
Abstract
Introduction
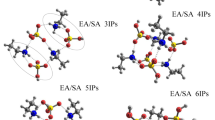
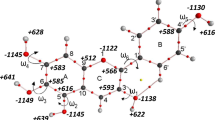
Enalapril maleate is an antihypertensive ethyl ester pro-drug with two crystalline forms. A network of hydrogen bonds in both polymorphs plays an important role on solid-state stability, charge transfer process and degradation reactions (when exposed to high humidity, temperature and/or pH changes).
Computational Procedures
Supramolecular arrangement was proposed by Hirshfeld surface using the CrystalExplorer17 software and quantum theory of atoms in molecules. The electronic structure properties were calculated using the functional hybrid M06-2X with 6–311++G** base function employing diffuse and polarization functions to improve the description of hydrogen atoms on intermolecular interactions. Also, the H+ charge transfer between enalapril and maleate molecules was performed using Car-Parrinello molecular dynamics with the Verlet algorithm. In both simulations, the temperature of the ionic system was maintained around 300 K using the Nosé-Hoover thermostat and the electronic system evolved without the use of the thermostat.
Results
This work evaluates the effect of maleate on the structural stability of enalapril maleate solid state. The electronic structural analysis points out a partially covalent character for N1-H∙∙∙O7 interaction; and the molecular dynamic showed a decentralized hydrogen on maleate driving a decomposition by charge transfer process while a centered hydrogen driving the stabilization. The charge transfer process and the mobility of the proton (H+) between enalapril and maleate molecules was demonstrated using supramolecular modeling analyses and molecular dynamics calculations.
















Similar content being viewed by others

Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Gomez HJ, Cirillo VJ, Irvin JD (1985) Enalapril. Drugs 30:13–24. https://doi.org/10.2165/00003495-198500301-00004
Smeets NJL, Schreuder MF, Dalinghaus M et al (2020) Pharmacology of enalapril in children: a review. Drug Discov Today 25:1957–1970. https://doi.org/10.1016/j.drudis.2020.08.005
Laurence B, Bruce C, Björn K (2011) Goodman & Gilman’s pharmacological basis of therapeutics, 12th edn. The McGraww-Hill Companies Inc., New York
Verbeeck RK, Kanfer I, Löbenberg R et al (2017) Biowaiver monographs for immediate-release solid oral dosage forms: enalapril. J Pharm Sci 106:1933–1943
Arafat T, Awad R, Hamad M et al (2005) Pharmacokinetics and pharmacodynamics profiles of enalapril maleate in healthy volunteers following determination of enalapril and enalaprilat by two specific enzyme immunoassays. J Clin Pharm Ther 30:319–328. https://doi.org/10.1111/j.1365-2710.2005.00646.x
Bernstein J (2020) Polymorphism in molecular crystals, 2nd edn. Oxford University Press
Ip DP, Brenner GS (1987) Enalapril maleate. Anal Profiles Drug Subs Excipients 16:207–243. https://doi.org/10.1016/S0099-5428(08)60557-2
Kiang Y-H, Huq A, Stephens PW, Xu W (2003) Structure determination of enalapril maleate form II from high-resolution x-ray powder diffraction data. J Pharm Sci 92:1844–1853. https://doi.org/10.1002/jps.10430
Davis MP, Mohara M, Shimura K, Korter TM (2020) Simulation and assignment of the terahertz vibrational spectra of enalapril maleate cocrystal polymorphs. J Phys Chem A 124:9793–9800. https://doi.org/10.1021/acs.jpca.0c08093
Ip DP, Brenner GS, Stevenson JM et al (1986) High resolution spectroscopic evidence and solution calorimetry studies on the polymorphs of enalapril maleate. Int J Pharm 28:183–191
Sherman BC (1996) Stable solid formulation of enalapril salt and process for preparation thereof. European Patent Office. Patent number: EP 1 442 746 A1
Rezende RLO, Santoro MIRM, Matos JR (2008) Stability and compatibility study on enalapril maleate using thermoanalytical techniques. J Therm Anal Calorim 93:881–886. https://doi.org/10.1007/s10973-007-8187-4
de Souza SMM, Melo Franco PIB, Leles MIG, da Conceição EC (2016) Evaluation of thermal stability of enalapril maleate tablets using thermogravimetry and differential scanning calorimetry. J Therm Anal Calorim 123:1943–1949. https://doi.org/10.1007/s10973-015-4648-3
Su L, Wang L, Yuan X et al (2021) Study on the stability and compatibility mechanism of enalapril maleate based on pKa and pH microenvironment. J Therm Anal Calorim 146:2161–2167. https://doi.org/10.1007/s10973-021-10596-7
Al-Omari MM, Abdelah MK, Badwan AA, Jaber AMY (2001) Effect of the drug-matrix on the stability of enalapril maleate in tablet formulations. J Pharm Biomed Anal 25:893–902
Bout MR, Vromans H (2022) Influence of commonly used excipients on the chemical degradation of enalapril maleate in its solid state: The role of condensed water. Eur J Pharm Sci 171:106–121. https://doi.org/10.1016/j.ejps.2022.106121
Stanisz B (2003) Evaluation of stability of enalapril maleate in solid phase. J Pharm Biomed Anal 31:375–380. https://doi.org/10.1016/S0731-7085(02)00325-4
Spackman MA, Jayatilaka D (2009) Hirshfeld surface analysis. CrystEngComm 11:19–32. https://doi.org/10.1039/b818330a
Spackman PR, Turner MJ, McKinnon JJ et al (2021) CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J Appl Crystallogr 54:1006–1011. https://doi.org/10.1107/S1600576721002910
Matta CF, Boyd RJ (2007) The quantum theory of atoms in molecules: from solid state to DNA and drug design. WILEY-VCH Verlag GmbH & Co, KGaA, Weinheim
Nanayakkara S, Kraka E (2019) A new way of studying chemical reactions: a hand-in-hand URVA and QTAIM approach. Phys Chem Chem Phys 21:15007–15018. https://doi.org/10.1039/c9cp01933b
Bader RFW (1998) A bond path: a universal indicator of bonded interactions. J Phys Chem A 102:7314–7323
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc 120:215–241. https://doi.org/10.1007/s00214-007-0310-x
Zhao Y, Truhlar DG (2006) A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J Chem Phys 125:194101. https://doi.org/10.1063/1.2370993
Frisch MJ, Trucks GW, Schlegel HB et al (2016) Gaussian 16. Revision E.01. Gaussian Inc, Wallingford CT
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33:580–592. https://doi.org/10.1002/jcc.22885
Hutter J (2012) Car-Parrinello molecular dynamics. Wiley Interdiscip Rev Comput Mol Sci 2:604–612. https://doi.org/10.1002/wcms.90
Pastore G, Smargiassi E, Buda F (1991) Theory of ab initio molecular-dynamics calculations. Phys Rev A (Coll Park) 44:6334–6347. https://doi.org/10.1103/PhysRevA.44.6334
Car R, Parrinello M (1985) unified approach for molecular dynamics and density-functional theory. Phys Rev Lett 55:2471–2474. https://doi.org/10.1103/PhysRevLett.55.2471
Alavi S (2009) Ab initio molecular dynamics. basic theory and advanced methods. Von Dominik Marx und Jürg Hutter. Angew Chem 121:9568–9569. https://doi.org/10.1002/ange.200904748
Verlet L (1967) Computer “experiments” on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys Rev 159:98–103. https://doi.org/10.1103/PhysRev.159.98
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865–3868. https://doi.org/10.1103/PhysRevLett.77.3865
Santin LG, Toledo EM, Carvalho-Silva VH et al (2016) Methanol solvation effect on the proton rearrangement of curcumin’s enol forms: an ab initio molecular dynamics and electronic structure viewpoint. J Phys Chem C 120:19923–19931. https://doi.org/10.1021/acs.jpcc.6b02393
Neto APV, Machado DFS, Lopes TO et al (2018) Explicit aqueous solvation treatment of epinephrine from Car-Parrinello molecular dynamics: effect of hydrogen bonding on the electronic absorption spectrum. J Phys Chem B 122:8439–8450. https://doi.org/10.1021/acs.jpcb.8b06110
Jabraoui H, Charpentier T, Gin S et al (2022) Behaviors of sodium and calcium ions at the borosilicate glass–water interface: gaining new insights through an ab initio molecular dynamics study. J Chem Phys 156:134501. https://doi.org/10.1063/5.0087390
Durlak P, Berski S, Latajka Z (2016) Theoretical studies on the molecular structure, conformational preferences, topological and vibrational analysis of allicin. Chem Phys Lett 644:5–13. https://doi.org/10.1016/j.cplett.2015.11.038
Latorre-Arca D, Soledad Larrechi M, Salavera D et al (2022) Quantitative and structural analysis of water association in water-lithium bromide-1,3-dimethylimidazolium chloride mixtures. J Mol Liq 368:120828. https://doi.org/10.1016/j.molliq.2022.120828
Vanderbilt D (1990) Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys Rev B 41:7892–7895. https://doi.org/10.1103/PhysRevB.41.7892
Nosé S (1984) A unified formulation of the constant temperature molecular dynamics methods. J Chem Phys 81:511–519. https://doi.org/10.1063/1.447334
Hoover WG (1985) Canonical dynamics: equilibrium phase-space distributions. Phys Rev A (Coll Park) 31:1695–1697. https://doi.org/10.1103/PhysRevA.31.1695
Kiang Y-H, Huq A, Stephens PW, Xu W (2003) Structure determination of enalapril maleate form II from high-resolution X-ray powder diffraction data. J Pharm Sci 92:1844–1853. https://doi.org/10.1002/jps.10430
Gu L, Strickley RG (1987) diketopiperazine formation, hydrolysis, and epimerization of the new dipeptide ang. Pharm Res 4:392–397
Nishio M (2011) The CH/π hydrogen bond in chemistry. Conformation, supramolecules, optical resolution and interactions involving carbohydrates. Phys Chem Chem Phys 13:13873–13900
Gautam D, Steiner T (2010) The weak hydrogen bond: In structural chemistry and biology. International Union of Crystallography Monographs on Crystallography (Oxford, 2001; online edn, Oxford Academic). https://doi.org/10.1093/acprof:oso/9780198509707.001.0001
Steiner T (2002) The hydrogen bond in solid state. Angew Chem Int 41:48–76
Grabowski SJ (2006) Hydrogen bonding : new insights. Springer
Jeffrey GA, Saenger W (1991) Hydrogen bonding in biological structures. Springer, Berlin Heidelberg
Rozas I, Alkorta I, Elguero J (2000) Behavior of ylides containing N, O, and C atoms as hydrogen bond acceptors. J Am Chem Soc 122:11154–11161. https://doi.org/10.1021/ja0017864
Costa RF, Aguiar ASN, Borges ID et al (2021) Effect of ortho- and para-chlorine substitution on hydroxychlorochalcone. J Mol Model 27:65. https://doi.org/10.1007/s00894-021-04670-y
Esrafili MD (2012) Investigation of H-bonding and halogen-bonding effects in dichloroacetic acid: DFT calculations of NQR parameters and QTAIM analysis. J Mol Model 18:5005–5016. https://doi.org/10.1007/s00894-012-1496-y
Hossain MdR, Hasan MdM, Nishat M et al (2021) DFT and QTAIM investigations of the adsorption of chlormethine anticancer drug on the exterior surface of pristine and transition metal functionalized boron nitride fullerene. J Mol Liq 323:114627. https://doi.org/10.1016/j.molliq.2020.114627
Nemati-Kande E, Karimian R, Goodarzi V, Ghazizadeh E (2020) Feasibility of pristine, Al-doped and Ga-doped boron nitride nanotubes for detecting SF4 gas: a DFT, NBO and QTAIM investigation. Appl Surf Sci 510:145490. https://doi.org/10.1016/j.apsusc.2020.145490
Rauk A (2001) Orbital interaction theory of organic chemistry. Wiley-Interscience
Shrivastav G, Khan TS, Agarwal M, Haider MA (2018) A Car-Parrinello molecular dynamics simulation study of the retro diels–alder reaction for partially saturated 2-pyrones in water. J Phys Chem C 122:11599–11607. https://doi.org/10.1021/acs.jpcc.8b00250
Frank I, Nadimi E (2021) Ab-initio molecular dynamics simulation of the electrolysis of nucleobases. Energies (Basel) 14:5021. https://doi.org/10.3390/en14165021
Durlak P, Latajka Z (2010) Car-Parrinello and path integral molecular dynamics study of the hydrogen bonds in 2-acetyl-1,8-dihydroxy-3,6-dimethylnaphthalene. Chem Phys Lett 499:56–61. https://doi.org/10.1016/j.cplett.2010.09.045
Stare J, Mavri J, Grdadolnik J et al (2011) Hydrogen bond dynamics of histamine monocation in aqueous solution: Car-Parrinello molecular dynamics and vibrational spectroscopy study. J Phys Chem B 115:5999–6010. https://doi.org/10.1021/jp111175e
Shekaari A, Jafari M (2021) Phase-transition behavior of (H 2 O) n =1−4 few-body systems from Car-Parrinello molecular dynamics. Phase Transitions 94:889–898. https://doi.org/10.1080/01411594.2021.1980564
Gao Z, Ma C, Lv G et al (2019) Car-Parrinello molecular dynamics study on the interaction between lignite and water molecules. Fuel 258:116189. https://doi.org/10.1016/j.fuel.2019.116189
Kiakojouri A, Nadimi E, Frank I (2020) Ab-initio molecular dynamics simulation of condensed-phase reactivity: the electrolysis of amino acids and peptides. Molecules 25:5415. https://doi.org/10.3390/molecules25225415
Schwermann C, Doltsinis NL (2020) Exciton transfer free energy from Car-Parrinello molecular dynamics. Phys Chem Chem Phys 22:10526–10535. https://doi.org/10.1039/C9CP06419B
Pliego JR (2022) Car-Parrinello molecular dynamics study of CuF, AgF, CuPF6 and AgPF6 in acetonitrile solvent and Cluster-Continuum calculation of the solvation free energy of Cu(I), Ag(I) and Li(I). J Mol Liq 359:119368. https://doi.org/10.1016/j.molliq.2022.119368
Acknowledgements
The authors are grateful to Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Amparo à Pesquisa de Goiás (FAPEG) and to Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for financial support. Also, the authors thank Prof. Dr. Ademir João Camargo of the Grupo de Química Teórica e Estrutural de Anápolis (Universidade Estadual de Goiás) for fruitful discussions. The data analysis to determine geometric parameters, RDF, MRT, among others, was performed through the GQTEA software, developed by him.
Funding
This work was supported by the Fundação de Amparo à Pesquisa do Estado de Goiás (FAPEG) and the Coordination for the Improvement of Higher Education Personnel (CAPES) through a scholarship for students and by the cluster of high-performance computers of the Grupo de Química Teórica e Estrutural de Anápolis.
Author information
Authors and Affiliations
Contributions
All authors contributed to the conception and design of the study and carried out the theoretical and analytical studies presented. The first version of the manuscript was written by Ana Carolina M. Lourenço, Lauriane G. Santin, James O. Fajemiroye, Solemar S. Oliveria, and Hamilton B. Napolitano. All authors commented and worked on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This paper belongs to the Topical Collection IX Symposium on Electronic Structure and Molecular Dynamics – IX SeedMol
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lourenço, A.C.M., Santin, L.G., Fajemiroye, J.O. et al. Studies on charge transfer of enalapril maleate: from solid-state to molecular dynamics. J Mol Model 29, 197 (2023). https://doi.org/10.1007/s00894-023-05597-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-023-05597-2