellular Smad effector proteins. BMP signaling is precisely regulated and perturbation of BMP signaling is connected to multiple diseases, including musculoskeletal diseases. In this review, we will summarize the recent progress in elucidation of BMP signal transduction, how overactive BMP signaling is involved in the pathogenesis of heterotopic ossification and Duchenne muscular dystrophy, and discuss possible therapeutic strategies for treatment of these diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bone morphogenetic proteins (BMPs) were first discovered and described by Marshall Urist as secreted proteins that guide proliferation and differentiation of mesenchymal cells of muscle into bone cells [1, 2]. Nowadays, BMPs are recognized to be multi-functional growth factors that belong to the transforming growth factor β (TGFβ) superfamily, which also includes TGFβs, growth and differentiation factors, activins and Müllerian inhibiting substance. All TGFβ family members are structurally related and are produced by cells as larger precursor proteins that are proteolytically processed into amino-terminal remnants and mature carboxy-terminal parts that bind to cell surface receptors. The mature parts have a characteristic cysteine knot structure. The TGFβ/BMP signaling pathway is essential for orchestration of embryonic development and maintenance of tissue homeostasis in adult animals [3].
More than 20 BMPs have been identified and characterized. Although BMPs were initially identified for their ability to induce bone formation [4], not all of the BMPs appear to be osteo-inductive. On the basis of phylogenetic analysis and sequence similarities, the osteo-inducing BMPs can be divided into three subgroups: the BMP2/4 subgroup, the BMP5/6/7/8 (OP) subgroup and the BMP9/10 subgroup [5]. All of the bone-inducing BMPs can induce mesenchymal stem cells to differentiate into osteoblasts in vitro [5]. However, studies using transgenic and knockout mice or animals with naturally occurring mutations in bone-inducing BMPs suggested that osteo-inductive BMPs are not only necessary for bone and cartilage formation but also play vital roles in heart and neural development (Table 1).
BMPs are morphogens and can induce different cell fates at different concentrations [6, 7]. They are not only required for establishment of dorsal–ventral pattern in embryogenesis but elicit a broad spectrum of biological activities in large variety of tissues, such as repair of bone fracture, maintenance of iron homeostasis, and so on [6–12]. Therefore, BMP signaling needs to be carefully regulated by positive and negative regulatory mechanisms to regulate the intensity and duration of the signaling response in a spatially controlled manner [13]. Perturbations of BMP signaling pathways contribute to progression of a variety of diseases including skeletal diseases, vascular diseases, tissue dystrophy, and cancer [5, 14]. This review will focus on the BMP signaling pathway in general and two different diseases that are linked with ectopic activity of BMPs, i.e., heterotopic ossifications (HO) and the muscle degeneration disease Duchenne muscular dystrophy (DMD).
BMP receptor signal pathway
BMP signaling pathway
Structure of BMPs and type I and II receptors
BMPs are structurally related cytokines that are found in all multi-cellular organisms. The crystal structure confirmed that the monomers of BMP7 and BMP2 share a common scaffold [15, 16]. Functional studies show that BMPs are highly conserved in evolution; in fact, Decapentaplegic (Dpp) and 60A, the Drosophila homologues of BMP2 and BMP7, were shown to induce bone formation in mammals [17] and human BMP4 can rescue dpp null dorsal–ventral patterning in Drosophila embryos [18]. Mature BMPs are dimeric proteins that can function either as homodimeric or heterodimeric complexes [19]. Most current knowledge of BMPs are based on studies from homodimeric BMPs, however, both homodimeric BMPs and heterodimeric BMPs are present in vivo, and exert multiple bio-functions [20].
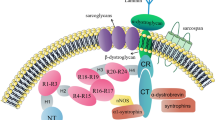
Like other members in the TGFβ family, BMPs signal across the plasma membrane by interacting and inducing complexes composed of type I and type II receptors that are endowed with intrinsic serine/threonine kinase activity (Fig. 1). In mammals, there are seven type I receptors, the BMPR-I group (ALK3 and ALK6), the ALK-I group (ALK1 and ALK2) and the TβR-I group (ALK4, ALK5 and ALK7) [5]. ALK1, -2, -3, and -6 have been shown to serve as BMP type I receptors. There are four type II receptors in mammals, i.e., BMPR-II, ActR-II and ActR-IIB and MISR-II, of which BMPR-II, ActR-II and ActR-IIB can serve as type II receptor for BMPs that are expressed in multiple tissues [5].

Schematic overview of BMP signaling. Upon formation of heteromeric complex composed of type II and type I receptors and the BMP dimers, FKBP12 is released from the type I receptors and released the phosphorylation site on type I receptor. Next, the type I receptor is phosphorylated by the type II receptor, which propagates the signal into the cells by phosphorylating the C-terminus of R-Smads. The phosphorylated R-Smads form a complex with the Co-Smad and are translocated into nucleus where they in collaboration with other transcription factors to regulate gene expression. The presence of membrane-tethered type III receptors on the membrane can enhance R-Smads phosphorylation. The cells can release the extracellular domain of the type III receptor, which is called the soluble form of type III receptors. The soluble form of type III receptors and other BMP antagonists such as Noggin and Chordin, repress BMP signaling through prohibiting BMP binding to its receptors. I-Smads repress BMP activity either by repressing complex of R-Smads/Co-Smads, or directly inactivate type I receptor activity. In the nucleus, phosphatases represses BMP activity by dephosphorylating R-Smads thereby promoting the exportation of R-Smads. In addition to R-Smads, BMP can also signal via MAPK (non-canonical BMP pathways) through activation of TAK1, which can further activate MAPK. MAPK will be transported into the nucleus, and activate some transcriptional factors, which can further initiate specific gene expression
Both type I and type II receptors are required for signal transduction [21]. The type II receptors are constitutively active and are responsible for activating type I receptors. The type I receptor contains a so-called L45 loop that extends from the kinase domain and which is required for interaction and activation of downstream receptor regulated Smads (R-Smads) [5]. The intracellular GS domain (glycine and serine-rich domain) of type I receptors located N-terminal to the serine-threonine kinase domain controls the kinase activity of type I receptors. The phosphorylation of serine and threonine residues in the GS domain by type II receptor activates the kinase activity of the type I receptor and initiates signal transduction mediated by the type I receptor [5]. Under normal circumstances, type I receptors can form oligomeric complexes with type II receptors in the absence of ligands. To prevent type I receptor activation independent of ligand stimulation, the negative regulator FKBP12 binds to the intracellular GS domain of type I receptors thereby preventing it from being phosphorylated in the absence of a ligand [22–24]. Upon ligand stimulation, FKBP12 dissociates from the type I receptors, thereby allowing the phosphorylation by type II receptors on serine and threonine residues in the GS domains. Mutations in the GS domain of type I receptors can lead to constitutive activation of the type I receptors [23, 25]. Notably, in contrast to other type II receptors, the BMPR-II contains a long C-terminal tail following the serine/threonine kinase domain [26]. The C-terminal tail is not involved in BMP-induced Smad signaling, however, in patients suffering from primary pulmonary hypertension (PPH), the C-terminal tail of BMPR-II was found to be truncated, suggesting a unique role for BMPR-II in Smad-independent signaling [27, 28]. Further studies revealed that BMPR-II through its long C-terminal tail mediates BMP-controlled cytoskeletal rearrangements [29, 30].
Smad protein-mediated BMP signaling
Upon formation and subsequent activation of a BMP ligand-receptor complex, the activated type I receptors phosphorylate receptor regulated Smad proteins (R-Smads) at their two C-terminal serine residues. ALK1, -2, -3, and -6 mediate the phosphorylation of R-Smad1, -5, and -8. The phosphorylated R-Smads can form complexes with the common mediated Smad (Co-Smad), Smad4, and translocate into the nucleus. In the nucleus, this Smad complex binds the DNA and in collaboration with co-activators and repressors and other transcription factors regulates the expression of specific genes [27].
Recently, the nuclear localized Smads were found to promote also miRNA maturation [31]. TGFβ and BMP stimulation promotes a rapid increase in expression of mature miR-21 through a post-transcriptional step; this process involves R-Smads but is Smad4 independent [31]. The Smad binding sequence on pri-miRNA, which is similar to the Smad binding element (SBE) normally present in the promoter region of TGFβ or BMP responsive genes, is required for R-Smads function in promoting mature miRNA processing [32].
Non-Smad BMP signaling
In addition to Smad proteins, BMPs are also able to transduce signals via Smad-independent signaling pathways, for example via ERK, p38, and JNK MAP kinases, small GTPases, and PI3K–Akt/PKPB pathways. BMPs can also activate TGFβ-activated kinase-1 (TAK1), a member of the MAP kinase kinase kinase family [33], which mediates the phosphorylation of p38, JNKs or ERK1/2 in various cell types [34–36]. Interestingly, the ERK1/2 MAPK kinase and TAK1 are important BMP-Smad signaling modulators. It has been demonstrated that both the Smad pathway and the P38/ERK MAPK pathway are required for BMP-induced osteoblast differentiation [37–39]. TAK1 was also shown as positive and negative regulator for Smad signaling. TAK1 was originally discovered as a BMP agonist that synergizes with Smad1/5 to induce ventralization in Xenopus embryos [40], however, TAK1 was also shown to interact with R-Smads and to interfere with R-Smads transactivation thereby repressing BMP-induced osteoblast differentiation [41]. Recently, TAK1 was found to promote Smad1/5/8 phosphorylation at C-terminal serine residues in chondrocytes and to be an essential regulator for BMP signaling in chondrogenesis in vitro and in vivo [42–44]. Therefore, BMP-induced TAK1 and its downstream MAP kinases might function as modulators for the canonical BMP-Smad pathway.
Modulators of the BMP signaling pathway
Given the important role BMP signaling is playing in a wide variety of biological processes, it has to be tightly regulated. This is achieved by both positive and negative regulation, which occurs at each step of the BMP/Smad signaling pathway. The expression, localization, and activation of BMP ligands, receptors, and Smads are intricately regulated, and this also involves the crosstalk with other signaling pathways [27, 39]. For instance, the Wnt, Notch, and FGF signaling pathways are reported either to be required or to promote BMP-induced osteoblast differentiation [39]. In the following section, we will first discuss the regulation of BMP/Smad signaling at the extracellular level, followed by intracellular BMP receptor/Smad-initiated responses and then the activity of Smads as nuclear effectors.
BMP antagonists
Numerous secreted proteins have been identified as BMP antagonists. BMP antagonists can directly bind to BMPs and thereby prohibit BMPs from binding to their receptors. All of these BMP antagonists have a cysteine-knot structure. Based on the size of cysteine-knots, the BMP antagonists can be divided into three subfamilies: the DAN subfamily (eight-membered ring) including USAG-1 and Sclerostin, the twisted gastrulation (Tsg) subfamily (nine-membered ring), and chordin and Noggin (ten-membered ring) [45, 46]. Detailed functional studies demonstrated that BMP antagonists selectively block the activity of specific BMPs. For instance, Noggin inhibits BMP2 and BMP4 but can not block BMP6 and BMP9 activity [47]. Chordin can bind to BMP2, -4, -7, but cannot interact with the other BMP-family proteins [48]. Sclerostin binds BMP6 and BMP7 and inhibits their activity [49].
BMP co-receptors
Currently a number of transmembrane and membrane-anchored proteins have been characterized as co-receptors or type III receptors, and function as modulators for TGFβ/BMP signaling. The repulsive guidance molecules (RGM) family, comprised of RGMa (also known as RGM), RGMb (also known as Dragon), RGMc (also denoted as Hfe2 or HJV), and RGMd (currently only found in fish [50]) form the first known BMP selective co-receptor family that can potentiate BMP signaling. RGM proteins are glycosylphosphatidylinositol (GPI)-anchored membrane proteins [50–53]. The mechanisms of how RGM proteins potentiate BMP signaling are still elusive. One possibility is that RGM proteins can interact with type I receptors and alter utilization of BMP type II receptors by BMP ligands [52, 54]. Recently, RGMs were suggested to enable association of neogenin with other BMP receptors (BMPRs) in lipid rafts of chondrocytes. The modes of receptors oligomerization could determine which downstream BMP signaling pathways are activated [55]. Neogenin and RGMc could facilitate the formation of membrane receptor complexes that deliver continuous Smad signaling, and are required for BMP-induced chondrogenesis in vitro and in vivo [56]. Betaglycan and endoglin were initially described as co-receptors for TGFβ, but have also been shown to function as co-receptors for BMP2, -4, -7, and BMP9, respectively [57–59]. Betaglycan can promote the binding of BMP ligands, BMP2, -4, -7, to ALK3 and ALK6 to enhance BMP signaling [57]. Endoglin is highly expressed in endothelial cells, which usually have little or no expression of betaglycan [60, 61]. The presence of endoglin in the endothelial cells may make endothelial cells more responsive to ALK1-mediated BMP9 signaling [59, 62]. Notably, cells can release the extracellular domain of the co-receptors by cleavage at the sites near their transmembrane regions [63–65]. The soluble forms of RGM proteins and endoglin, which only contain the extracellular domain of these receptors, were established as BMP inhibitors in recent studies [11, 66] as they could possibly compete with membrane-receptors for binding to BMP ligands. Moreover, Xenopus BAMBI and its mammalian homolog Nma have been identified as pseudo-receptors that contain extracellular domains structurally related to that of type I receptors. They lack the intracellular kinase domain, and as a consequence function as repressors of BMP signaling to prevent the formation of active receptor complexes [67, 68].
Intracellular regulation of BMP signaling
The inhibitory Smads (I-Smad) comprise Smad6 and Smad7, which serve as inhibitors for the Smad signaling pathways. Smad6 mainly targets BMP signaling while Smad7 represses both TGFβ and BMP signaling [69–71]. Smad6 has been shown to compete with R-Smads for interaction with Smad4, and can recruit the transcriptional corepressor CtBP to repress BMP-induced transcription [69, 72]. Smad7 represses R-Smad phosphorylation. In addition, it can recruit phosphatases that can mediate type I receptor dephosphorylation and inactivation [70, 73].
The ubiquitin system also actively participates in the regulation of BMP signaling. Smad7 can recruit Smurf E3 ubiquitin ligases and mediate the turnover of activated type I receptors [74, 75]. In addition, Smurfs can directly interact with R-Smads and promote their degradation [76, 77]. The deubiquitinating enzyme (DUB) UCH37 can bind to Smad7. Then it can deubiquitinate and stabilize type I receptors and hence function as agonist for TGFβ signaling [78].
Smad anchor for receptor activation (SARA) protein can enhance TGFβ signaling by recruiting and presenting non-phosphorylated R-Smads to active membrane type I receptors [79]. Recently, endofin was characterized as a protein acting similarly to SARA in BMP signaling, which can recruit nonphosphorylated Smad1, enhance Smad1 phosphorylation, and the subsequent nuclear translocation of Smad1 [80].
Since the BMP signaling is transduced by phosphorylated C-terminal R-Smads, phosphatases for R-Smads could function as repressor for BMP signaling. The phosphatase proteins, small C-terminal domain phosphatase (SCP1/2) and protein phosphatase magnesium-dependent 1A (PPM1A), can efficiently dephosphorylate the C-terminal domain of Smad1 in the nucleus and attenuate BMP signaling [81, 82]. Recently, SCP1 was established to repress BMP-induced osteoblast differentiation [83]. In contrast, PP2A, which can dephosphorylate R-Smads at their linker region, was shown to enhance canonical Smad signaling [84].
In the nucleus, the activated Smad complexes interact with other transcription factors to control gene expression. Histone deacetylases (HDACs), chromatin modulators, function as negative regulators for BMP signaling. c-Ski and Twist-1 are well-studied negative regulators for BMP signaling, which interact with Smad4 and recruit HDACs to the Smad complex thereby repressing its transcriptional activity and antagonize BMP signaling. Both c-Ski and Twist-1 can inhibit BMP-induced osteoblast differentiation [85–88].
Endocytosis and BMP signaling
Formation of the ligand-receptor complex can initiate endocytosis of active ligands and receptors. Endocytosis-mediated internalization of receptors cannot only control receptor density, thus modulating signaling activity, but is also required for signal transduction in some situations [89, 90].
It was proposed that BMP type I receptors internalization is mediated by clathrin-mediated endocytosis, which is required for continuation of Smad signaling [91]. Endofin, the SARA-like protein in BMP signaling located in the endosome derived from clathrin-coated pits, promotes the BMP-Smad signaling [80]. However, the interaction of Smad7–Smurf2 complexes that are present in lipid raft caveolae to the type I receptors can promote their rapid turnover and repress signaling [89].
Negative feedback loops for BMP signaling
In the previous section, we have discussed various negative regulatory mechanisms for BMP signaling. Multiple negative regulators have been demonstrated to be direct target genes of BMP signaling. For instance, Noggin/Chordin and I-Smads are all well established as direct BMP target genes [92–96]. Endofin contains a protein-phosphatase-binding motif. Depending on the amount of nonphosphorylated Smad1 in the cells, endofin can also function as a BMP inhibitor by recruiting phosphatases to inactivate type I receptors [80]. The activation of BMP signaling can also result in degradation of type I receptors via endocytosis [89]. These mechanisms establish auto-regulatory negative feedback loops for BMP signaling to exert spatial–temporal control over its multiple activities.
In the above sections, we have summarized current research results on BMP signaling. BMPs were originally discovered as bone inducers and repressors of myogenesis [1, 4, 97]. In the following sections, we will discuss the roles of BMP signaling in the progression of two representative bone and skeletal muscle diseases: heterotopic ossification (HO) and Duchenne muscular dystrophy (DMD).
BMP signaling in heterotopic ossification
Heterotopic ossification (HO) is defined as bone formation at aberrant locations outside the skeleton; mature bone tissue can be found in the soft tissue where bone normally does not exist. The presence of HO might cause joint stiffness, limited range of motion, swelling and pain, and can even result in severe functional limitations [98]. HO was first clearly described in 1883, and then in 1918, Déjerine and Ceillier found that soldiers in World War I with spinal cord trauma frequently acquired HO. Nowadays, it is well described in multiple clinical reports, for example, patients who have total hip arthroplasty or injury at spinal cord are at risk of developing HO [99]. A few years ago, Charmers and colleagues proposed that osteogenic precursor cells, inducing agents and permissive microenvironments are essential conditions for ectopic bone formation [100]. Among all the discovered osteo-inductive growth factors, BMPs are considered important growth factors involved in bone formation; the ability to induce bone when implanted at ectopic sites in rats led to their discovery [1, 4, 101–103]. Besides the trauma-induced HO, there is also a hereditary form of HO called fibrodysplasia ossificans progressiva (FOP).
Fibrodysplasia ossificans progressiva
Fibrodysplasia ossificans progressiva (FOP) has an incidence of 1 in 2 million. Patients develop progressive heterotopic ossification (HO) in the soft tissues either as a result of trauma or spontaneously. Children born with FOP appear normal at birth apart from deformed great toes [25]. Before the age of ten, FOP patients develop painful and highly inflammatory soft tissue swellings, which can transform into bone [104]. The occurrence of ectopic bone usually follows a fixed pattern: starting from the neck, then in the shoulders, arms, chest areas, and finally in the feet. The development of ectopic bone formation in FOP patients occurs through an endochondral ossification pathway. A histological examination identified several stages of the FOP lesion formation: lymphocyte infiltration, degradation of muscle tissue, fibroproliferative and highly angiogenic stages, cartilage and finally formation of bone [105]. Minor trauma to soft tissue can initiate painful ectopic bone formation in FOP patients, but sometimes bone formation seems to occur spontaneously without detectable trauma [106, 107]. Surgical resection to remove the ectopic bone tissue is not an option for treatment of the FOP patients as the surgical trauma induces the formation of new heterotopic bone [107].
In 1997, Shafritz reported that BMP4 is overexpressed in lymphoblastoid cells and lesional cells of FOP patients [108]. The BMP4 antagonist Noggin is a direct target gene for BMP signaling. However, BMP4-induced Noggin expression in lymphocytes of FOP patients was found to be attenuated compared to the control lymphocytes [108, 109], implying the dysregulation of BMP4–Noggin negative feedback loop in FOP patients. However, until now, only transgenic mice expressing BMP4 under the control of the neuron-specific enolase (NSE) promoter developed a FOP-like phenotype [102]. Others reported that BMP4 transgenic mice either died at birth or failed to develop a FOP-like disorder [110–113]. Moreover, the FOP lymphocytes displayed higher expression of ALK3 and a defect in endocytosis-dependent degradation of BMP type I receptors, which could result in constitutively high expression of ALK3 on the membrane [114]. In 2006, the gene responsible for the FOP disease was identified as the ALK2 gene encoding a BMP type I receptor. The classic FOP-associated ALK2 mutation is R206H; this residue is located in the GS domain and interferes with the binding of the negative regulator FKBP12, which results in ALK2 activation in the absence of BMP ligands [25, 115–117]. Recently, an ALK2 R206H knock in mice was reported to have FOP symptoms, including malformed first digits in the hind limbs and postnatal extraskeletal bone formation [118]. These results further supported that mutant ALK2, which can sensitize mesenchymal cells to undergo BMP-induced osteoblast differentiation and bone formation in vitro, caused FOP [118].
The mutated ALK2 in FOP patients that leads to elevated BMP signaling plays a pivotal role in ectopic bone formation in the FOP patients. However, transgenic mice with global postnatal expression of constitutively activated (CA)-ALK2 (induced without inflammation) do not develop ectopic bone. CA-ALK2 in combination with local inflammation mediated by adenoviral infection induced bone formation in skeletal muscle, joint fusion, and functional impairment [119]. Moreover, mice treated with the anti-inflammatory drug dexamethasone showed significantly reduced ectopic bone formation induced by adenoviral infection in the skeletal muscle of CA-ALK2 transgenic mice [119]. Many studies on FOP patients support the important role of inflammation in disease progression [120, 121]. A clinical study found that bone-marrow transplantation from a normal donor in a FOP patient, which received immunosuppression, ameliorated the activation of ectopic bone formation [122]. Thus, hematopoietic cells may contribute to ectopic bone formation.
For a long time, osteoprogenitor cells for ectopic bone were considered to be the mesenchymal stem cells residing in the skeletal muscles which have the potential to differentiate into multiple mesenchymal lineages [101, 123–126]. In 2010, Medici and colleagues [127] showed an endothelial origin of up to 50 % of the heterotopic cartilage and bone in both FOP patients and the CA-ALK2 transgenic FOP mouse model. In vitro, CA-ALK2 or TGFβ and BMP4 stimulation is able to induce endothelial-to-mesenchymal transition (Endo-MT). CA-ALK2 or TGFβ and BMP4 stimulation induce the expression of transcription factors Snail/Slug/Twist/ZEB-1/Sip-1, all of which are important for epithelial-to-mesenchymal (EMT) transition, and convert mature endothelial cells into mesenchymal stem cell-like cells, which subsequently differentiate into chondrocytes, osteoblasts, or adipocytes under the appropriate differentiation conditions [127] (Fig. 2). In addition, the increased number of circulating osteoprogenitor cells of hematopoietic origin, were reported to associate with active HO formation in patients with FOP, and to be present in the pre-osseous fibroproliferative lesions. Therefore, circulating osteoprogenitor cells are another group of osteoprogenitor cells that can contribute to HO in susceptible host tissue [128].

Diagram illustrating endothelial-to-mesenchymal transition (Endo-MT) and its role in the heterotopic bone formation in FOP patients. In the endothelial cells, TGFβ or BMP4 induces expression of transcription factors for mesoderm induction including Snail/Slug/Twist/ZEB-1/Sip-1, and reprogram endothelial cells into multipotent mesoderm cells through Endo-MT transition. The multipotent mesoderm cells can be further differentiated into osteoblasts, chondrocytes, and adipocytes under proper differentiation conditions. The type I receptors ALK2 and ALK5 participate in the process. In FOP patients, the mutant ALK2 can directly induce Endo-MT and convert endothelial cells into multipotent mesoderm cells. Then, under pathogenic inflammatory conditions in FOP patients, the mesoderm cells differentiate into cartilage, which can be further developed into heterotopic bone through endochondral ossification
Elevated BMP signaling due to a defect in the auto-regulatory feedback loop was already confirmed in the lymphocytes from FOP patients [129]. Lymphocytes are important responsive cells in inflammation and therefore it could also be interesting to determine the role of lymphocytes with dysregulated BMP activity in inflammation and whether they can promote bone formation. Moreover, the defective negative regulatory loop of BMP signaling is mainly observed in lymphocytes from FOP patients. It is not clear whether the negative regulation mechanism is also defective in osteoprogenitor cells in FOP patients. It could be interesting to investigate negative regulatory mechanisms in the osteoprogenitor cells from the FOP patients, and whether mutated ALK2 could lead to defective negative regulatory mechanisms in lymphocytes and other cell types.
Mutated ALK2 from FOP patients can directly convert mature endothelial cells into multi-potent mesenchymal cells, and sensitize mesenchymal cells to BMP-induced osteoblast differentiation [117, 127], thus making ALK2 a putative therapeutic target to prevent HO in FOP patients. LDN-193189, a specific BMP type I receptor kinase inhibitor, with the highest potency towards ALK2 kinase activity, was reported to decrease HO in CA-ALK2 transgenic mice [119]. Although in CA-ALK2 transgenic mice, LDN-193189 treatment showed no toxicity on mice growth, behavior, or bone density [119], further investigation is required before LDN-193189 can be applied to FOP patients. LDN-193189 is a potent BMP receptor inhibitor that significantly reduces ALK2 activity, but also other kinase activities at high dose [130, 131]. Recently, BMP signaling was found to be required for muscle regeneration, as discussed in more detail below, and therefore complete inhibition of ALK2-dependent BMP signaling should be avoided. The ideal kinase inhibitor for treatment of FOP without possible on-target side-effects in other tissues is one that specifically targets mutated ALK2, inhibiting the extra activity of ALK2 derived from the FOP allele, while also not affecting ALK1, ALK3, ALK6, and wild-type ALK2 kinase activity. Nowadays, genetic tools, including antisense therapy and RNA interference, have already been developed to modify specific gene or protein expression [132, 133]. Kaplan and colleagues have successfully employed allele-specific RNAi (ASP-RNAi) technique to reduce disease-causing ALK2 activity without inhibition of the normal ALK2 allele in FOP cells [134]. In the future, other genetic tools to specifically modulate mutant ALK2 expression in FOP patients might be good alternatives for treatment of disease.
Acquired form of heterotopic ossification
As mentioned before, FOP is a very rare genetic disease characterized by progressive heterotopic ossification (HO) induced by an activated ALK2 mutant. The most common form of HO is the acquired form, which is either induced by trauma or linked to damage in the nervous system (spinal cord or brain). Post-traumatic HO is caused by injuries at soft tissue at any site. The most common post-traumatic HO is observed after severe long bone fracture or in the hip after total hip arthroplasty [135]. More than 50 % of patients develop HO after total hip arthroplasty [136, 137]. The damaged muscle is another area at high risk of developing HO [101, 102, 138]. Recently, HO was unexpectedly discovered in end-stage valvular heart disease [139–142]. In spinal cord injured patients, the incidence of HO is between 20 and 25 %, while in closed brain injury HO occurs in 10–20 % of patients [99]. Patients with brain injury could develop peri-articular HO especially in the hip and elbow joint [143]. For patients with spinal cord injury, the HO is mostly observed in the hip region [144]. Until now, it is not well understood how the injuries in the nervous system lead to HO in the hip or elbow joint. In the following section, we will mainly discuss post-traumatic HO.
As mentioned, inflammatory conditions have been reported to be important for the progression of the disease in FOP [120, 145]. Like in FOP, inflammation is involved in the formation of ectopic bone in acquired HO. The pro-inflammatory cytokine, TNFα can stimulate the expression of BMP2, an important bone inducer in endothelial cells [146]. In addition, TNFα can augment the recruitment and differentiation of muscle-residing stroma cells (mrSCs) to enhance bone formation [125]. So the nonsteroidal anti-inflammatory drugs (NSAIDS) are important therapies for reducing the risk of HO. The anti-inflammatory agent Indomethacin is already commonly used as treatment of patients after acetabular fracture to prevent the possible occurrence of HO [147].
BMPs may mediate the induction of acquired HO. In the valve where HO was identified, BMP2 and BMP4 were found to be expressed by myofibroblasts and preosteoblasts in areas adjacent to B- and T-lymphocyte infiltrations [139]. Moreover, multiple studies showed that BMP2, BMP4, and BMP9-induced HO in skeletal muscle by intramuscular injections [101, 103, 138].
Tie2+ progenitor cells are discovered as major osteoprogenitor cells that respond to an inflammatory stimulation and further differentiate into heterotopic bones in BMP2 and BMP4-induced HO in the skeletal muscle [138]. Tie2 is a hallmark for endothelium cells, suggesting an important role of endothelial cells in contributing to HO [127, 138]. Interestingly, another report identified a group of non-endothelium Tie2+ cells residing in the interstitium of skeletal muscle and other tissues, displaying multi-potent ability to differentiate into mesoderm linage cells including osteoblasts and adipocytes [148]. Instead of other Tie2+ cells from the endothelium, it is this Tie2+ cell population of non-endothelium origin that are responsive to BMP2-induced HO in the skeletal muscle [148]. Medici et al. [127] discovered that multipotent cells derived from endothelial cells still expressed Tie2. Therefore, it would be interesting to investigate whether these Tie2+ multipotent mesenchymal cells might be converted from endothelial cells, or whether the niche holding these cells could facilitate transition of endothelial cells into multipotent mesenchymal cells in vivo. In addition to Tie2+ cells, mrSCs also contributed to the HO induced by BMP2 and BMP9 in the damaged muscle [101]. In addition, circulating osteogenic precursor cells are considered as a group of possible osteoprogenitor cells for HO. Circulating osteogenic cells were discovered to home to sites of vascular injury and were associated with HO formation in the heart valve [149].
Interestingly, in an in vitro study, BMP9 appeared to be more potent for inducing differentiation of mrSCs into osteoblasts than BMP2 [101]. In vivo, BMP2 can induce ectopic bone formation in the skeletal muscle with or without cardiotoxin (CTX)-induced muscle damage, whereas BMP9 only induced ectopic bone formation in CTX-induced damaged muscle [101]. Thus, it seems that BMP2 and BMP9 are not functionally equivalent to induce bone formation in the skeletal muscle [101, 150]. In vivo, BMP2 is secreted at the bone fracture area and is required for the initiation of fracture healing [12, 151]. BMP9 appears to function mainly in angiogenesis [152]. Therefore it would be interesting to investigate whether BMP2 and BMP9 play different roles in the initiation of bone formation in the muscle, or have different roles in the inflammatory reaction. A recent study suggested a group of non-endothelial Tie2+ cells as osteoprogenitor cells responsive to BMP2 to induce bone formation in skeletal muscle [148]. Further research might focus on the responsiveness of these cells to BMP2 and BMP9 stimulation in normal muscle or CTX injured muscle.
Different from FOP patients, in whom the ectopic bone is impossible to be removed by surgical operation, the only effective treatment of symptomatic established HO is surgical resection of ectopic bone tissue. To prevent the possible relapse of HO, it is prudent to avoid soft-tissue trauma in the operation room. Gentle handling of tissues includes complete wound lavage and removal of all bone debris and reaming was suggested to decrease the risk of HO after surgery [153, 154]. Radiation and usage of NSAIDS are used to further decrease the chance of getting HO [154]. Radiating pluripotential mesenchymal cells, the possible osteoprogenitor cells for HO, may effectively prevent the formation of HO [155]. Unfortunately, the NSAID therapy, while reasonably effective, has side-effects, most notably gastrointestinal ulceration, decreased platelet aggregation and renal toxicity [154, 156]. Since BMPs are well-established inducers of the HO, BMP inhibitors might turn out to be useful in the prevention of HO in the future.
BMP signaling in muscle regeneration and DMD disease
BMP signaling in muscle regeneration
In damaged regenerating muscle, BMPs, such as BMP2, BMP4, and BMP9, can potently induce bone formation [138]. Therefore one may think of the use of BMP inhibitors to repress HO in the skeletal muscle. However, recently studies [157, 158] on muscle regeneration after muscle damage suggested an essential role of BMP signaling in muscle regeneration [159].
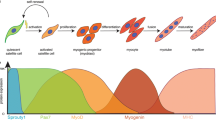
Muscle regeneration is comprised of three steps: upon muscle damage, quiescent muscle stem cells or satellite cells (characterized by Pax7+, MyoD−), which reside between the basement membrane and sarcolemma of individual muscle fibers, are activated; activated satellite cells (Pax7+, MyoD+) proliferate and subsequently either differentiate into myoblasts (Pax7−, MyoD+, Myog−), which initiate myogenic differentiation (Pax7−, MyoD+, Myog+) and fuse to repair damaged fibers or form nascent muscle fibers. Part of the activated satellite cells convert back to quiescent satellite cells (Pax7+, MyoD−) thereby self-renewing the satellite cell pool [160]. Together with satellite cells, mrSC, fibroblasts, and immune cells also participate in the regeneration process. Following acute tissue injury, inflammatory cells, fibroblasts, and mrSC migrate to the injured areas to restore tissue homeostasis. The inflammatory cells remove the damaged or dead fibers, which are then replaced by the activated satellite cells and the mrSCs [161]. In addition, fibroblasts and inflammatory cells stimulate satellite cell activation by secreting stimulatory growth factors/cytokines, such as HGF, FGF, and IGF [162]. The relevance of these different cell populations in muscle regeneration has recently been shown by several studies. Ablation of satellite cells, muscle fibroblasts, or macrophages all resulted in impaired muscle regeneration in mice [163–166].
Upon muscle damage, ALK3 expression is elevated in the activated satellite cells, and Smad1/5/8 phosphorylation is detected in the nucleus of the activated satellite cells, implying the activation of the BMP pathway [157]. In contrast, BMP antagonist Noggin is expressed in satellite cells committed to myogenic differentiation. The use of BMP inhibitors to repress BMP signaling in the regenerating muscle by means of dorsomorphin or a soluble ALK3 extracellular domain ligand trap, resulted in smaller regenerated myofibers and fibrosis; in vitro either inhibition of Noggin or exogenous BMP4 stimulated satellite cells division and repressed satellite cell differentiation, whereas addition of Noggin or soluble ALK3 had the opposite effect. Therefore, it was hypothesized that BMP signaling is required for the maintenance of the pool of activated satellite cells [157]. Clever’s study on Id1+/−; Id3−/− mutant mice suggested that Id1/3, which are direct target genes of the BMP-induced Smad pathway, mediate BMPs inhibitory effect on muscle differentiation. The Id1+/−; Id3−/− mutant mice displayed delayed and reduced skeletal muscle regeneration, characterized by a decreased number of activated satellite cells after injury [167]. BMP signaling repressed differentiation of satellite cells into myotubes. Therefore, the activity of BMP signaling should be switched off when enough satellite cells have been generated. The mechanisms controlling the timing that induce the satellite cells to switch from proliferation to differentiation are not well deciphered. But BMP antagonist Noggin and Chordin are elevated upon differentiation, which could repress the endogenous BMP signaling in activated satellite cells, and initiate the differentiation program [130, 157, 158].
Activation of BMP signaling is necessary for maintenance of activated satellite cells in the damaged muscle. However, BMP2, BMP4, and BMP9 have been demonstrated to induce ectopic bone formation in the damaged muscle. Therefore one might be prudent when using BMP inhibitors to repress HO occurrence in skeletal muscle as they may disturb the muscle regeneration. One option to overcome this complication could be to use the inhibitors that could specifically target the osteoprogenitor cells for ectopic bone cells. Another possibility is to inhibit the activity of receptors that are involved in the HO process, but not in the muscle regeneration process. Up to now, ALK3 is the only BMP type I receptor discovered to be involved in the muscle regeneration process [157] and it is not known if the other BMP receptors are involved in this process. It is not known whether ALK3 is also actively involved in HO occurrence in the skeletal muscle, like ALK2. Further research should therefore be focused on the specific and/or overlapping functions of different BMP type I receptors in muscle regeneration and HO.
BMP signaling in DMD disease
BMP signaling is important in balancing the satellite cell proliferation and differentiation program. Dysregulated BMP signaling might be linked with progression of muscle diseases. DMD disease is one of the diseases in which elevated BMP signaling in the satellite cells might exacerbate the disease [168].
DMD disease is a recessive X-linked form of muscular dystrophy that results in muscle degeneration. The disease is caused by mutations in the DMD gene [169], encoding the dystrophin protein that connects the cytoskeleton of muscle fibers to the underlying basal lamina. The absence of functional dystrophin in the myofiber leads to membrane damage, which results in increased calcium-influx and subsequent muscle fiber breakdown in DMD patients [170, 171]. Due to the constitutive muscle fiber damage, DMD patients suffer from chronic inflammation, in which infiltrated inflammatory cells and persistently activated fibroblasts stimulate fibrosis [161]. In addition to fibroblasts, muscle fibers in DMD patients are replaced by adipose tissue, although the underlying molecular mechanism is unknown [172]. Furthermore, the muscle’s regenerative capacity may be exhausted under chronic inflammatory conditions. Although the mechanism is not known, the continuous activation of satellite cells may lead to depletion of the satellite cell population. In addition, myoblasts isolated from DMD patients show proliferation and/or differentiation defects, which may further contribute to the decline in muscle regeneration [173, 174]. Moreover, myoblast to myofibroblast transdifferentiation has been reported to be partially causal for muscle fibrosis and may also further contribute to impaired muscle repair in DMD muscle [175].
To find the underlying molecular mechanism for the inefficient differentiation of DMD myoblasts, Sterrenburg and colleagues performed a microarray assay to compare expression profiles in DMD myoblasts and healthy myoblasts [168]. BMP4 expression was found to be significantly higher expressed in DMD cultures compared to myoblasts of healthy individuals [168]. BMP4 can maintain satellite cells in a proliferative state and inhibit myogenic differentiation [157]. In vitro, BMP4 was shown to inhibit both MyoD and myogenin, muscle-specific transcription factors that regulate differentiation of satellite cells into skeletal muscles. The elevated level of BMP4 in DMD myoblasts could partially explain the inefficiency of satellite cells to form new muscle fibers in DMD patients [176].
Muscle fibrosis is a prominent pathological symptom in DMD patients. The TGFβ signaling pathway has already been established as a key factor involved in fibrosis in DMD patients [177]. Multiple studies have demonstrated that BMP7 can reduce TGFβ-induced renal fibrosis and cardiac fibrosis [178–180]. Recently, BMP6 was discovered to attenuate TGFβ signaling in Dupuytren’s fibroblasts, and inhibit the fibrotic response [181]. However, there are no reports that BMP signaling affects TGFβ-induced muscle fibrosis. It is not known whether elevated BMP4 signaling contributes to the chronic inflammatory reaction in damaged skeletal muscle and subsequent fibrosis in DMD patients. A recent study suggested that BMP4 can induce EndoMT [127]. EndoMT transition has been demonstrated to contribute to cardiac fibrosis [180]. Therefore BMP4 might possibly play a role in muscle fibrosis through induction of EndoMT. It would be interesting to investigate the exact role for BMP signaling in muscle fibrosis.
As mentioned above, adipose tissue also replaces muscle fibers in dystrophic muscle of DMD patients [172]. A group of Tie2+ cells residing between skeletal muscle and endothelium was established as multipotent cells, and could differentiate into adipocytes in vitro [148]. BMP4 is able to induce mesenchymal stem cells into adipocyte-lineage cells in vitro [182]. Therefore BMP4 might also contribute to the accumulation of adipocytes in DMD patients.
To validate whether BMP signaling indeed contributes to the progression of DMD disease, we administrated dystrophin-deficient mdx mice with the BMP antagonist Noggin [130]. We observed enhanced MyoD and myogenin expression in the mice treated with Noggin, suggesting improved muscle regeneration, characterized by improved muscle histology [130], but could not detect a decrease in the inflammatory response. These results suggested that inhibition of BMP signaling might be beneficial for improving muscle regeneration in DMD patients. However, considering the importance of BMP signaling during muscle regeneration in healthy muscle, a potential beneficial effect of BMP antagonists in dystrophic muscle is likely to be dose-dependent, and complete repression of BMP signaling may even be detrimental. Therefore, the dose-dependent effects of such approaches should be assessed in more detail in animal models of DMD. In addition, one has to keep in mind that improving muscle regeneration will not result in improved muscle function in DMD muscle, since the primary genetic defect remains. Therefore, future DMD therapies should aim at both restoring dystrophin function and improve the muscle condition by counteracting fibrosis and improving muscle regeneration.
Conclusions
In this review, we have summarized the current progress of research on HO and DMD diseases that are related to elevated BMP signaling. The critical involvement of overactive BMP signaling in ectopic bone formation in HO patients is well established. Whether deregulated BMP signaling also contributes to DMD pathology by repressing muscle regeneration needs more investigation. Inhibition of excessive BMP signaling might be a promising therapeutic approach for treatment of these diseases, especially in FOP patient and DMD patients, in which the surgical treatment is impossible.
BMPs have been well established as crucial cytokines that control multiple biological phenomena, either during embryonic development or to control postnatal tissue homeostasis. Therefore anti-BMP treatment in the above-mentioned diseases should be considered with care to prevent the possible on-target side-effects and dose-dependent effects of such treatments should be determined in animal models. The BMP type I receptor kinase inhibitors dorsomorphin and LDN-193189 have shown to inhibit at high dose both BMP and TGFβ activity. The BMP antagonist Noggin is not so stable in vivo and was shown not to be able to inhibit BMP6 and BMP9 [47]. The soluble receptors and the neutralizing antibodies can only target the extracellular BMPs, therefore they would not be so beneficial for treatment of FOP patients who have mutated ALK2. Currently, ASP-RNAi technique has been successfully applied to specifically decrease mutant ALK2 allele activity and restore normal BMP activity in FOP cells [134]. In the future, other genetic tools, including antisense therapy and mi-RNA can be employed to decrease BMP activity.
Abbreviations
- HO:
-
Heterotopic ossification
- DMD:
-
Duchenne muscular dystrophy
- BMP:
-
Bone morphogenetic protein
- TGFβ:
-
Transforming growth factor-β
- Dpp:
-
Decapentaplegic
- R-Smad:
-
Receptor regulated Smad
- SBE:
-
Smad binding element
- TAK1:
-
TGFβ-activated kinase-1
- RGM:
-
Repulsive guidance molecule
- GPI:
-
Glycosylphosphatidylinositol
- I-Smad:
-
Inhibitory Smad
- DUB:
-
Deubiquitinating enzyme
- SARA:
-
Smad anchor for receptor activation
- SCP:
-
Small C-terminal domain phosphatase
- PPM1A:
-
Protein phosphatase magnesium-dependent 1A
- HDAC:
-
Histone deacetylase
- NSE:
-
Neuron-specific enolase
- CA:
-
Constitutively activated
- Endo-MT:
-
Endothelial-to-mesenchymal transition
- EMT:
-
Epithelial-to-mesenchymal transition
- ASP-RNAi:
-
Allele-specific RNAi
- mrSC:
-
Muscle-residing stroma cell
- NSAIDS:
-
Nonsteroidal anti-inflammatory drugs
- CTX:
-
Cardiotoxin
- TNFα:
-
Tumor necrosis factor-α
References
Urist MR (1965) Bone: formation by autoinduction. Science 150:893–899
Urist MR, Strates BS (1971) Bone morphogenetic protein. J Dent Res 50:1392–1406
Derynck R, Miyazono K (2008) TGF-β and the TGF-β family. In: Derynck R, Miyazono K (eds) The TGF-β family. Cold Spring Harbor Press, New York, pp 29–44
Wozney JM, Rosen V, Celeste AJ, Mitsock LM, Whitters MJ, Kriz RW, Hewick RM, Wang EA (1988) Novel regulators of bone formation: molecular clones and activities. Science 242:1528–1534
Miyazono K, Kamiya Y, Morikawa M (2010) Bone morphogenetic protein receptors and signal transduction. J Biochem 147:35–51
Dale L, Wardle FC (1999) A gradient of BMP activity specifies dorsal-ventral fates in early Xenopus embryos. Semin Cell Dev Biol 10:319–326
Peluso CE, Umulis D, Kim YJ, O’Connor MB, Serpe M (2011) Shaping BMP morphogen gradients through enzyme-substrate interactions. Dev Cell 21:375–383
Chen H, Shi S, Acosta L, Li W, Lu J, Bao S, Chen Z, Yang Z, Schneider MD, Chien KR, Conway SJ, Yoder MC, Haneline LS, Franco D, Shou W (2004) BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 131:2219–2231
Zhao GQ, Liaw L, Hogan BL (1998) Bone morphogenetic protein 8A plays a role in the maintenance of spermatogenesis and the integrity of the epididymis. Development 125:1103–1112
Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP (2009) Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet 41:478–481
Andriopoulos B Jr, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, Knutson MD, Pietrangelo A, Vukicevic S, Lin HY, Babitt JL (2009) BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet 41:482–487
Onishi T, Ishidou Y, Nagamine T, Yone K, Imamura T, Kato M, Sampath TK, ten Dijke P, Sakou T (1998) Distinct and overlapping patterns of localization of bone morphogenetic protein (BMP) family members and a BMP type II receptor during fracture healing in rats. Bone 22:605–612
Itoh S, ten Dijke P (2007) Negative regulation of TGF-β receptor/Smad signal transduction. Curr Opin Cell Biol 19:176–184
Kim M, Choe S (2011) BMPs and their clinical potentials. BMB Rep 44:619–634
Griffith DL, Keck PC, Sampath TK, Rueger DC, Carlson WD (1996) Three-dimensional structure of recombinant human osteogenic protein 1: structural paradigm for the transforming growth factor β superfamily. Proc Natl Acad Sci USA 93:878–883
Scheufler C, Sebald W, Hulsmeyer M (1999) Crystal structure of human bone morphogenetic protein-2 at 2.7 A resolution. J Mol Biol 287:103–115
Sampath TK, Rashka KE, Doctor JS, Tucker RF, Hoffmann FM (1993) Drosophila transforming growth factor β superfamily proteins induce endochondral bone formation in mammals. Proc Natl Acad Sci USA 90:6004–6008
Padgett RW, Wozney JM, Gelbart WM (1993) Human BMP sequences can confer normal dorsal-ventral patterning in the Drosophila embryo. Proc Natl Acad Sci USA 90:2905–2909
Israel DI, Nove J, Kerns KM, Kaufman RJ, Rosen V, Cox KA, Wozney JM (1996) Heterodimeric bone morphogenetic proteins show enhanced activity in vitro and in vivo. Growth Factors 13:291–300
Guo J, Wu G (2012) The signaling and functions of heterodimeric bone morphogenetic proteins. Cytokine Growth Factor Rev 23:61–67
Wrana JL, Attisano L, Wieser R, Ventura F, Massagué J (1994) Mechanism of activation of the TGF-β receptor. Nature 370:341–347
Wang T, Li BY, Danielson PD, Shah PC, Rockwell S, Lechleider RJ, Martin J, Manganaro T, Donahoe PK (1996) The immunophilin FKBP12 functions as a common inhibitor of the TGFβ family type I receptors. Cell 86:435–444
Chen YG, Liu F, Massague J (1997) Mechanism of TGFβ receptor inhibition by FKBP12. EMBO J 16:3866–3876
Huse M, Chen YG, Massagué J, Kuriyan J (1999) Crystal structure of the cytoplasmic domain of the type I TGFβ receptor in complex with FKBP12. Cell 96:425–436
Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS (2006) A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 38:525–527
Rosenzweig BL, Imamura T, Okadome T, Cox GN, Yamashita H, ten Dijke P, Heldin CH, Miyazono K (1995) Cloning and characterization of a human type II receptor for bone morphogenetic proteins. Proc Natl Acad Sci USA 92:7632–7636
Miyazono K, Maeda S, Imamura T (2005) BMP receptor signaling: transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev 16:251–263
Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA (2000) Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 67:737–744
Foletta VC, Lim MA, Soosairajah J, Kelly AP, Stanley EG, Shannon M, He W, Das S, Massague J, Bernard O (2003) Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J Cell Biol 162:1089–1098
Lee-Hoeflich ST, Causing CG, Podkowa M, Zhao X, Wrana JL, Attisano L (2004) Activation of LIMK1 by binding to the BMP receptor, BMPRII, regulates BMP-dependent dendritogenesis. EMBO J 23:4792–4801
Davis BN, Hilyard AC, Lagna G, Hata A (2008) SMAD proteins control DROSHA-mediated microRNA maturation. Nature 454:56–61
Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A (2010) Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol Cell 39:373–384
Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K (1995) Identification of a member of the MAPKKK family as a potential mediator of TGF-β signal transduction. Science 270:2008–2011
Moriguchi T, Kuroyanagi N, Yamaguchi K, Gotoh Y, Irie K, Kano T, Shirakabe K, Muro Y, Shibuya H, Matsumoto K, Nishida E, Hagiwara M (1996) A novel kinase cascade mediated by mitogen-activated protein kinase kinase 6 and MKK3. J Biol Chem 271:13675–13679
Shirakabe K, Yamaguchi K, Shibuya H, Irie K, Matsuda S, Moriguchi T, Gotoh Y, Matsumoto K, Nishida E (1997) TAK1 mediates the ceramide signaling to stress-activated protein kinase/c-Jun N-terminal kinase. J Biol Chem 272:8141–8144
Reimann T, Hempel U, Krautwald S, Axmann A, Scheibe R, Seidel D, Wenzel KW (1997) Transforming growth factor-β1 induces activation of Ras, Raf-1, MEK and MAPK in rat hepatic stellate cells. FEBS Lett 403:57–60
Gallea S, Lallemand F, Atfi A, Rawadi G, Ramez V, Spinella-Jaegle S, Kawai S, Faucheu C, Huet L, Baron R, Roman-Roman S (2001) Activation of mitogen-activated protein kinase cascades is involved in regulation of bone morphogenetic protein-2-induced osteoblast differentiation in pluripotent C2C12 cells. Bone 28:491–498
Jun JH, Yoon WJ, Seo SB, Woo KM, Kim GS, Ryoo HM, Baek JH (2010) BMP2-activated Erk/MAP kinase stabilizes Runx2 by increasing p300 levels and histone acetyltransferase activity. J Biol Chem 285:36410–36419
Chen G, Deng C, Li YP (2012) TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci 8:272–288
Shibuya H, Iwata H, Masuyama N, Gotoh Y, Yamaguchi K, Irie K, Matsumoto K, Nishida E, Ueno N (1998) Role of TAK1 and TAB 1 in BMP signaling in early Xenopus development. EMBO J 17:1019–1028
Hoffmann A, Preobrazhenska O, Wodarczyk C, Medler Y, Winkel A, Shahab S, Huylebroeck D, Gross G, Verschueren K (2005) Transforming growth factor-β-activated kinase-1 (TAK1), a MAP3K, interacts with Smad proteins and interferes with osteogenesis in murine mesenchymal progenitors. J Biol Chem 280:27271–27283
Shim JH, Greenblatt MB, Xie M, Schneider MD, Zou W, Zhai B, Gygi S, Glimcher LH (2009) TAK1 is an essential regulator of BMP signalling in cartilage. EMBO J 28:2028–2041
Greenblatt MB, Shim JH, Glimcher LH (2010) TAK1 mediates BMP signaling in cartilage. Ann N Y Acad Sci 1192:385–390
Gunnell LM, Jonason JH, Loiselle AE, Kohn A, Schwarz EM, Hilton MJ, O’Keefe RJ (2010) TAK1 regulates cartilage and joint development via the MAPK and BMP signaling pathways. J Bone Miner Res 25:1784–1797
Vitt UA, Hsu SY, Hsueh AJ (2001) Evolution and classification of cystine knot-containing hormones and related extracellular signaling molecules. Mol Endocrinol 15:681–694
Yanagita M (2005) BMP antagonists: their roles in development and involvement in pathophysiology. Cytokine Growth Factor Rev 16:309–317
Song K, Krause C, Shi S, Patterson M, Suto R, Grgurevic L, Vukicevic S, van Dinther M, Falb D, ten Dijke P, Alaoui-Ismaili MH (2010) Identification of a key residue mediating bone morphogenetic protein (BMP)-6 resistance to noggin inhibition allows for engineered BMPs with superior agonist activity. J Biol Chem 285:12169–12180
Lowery JW, de Caestecker MP (2010) BMP signaling in vascular development and disease. Cytokine Growth Factor Rev 21:287–298
Kusu N, Laurikkala J, Imanishi M, Usui H, Konishi M, Miyake A, Thesleff I, Itoh N (2003) Sclerostin is a novel secreted osteoclast-derived bone morphogenetic protein antagonist with unique ligand specificity. J Biol Chem 278:24113–24117
Camus LM, Lambert LA (2007) Molecular evolution of hemojuvelin and the repulsive guidance molecule family. J Mol Evol 65:68–81
Samad TA, Rebbapragada A, Bell E, Zhang Y, Sidis Y, Jeong SJ, Campagna JA, Perusini S, Fabrizio DA, Schneyer AL, Lin HY, Brivanlou AH, Attisano L, Woolf CJ (2005) DRAGON, a bone morphogenetic protein co-receptor. J Biol Chem 280:14122–14129
Babitt JL, Zhang Y, Samad TA, Xia Y, Tang J, Campagna JA, Schneyer AL, Woolf CJ, Lin HY (2005) Repulsive guidance molecule (RGMa), a DRAGON homologue, is a bone morphogenetic protein co-receptor. J Biol Chem 280:29820–29827
Xia Y, Sidis Y, Mukherjee A, Samad TA, Brenner G, Woolf CJ, Lin HY, Schneyer A (2005) Localization and action of Dragon (repulsive guidance molecule b), a novel bone morphogenetic protein coreceptor, throughout the reproductive axis. Endocrinology 146:3614–3621
Xia Y, Yu PB, Sidis Y, Beppu H, Bloch KD, Schneyer AL, Lin HY (2007) Repulsive guidance molecule RGMa alters utilization of bone morphogenetic protein (BMP) type II receptors by BMP2 and BMP4. J Biol Chem 282:18129–18140
Nohe A, Hassel S, Ehrlich M, Neubauer F, Sebald W, Henis YI, Knaus P (2002) The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J Biol Chem 277:5330–5338
Zhou Z, Xie J, Lee D, Liu Y, Jung J, Zhou L, Xiong S, Mei L, Xiong WC (2010) Neogenin regulation of BMP-induced canonical Smad signaling and endochondral bone formation. Dev Cell 19:90–102
Kirkbride KC, Townsend TA, Bruinsma MW, Barnett JV, Blobe GC (2008) Bone morphogenetic proteins signal through the transforming growth factor-β type III receptor. J Biol Chem 283:7628–7637
Lee NY, Kirkbride KC, Sheu RD, Blobe GC (2009) The transforming growth factor-β type III receptor mediates distinct subcellular trafficking and downstream signaling of activin-like kinase (ALK)3 and ALK6 receptors. Mol Biol Cell 20:4362–4370
Scharpfenecker M, van Dinther M, Liu Z, van Bezooijen RL, Zhao Q, Pukac L, Lowik CW, ten Dijke P (2007) BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J Cell Sci 120:964–972
Fafeur V, Terman BI, Blum J, Bohlen P (1990) Basic FGF treatment of endothelial cells down-regulates the 85-kDa TGFβ receptor subtype and decreases the growth inhibitory response to TGF-β1. Growth Factors 3:237–245
Cheifetz S, Bellon T, Cales C, Vera S, Bernabeu C, Massagué J, Letarte M (1992) Endoglin is a component of the transforming growth factor-β receptor system in human endothelial cells. J Biol Chem 267:19027–19030
David L, Mallet C, Mazerbourg S, Feige JJ, Bailly S (2007) Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 109:1953–1961
Andersson S, Davis DL, Dahlback H, Jornvall H, Russell DW (1989) Cloning, structure, and expression of the mitochondrial cytochrome P-450 sterol 26-hydroxylase, a bile acid biosynthetic enzyme. J Biol Chem 264:8222–8229
Lopez-Casillas F, Cheifetz S, Doody J, Andres JL, Lane WS, Massagué J (1991) Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-β receptor system. Cell 67:785–795
Kuninger D, Kuns-Hashimoto R, Kuzmickas R, Rotwein P (2006) Complex biosynthesis of the muscle-enriched iron regulator RGMc. J Cell Sci 119:3273–3283
Castonguay R, Werner ED, Matthews RG, Presman E, Mulivor AW, Solban N, Sako D, Pearsall RS, Underwood KW, Seehra J, Kumar R, Grinberg AV (2011) Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J Biol Chem 286:30034–30046
Balemans W, Van Hul W (2002) Extracellular regulation of BMP signaling in vertebrates: a cocktail of modulators. Dev Biol 250:231–250
Onichtchouk D, Chen YG, Dosch R, Gawantka V, Delius H, Massagué J, Niehrs C (1999) Silencing of TGF-β signalling by the pseudoreceptor BAMBI. Nature 401:480–485
Hata A, Lagna G, Massagué J, Hemmati-Brivanlou A (1998) Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev 12:186–197
Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P (1997) Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature 389:631–635
Casellas R, Brivanlou AH (1998) Xenopus Smad7 inhibits both the activin and BMP pathways and acts as a neural inducer. Dev Biol 198:1–12
Lin X, Liang YY, Sun B, Liang M, Shi Y, Brunicardi FC, Feng XH (2003) Smad6 recruits transcription corepressor CtBP to repress bone morphogenetic protein-induced transcription. Mol Cell Biol 23:9081–9093
Valdimarsdottir G, Goumans MJ, Itoh F, Itoh S, Heldin CH, ten Dijke P (2006) Smad7 and protein phosphatase 1α are critical determinants in the duration of TGF-β/ALK1 signaling in endothelial cells. BMC Cell Biol 7:16
Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL (2000) Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGFβ receptor for degradation. Mol Cell 6:1365–1375
Murakami G, Watabe T, Takaoka K, Miyazono K, Imamura T (2003) Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol Biol Cell 14:2809–2817
Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH (1999) A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 400:687–693
Zhang Y, Chang C, Gehling DJ, Hemmati-Brivanlou A, Derynck R (2001) Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc Natl Acad Sci USA 98:974–979
Wicks SJ, Haros K, Maillard M, Song L, Cohen RE, Dijke PT, Chantry A (2005) The deubiquitinating enzyme UCH37 interacts with Smads and regulates TGF-β signalling. Oncogene 24:8080–8084
Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL (1998) SARA, a FYVE domain protein that recruits Smad2 to the TGFβ receptor. Cell 95:779–791
Shi W, Chang C, Nie S, Xie S, Wan M, Cao X (2007) Endofin acts as a Smad anchor for receptor activation in BMP signaling. J Cell Sci 120:1216–1224
Knockaert M, Sapkota G, Alarcon C, Massague J, Brivanlou AH (2006) Unique players in the BMP pathway: small C-terminal domain phosphatases dephosphorylate Smad1 to attenuate BMP signaling. Proc Natl Acad Sci USA 103:11940–11945
Duan X, Liang YY, Feng XH, Lin X (2006) Protein serine/threonine phosphatase PPM1A dephosphorylates Smad1 in the bone morphogenetic protein signaling pathway. J Biol Chem 281:36526–36532
Kokabu S, Ohte S, Sasanuma H, Shin M, Yoneyama K, Murata E, Kanomata K, Nojima J, Ono Y, Yoda T, Fukuda T, Katagiri T (2011) Suppression of BMP-Smad signaling axis-induced osteoblastic differentiation by small C-terminal domain phosphatase 1, a Smad phosphatase. Mol Endocrinol 25:474–481
Bengtsson L, Schwappacher R, Roth M, Boergermann JH, Hassel S, Knaus P (2009) PP2A regulates BMP signalling by interacting with BMP receptor complexes and by dephosphorylating both the C-terminus and the linker region of Smad1. J Cell Sci 122:1248–1257
Takeda M, Mizuide M, Oka M, Watabe T, Inoue H, Suzuki H, Fujita T, Imamura T, Miyazono K, Miyazawa K (2004) Interaction with Smad4 is indispensable for suppression of BMP signaling by c-Ski. Mol Biol Cell 15:963–972
Wang W, Mariani FV, Harland RM, Luo K (2000) Ski represses bone morphogenic protein signaling in Xenopus and mammalian cells. Proc Natl Acad Sci USA 97:14394–14399
Luo K (2003) Negative regulation of BMP signaling by the ski oncoprotein. J Bone Joint Surg Am 85-A(Suppl 3):39–43
Hayashi M, Nimura K, Kashiwagi K, Harada T, Takaoka K, Kato H, Tamai K, Kaneda Y (2007) Comparative roles of Twist-1 and Id1 in transcriptional regulation by BMP signaling. J Cell Sci 120:1350–1357
Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL (2003) Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat Cell Biol 5:410–421
Ceresa BP, Schmid SL (2000) Regulation of signal transduction by endocytosis. Curr Opin Cell Biol 12:204–210
Hartung A, Bitton-Worms K, Rechtman MM, Wenzel V, Boergermann JH, Hassel S, Henis YI, Knaus P (2006) Different routes of bone morphogenic protein (BMP) receptor endocytosis influence BMP signaling. Mol Cell Biol 26:7791–7805
Takase M, Imamura T, Sampath TK, Takeda K, Ichijo H, Miyazono K, Kawabata M (1998) Induction of Smad6 mRNA by bone morphogenetic proteins. Biochem Biophys Res Commun 244:26–29
Ishida W, Hamamoto T, Kusanagi K, Yagi K, Kawabata M, Takehara K, Sampath TK, Kato M, Miyazono K (2000) Smad6 is a Smad1/5-induced Smad inhibitor. Characterization of bone morphogenetic protein-responsive element in the mouse Smad6 promoter. J Biol Chem 275:6075–6079
Benchabane H, Wrana JL (2003) GATA- and Smad1-dependent enhancers in the Smad7 gene differentially interpret bone morphogenetic protein concentrations. Mol Cell Biol 23:6646–6661
Gazzerro E, Gangji V, Canalis E (1998) Bone morphogenetic proteins induce the expression of noggin, which limits their activity in cultured rat osteoblasts. J Clin Invest 102:2106–2114
Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425:577–584
Katagiri T, Yamaguchi A, Komaki M, Abe E, Takahashi N, Ikeda T, Rosen V, Wozney JM, Fujisawa-Sehara A, Suda T (1994) Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J Cell Biol 127:1755–1766
Vanden Bossche L, Vanderstraeten G (2005) Heterotopic ossification: a review. J Rehabil Med 37:129–136
Shehab D, Elgazzar AH, Collier BD (2002) Heterotopic ossification. J Nucl Med 43:346–353
Chalmers J, Gray DH, Rush J (1975) Observations on the induction of bone in soft tissues. J Bone Jt Surg Br 57:36–45
Leblanc E, Trensz F, Haroun S, Drouin G, Bergeron E, Penton CM, Montanaro F, Roux S, Faucheux N, Grenier G (2011) BMP-9-induced muscle heterotopic ossification requires changes to the skeletal muscle microenvironment. J Bone Miner Res 26:1166–1177
Kan L, Hu M, Gomes WA, Kessler JA (2004) Transgenic mice overexpressing BMP4 develop a fibrodysplasia ossificans progressiva (FOP)-like phenotype. Am J Pathol 165:1107–1115
Glaser DL, Economides AN, Wang L, Liu X, Kimble RD, Fandl JP, Wilson JM, Stahl N, Kaplan FS, Shore EM (2003) In vivo somatic cell gene transfer of an engineered Noggin mutein prevents BMP4-induced heterotopic ossification. J Bone Jt Surg Am 85-A:2332–2342
Cohen RB, Hahn GV, Tabas JA, Peeper J, Levitz CL, Sando A, Sando N, Zasloff M, Kaplan FS (1993) The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. A study of forty-four patients. J Bone Jt Surg Am 75:215–219
Shore EM, Kaplan FS (2008) Insights from a rare genetic disorder of extra-skeletal bone formation, fibrodysplasia ossificans progressiva (FOP). Bone 43:427–433
Kaplan FS, Le Merrer M, Glaser DL, Pignolo RJ, Goldsby RE, Kitterman JA, Groppe J, Shore EM (2008) Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol 22:191–205
Connor JM, Evans DA (1982) Fibrodysplasia ossificans progressiva. The clinical features and natural history of 34 patients. J Bone Jt Surg Br 64:76–83
Shafritz AB, Shore EM, Gannon FH, Zasloff MA, Taub R, Muenke M, Kaplan FS (1996) Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. N Engl J Med 335:555–561
Ahn J, Serrano de la Pena L, Shore EM, Kaplan FS (2003) Paresis of a bone morphogenetic protein-antagonist response in a genetic disorder of heterotopic skeletogenesis. J Bone Jt Surg Am 85-A:667–674
Blessing M, Nanney LB, King LE, Jones CM, Hogan BL (1993) Transgenic mice as a model to study the role of TGF-β-related molecules in hair follicles. Genes Dev 7:204–215
Bellusci S, Henderson R, Winnier G, Oikawa T, Hogan BL (1996) Evidence from normal expression and targeted misexpression that bone morphogenetic protein (Bmp-4) plays a role in mouse embryonic lung morphogenesis. Development 122:1693–1702
Tsumaki N, Nakase T, Miyaji T, Kakiuchi M, Kimura T, Ochi T, Yoshikawa H (2002) Bone morphogenetic protein signals are required for cartilage formation and differently regulate joint development during skeletogenesis. J Bone Miner Res 17:898–906
Zhao X, Zhang Z, Song Y, Zhang X, Zhang Y, Hu Y, Fromm SH, Chen Y (2000) Transgenically ectopic expression of Bmp4 to the Msx1 mutant dental mesenchyme restores downstream gene expression but represses Shh and Bmp2 in the enamel knot of wild type tooth germ. Mech Dev 99:29–38
de la Pena LS, Billings PC, Fiori JL, Ahn J, Kaplan FS, Shore EM (2005) Fibrodysplasia ossificans progressiva (FOP), a disorder of ectopic osteogenesis, misregulates cell surface expression and trafficking of BMPRIA. J Bone Miner Res 20:1168–1176
Song GA, Kim HJ, Woo KM, Baek JH, Kim GS, Choi JY, Ryoo HM (2010) Molecular consequences of the ACVR1(R206H) mutation of fibrodysplasia ossificans progressiva. J Biol Chem 285:22542–22553
Groppe JC, Wu J, Shore EM, Kaplan FS (2011) In vitro analyses of the dysregulated R206H ALK2 kinase-FKBP12 interaction associated with heterotopic ossification in FOP. Cells Tissues Organs 194:291–295
van Dinther M, Visser N, de Gorter DJ, Doorn J, Goumans MJ, de Boer J, ten Dijke P (2010) ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. J Bone Miner Res 25:1208–1215
Chakkalakal SA, Zhang D, Culbert AL, Convente MR, Caron RJ, Wright AC, Maidment AD, Kaplan FS, Shore EM (2012) An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J Bone Miner Res (in press)
Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, Hong DW, McManus PM, Katagiri T, Sachidanandan C, Kamiya N, Fukuda T, Mishina Y, Peterson RT, Bloch KD (2008) BMP type I receptor inhibition reduces heterotopic ossification. Nat Med 14:1363–1369
Pignolo RJ, Shore EM, Kaplan FS (2011) Fibrodysplasia ossificans progressiva: clinical and genetic aspects. Orphanet J Rare Dis 6:80
Kaplan FS, Lounev VY, Wang H, Pignolo RJ, Shore EM (2011) Fibrodysplasia ossificans progressiva: a blueprint for metamorphosis. Ann N Y Acad Sci 1237:5–10
Kaplan FS, Glaser DL, Shore EM, Pignolo RJ, Xu M, Zhang Y, Senitzer D, Forman SJ, Emerson SG (2007) Hematopoietic stem-cell contribution to ectopic skeletogenesis. J Bone Jt Surg Am 89:347–357
Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FM (2010) Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol 12:153–163
Grenier G, Scime A, Le Grand F, Asakura A, Perez-Iratxeta C, Andrade-Navarro MA, Labosky PA, Rudnicki MA (2007) Resident endothelial precursors in muscle, adipose, and dermis contribute to postnatal vasculogenesis. Stem Cells 25:3101–3110
Glass GE, Chan JK, Freidin A, Feldmann M, Horwood NJ, Nanchahal J (2011) TNF-α promotes fracture repair by augmenting the recruitment and differentiation of muscle-derived stromal cells. Proc Natl Acad Sci USA 108:1585–1590
Asakura A, Seale P, Girgis-Gabardo A, Rudnicki MA (2002) Myogenic specification of side population cells in skeletal muscle. J Cell Biol 159:123–134
Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR (2010) Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 16:1400–1406
Suda RK, Billings PC, Egan KP, Kim JH, McCarrick-Walmsley R, Glaser DL, Porter DL, Shore EM, Pignolo RJ (2009) Circulating osteogenic precursor cells in heterotopic bone formation. Stem Cells 27:2209–2219
Kaplan FS, Fiori J, De La Peña LS, Ahn J, Billings PC, Shore EM (2006) Dysregulation of the BMP-4 signaling pathway in fibrodysplasia ossificans progressiva. Ann N Y Acad Sci 1068:54–65
Shi S, Hoogaars WM, de Gorter DJ, van Heiningen SH, Lin HY, Hong CC, Kemaladewi DU, Aartsma-Rus A, ten Dijke P, t’ Hoen PA (2011) BMP antagonists enhance myogenic differentiation and ameliorate the dystrophic phenotype in a DMD mouse model. Neurobiol Dis 41:353–360
Vogt J, Traynor R, Sapkota GP (2011) The specificities of small molecule inhibitors of the TGFβ and BMP pathways. Cell Signal 23:1831–1842
Aartsma-Rus A, van Ommen GJ (2007) Antisense-mediated exon skipping: a versatile tool with therapeutic and research applications. RNA 13:1609–1624
Rodriguez-Lebron E, Paulson HL (2006) Allele-specific RNA interference for neurological disease. Gene Ther 13:576–581
Kaplan J, Kaplan FS, Shore EM (2012) Restoration of normal BMP signaling levels and osteogenic differentiation in FOP mesenchymal progenitor cells by mutant allele-specific targeting. Gene Ther (in press)
Thomas BJ (1992) Heterotopic bone formation after total hip arthroplasty. Orthop Clin North Am 23:347–358
Riegler HF, Harris CM (1976) Heterotopic bone formation after total hip arthroplasty. Clin Orthop Relat Res 117:209–216
Back DL, Smith JD, Dalziel RE, Young DA, Shimmin A (2007) Incidence of heterotopic ossification after hip resurfacing. ANZ J Surg 77:642–647
Lounev VY, Ramachandran R, Wosczyna MN, Yamamoto M, Maidment AD, Shore EM, Glaser DL, Goldhamer DJ, Kaplan FS (2009) Identification of progenitor cells that contribute to heterotopic skeletogenesis. J Bone Jt Surg Am 91:652–663
Mohler ER 3rd, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS (2001) Bone formation and inflammation in cardiac valves. Circulation 103:1522–1528
Feldman T, Glagov S, Carroll JD (1993) Restenosis following successful balloon valvuloplasty: bone formation in aortic valve leaflets. Cathet Cardiovasc Diagn 29:1–7
Fernandez Gonzalez AL, Montero JA, Martinez Monzonis A, Gil O, Alemany P (1997) Osseous metaplasia and hematopoietic bone marrow in a calcified aortic valve. Tex Heart Inst J 24:232
Arumugam SB, Sankar NM, Cherian KM (1995) Osseus metaplasia with functioning marrow in a calcified aortic valve. J Card Surg 10:610–611
Moore TJ (1993) Functional outcome following surgical excision of heterotopic ossification in patients with traumatic brain injury. J Orthop Trauma 7:11–14
Garland DE (1988) Clinical observations on fractures and heterotopic ossification in the spinal cord and traumatic brain injured populations. Clin Orthop Relat Res 233:86–101
Kaplan FS, Groppe J, Pignolo RJ, Shore EM (2007) Morphogen receptor genes and metamorphogenes: skeleton keys to metamorphosis. Ann N Y Acad Sci 1116:113–133
Csiszar A, Smith KE, Koller A, Kaley G, Edwards JG, Ungvari Z (2005) Regulation of bone morphogenetic protein-2 expression in endothelial cells: role of nuclear factor-kappaB activation by tumor necrosis factor-alpha, H2O2, and high intravascular pressure. Circulation 111:2364–2372
McLaren AC (1990) Prophylaxis with indomethacin for heterotopic bone. After open reduction of fractures of the acetabulum. J Bone Jt Surg Am 72:245–247
Wosczyna MN, Biswas AA, Cogswell CA, Wosczyna MN, Biswas AA, Cogswell CA, Goldhamer DJ (2012) Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J Bone Miner Res 72:1004–1017
Egan KP, Kim JH, Mohler ER 3rd, Pignolo RJ (2011) Role for circulating osteogenic precursor cells in aortic valvular disease. Arterioscler Thromb Vasc Biol 31:2965–2971
Shore EM (2011) Osteoinductive signals and heterotopic ossification. J Bone Miner Res 26:1163–1165
Tsuji K, Bandyopadhyay A, Harfe BD, Cox K, Kakar S, Gerstenfeld L, Einhorn T, Tabin CJ, Rosen V (2006) BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat Genet 38:1424–1429
David L, Mallet C, Keramidas M, Lamande N, Gasc JM, Dupuis-Girod S, Plauchu H, Feige JJ, Bailly S (2008) Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ Res 102:914–922
Hierton C, Blomgren G, Lindgren U (1983) Factors associated with heterotopic bone formation in cemented total hip prostheses. Acta Orthop Scand 54:698–702
Chao ST, Joyce MJ, Suh JH (2007) Treatment of heterotopic ossification. Orthopedics 30:457–464 (quiz 465-6)
Coventry MB, Scanlon PW (1981) The use of radiation to discourage ectopic bone. A nine-year study in surgery about the hip. J Bone Jt Surg Am 63:201–208
Sell S, Willms R, Jany R, Esenwein S, Gaissmaier C, Martini F, Bruhn G, Burkhardsmaier F, Bamberg M, Kusswetter W (1998) The suppression of heterotopic ossifications: radiation versus NSAID therapy—a prospective study. J Arthroplasty 13:854–859
Ono Y, Calhabeu F, Morgan JE, Katagiri T, Amthor H, Zammit PS (2011) BMP signalling permits population expansion by preventing premature myogenic differentiation in muscle satellite cells. Cell Death Differ 18:222–234
Friedrichs M, Wirsdoerfer F, Flohe SB, Schneider S, Wuelling M, Vortkamp A (2011) BMP signaling balances proliferation and differentiation of muscle satellite cell descendants. BMC Cell Biol 12:26
Ruschke K, Hiepen C, Becker J, Knaus P (2012) BMPs are mediators in tissue crosstalk of the regenerating musculoskeletal system. Cell Tissue Res 347:521–544
Charge SB, Rudnicki MA (2004) Cellular and molecular regulation of muscle regeneration. Physiol Rev 84:209–238
Mann CJ, Perdiguero E, Kharraz Y, Aguilar S, Pessina P, Serrano AL, Munoz-Canoves P (2011) Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle 1:21
Karalaki M, Fili S, Philippou A, Koutsilieris M (2009) Muscle regeneration: cellular and molecular events. In Vivo (Athens, Greece) 23:779–796
Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G (2011) Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 138:3625–3637
Lepper C, Partridge TA, Fan CM (2011) An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 138:3639–3646
Sambasivan R, Yao R, Kissenpfennig A, Van Wittenberghe L, Paldi A, Gayraud-Morel B, Guenou H, Malissen B, Tajbakhsh S, Galy A (2011) Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 138:3647–3656
Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B (2007) Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med 204:1057–1069
Clever JL, Sakai Y, Wang RA, Schneider DB (2010) Inefficient skeletal muscle repair in inhibitor of differentiation knockout mice suggests a crucial role for BMP signaling during adult muscle regeneration. Am J Physiol Cell Physiol 298:C1087–C1099
Sterrenburg E, van der Wees CG, White SJ, Turk R, de Menezes RX, van Ommen GJ, den Dunnen JT, t’ Hoen PA (2006) Gene expression profiling highlights defective myogenesis in DMD patients and a possible role for bone morphogenetic protein 4. Neurobiol Dis 23:228–236
Hoffman EP, Brown RH Jr, Kunkel LM (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51:919–928
Ohlendieck K, Matsumura K, Ionasescu VV, Towbin JA, Bosch EP, Weinstein SL, Sernett SW, Campbell KP (1993) Duchenne muscular dystrophy: deficiency of dystrophin-associated proteins in the sarcolemma. Neurology 43:795–800
Blake DJ, Weir A, Newey SE, Davies KE (2002) Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev 82:291–329
Haslett JN, Sanoudou D, Kho AT, Bennett RR, Greenberg SA, Kohane IS, Beggs AH, Kunkel LM (2002) Gene expression comparison of biopsies from Duchenne muscular dystrophy (DMD) and normal skeletal muscle. Proc Natl Acad Sci USA 99:15000–15005
Blau HM, Webster C, Pavlath GK (1983) Defective myoblasts identified in Duchenne muscular dystrophy. Proc Natl Acad Sci USA 80:4856–4860
Blau HM, Webster C, Pavlath GK, Chiu CP (1985) Evidence for defective myoblasts in Duchenne muscular dystrophy. Adv Exp Med Biol 182:85–110
Li Y, Huard J (2002) Differentiation of muscle-derived cells into myofibroblasts in injured skeletal muscle. Am J Pathol 161:895–907
Kottlors M, Kirschner J (2010) Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res 340:541–548
Bernasconi P, Torchiana E, Confalonieri P, Brugnoni R, Barresi R, Mora M, Cornelio F, Morandi L, Mantegazza R (1995) Expression of transforming growth factor-β1 in dystrophic patient muscles correlates with fibrosis. Pathogenetic role of a fibrogenic cytokine. J Clin Invest 96:1137–1144
Morrissey J, Hruska K, Guo G, Wang S, Chen Q, Klahr S (2002) Bone morphogenetic protein-7 improves renal fibrosis and accelerates the return of renal function. J Am Soc Nephrol JASN 13(Suppl 1):S14–S21
Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R (2003) BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 9:964–968
Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R (2007) Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 13:952–961
Krause C, Kloen P, ten Dijke P (2011) Elevated transforming growth factor β and mitogen-activated protein kinase pathways mediate fibrotic traits of Dupuytren’s disease fibroblasts. Fibrogenesis Tissue Repair 4:14
Tang QQ, Otto TC, Lane MD (2004) Commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc Natl Acad Sci USA 101:9607–9611
Zhang H, Bradley A (1996) Mice deficient for BMP2 are nonviable and have defects in amnion/chorion and cardiac development. Development 122:2977–2986
Schlange T, Andree B, Arnold HH, Brand T (2000) BMP2 is required for early heart development during a distinct time period. Mech Dev 91:259–270
Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, Tabin CJ (2006) Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet 2:e216
King JA, Marker PC, Seung KJ, Kingsley DM (1994) BMP5 and the molecular, skeletal, and soft-tissue alterations in short ear mice. Dev Biol 166:112–122
Jena N, Martin-Seisdedos C, McCue P, Croce CM (1997) BMP7 null mutation in mice: developmental defects in skeleton, kidney, and eye. Exp Cell Res 230:28–37
Zhao GQ, Hogan BL (1996) Evidence that mouse Bmp8a (Op2) and Bmp8b are duplicated genes that play a role in spermatogenesis and placental development. Mech Dev 57:159–168
Miller AF, Harvey SA, Thies RS, Olson MS (2000) Bone morphogenetic protein-9. An autocrine/paracrine cytokine in the liver. J Biol Chem 275:17937–17945
Acknowledgments
Research in our laboratories on BMP signaling is supported by IOP Genomics Grant IGE07001, The Netherlands Organization for Scientific Research and the Centre for Biomedical Genetics.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Shi, S., de Gorter, D.J.J., Hoogaars, W.M.H. et al. Overactive bone morphogenetic protein signaling in heterotopic ossification and Duchenne muscular dystrophy. Cell. Mol. Life Sci. 70, 407–423 (2013). https://doi.org/10.1007/s00018-012-1054-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-012-1054-x