
Abstract
Future wheat production faces considerable challenges, such as how to ensure on-farm yield gains across agricultural environments that are increasingly challenged by factors such as soil erosion, environmental change and rapid changes in crop pest and disease profiles. Within the context of crop improvement, the ability to identify, track and deploy specific combinations of genes tailored for improved crop performance in target environments will play an important role in ensuring future sustainable wheat production. In this chapter, a range of germplasm resources and populations are reviewed can be exploited for genetic locus discovery, characterisation and functional analysis in wheat. These include experimental populations constructed from two or more parents, association mapping panels and artificially mutated populations. Efficient integration of the knowledge gained from exploiting such resources with other emerging breeding approaches and technologies, such as high-throughput field phenotyping, multi-trait ensemble phenotypic weighting and genomic selection, will help underpin future breeding for improved crop performance, quality and resilience.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Multi-parent populations
- Plant genetic diversity
- Sustainable crop production
- Nested association mapping (NAM)
- Multi-parent advanced generation intercross (MAGIC)
- Targeting Induced Local Lesions in Genomes (TILLING)
15.1 Gene Discovery in the Context of Wheat Improvement and Breeding
If you compare two bread wheat (Triticum aestivum L.) cultivars, the chances are that you will find differences between them—and lots of them. Whether these differences are for agronomic traits, such as resistance to disease, for quality traits such as those important for bread making, or for a range of morphological traits such as those used to uniquely ‘describe’ a variety during varietal registration (Jones et al. 2013), such variation is abundant. It is the heritable component of these observable differences that is exploited via breeding to deliver new improved wheat varieties and deals with the complexities of pleiotropic effects resulting from the process. The question as to how best to do this is not a straightforward one. To give a simplified example, phenotypic selection for underlying combinations of genes and alleles that result in increased grain number per ear may result in fewer ears overall. Similarly, increasing the grain protein is often associated with a reduction in overall grain yield in wheat (Simmonds 1995; White et al. 2022) and other crop species (e.g. Dudley 2007), and increasing leaf size is thought to result in larger, but less dense stomata (Zanella et al. 2022). As the principal breeding target, grain yield represents the sum of all interacting genetic/epigenetic, environmental and management factors that occur from sowing to harvest. Selection for grain yield works well, with breeders having consistently delivered ~ 1% genetic gains per year in wheat yield potential over recent decades (e.g. Mackay et al. 2011). To some extent, wheat breeding practices focus on delivering performance under the assessment criteria and carefully managed growth conditions used by national bodies to determine subsets of the ‘best’ varieties marketed at a given time. In the United Kingdom (UK), for example, the annual AHDB ‘Recommended List’ provides performance data for such varietal subsets to help farmers choose which varieties to grow (www.ahdb.co.uk/knowledge-library/reccommended-lists-for-cereals-and-oilseeds-rl). However, on-farm wheat yields are increasingly falling behind the genetic potential of the varieties grown. Termed the ‘yield-gap’, and observed in wheat growing areas across the world (Senapati et al. 2022), this is likely to be due to the cost–benefit and practical considerations and trade-offs that take place under commercial farm conditions. Future wheat production will face additional challenges such as environmental change, soil degradation, increasing energy and input costs, and the effects of political conflict or instability. Thus, wheat genetic improvement will increasingly need to focus on yield stability under sub-optimal, fluctuating or unpredictable growth environments—delivered within the context of more sustainable food production systems. As the development of new wheat varieties is a relatively lengthy process (typically taking around 10 years), all available tools must be exploited to meet these challenges. As underpinning technologies advance, the ability to identify specific wheat genes or genetic loci, and understand how they function and interact within the context of crop performance, will play an increasingly important role towards delivering future wheat.
15.2 Genetic Variation in Hexaploid Bread Wheat: Luck, Bottlenecks and Breeding
If the foundation of gene discovery is heritable variation, then before exploring the germplasm and genomic resources currently in use to accelerate gene discovery and functional analysis in wheat, it is first worth briefly considering the history behind current wheat genetic variation. Collectively, the natural genetic variation present in modern day wheat represents the culmination of the speciation, domestication and breeding events and processes that have occurred in its past. Human selection and interventions have affected the wheat genome and the variation it contains, starting from its first origins in Neolithic farmers’ fields, up to the current day. However, variation at the DNA level is not so evenly distributed across the bread wheat genome. To some extent, this is due to the order, age and nature of the polyploidisation events that occurred during its speciation. The bread wheat genome is hexaploid (2n = 6x = 42), which means it consists of three subgenomes that have merged via inter-species hybridisation events during its evolutionary history (reviewed by Levy and Feldman 2022; Fig. 15.1). Notably, the most recent event was a spontaneous hybridisation around 9000 years ago between the tetraploid progenitor of pasta wheat (the AA and BB subgenome donor) and a diploid wild wheat relative that grew alongside it called ‘goat grass’ (Aegilops tauschii Coss., DD subgenome donor) to create hexaploid bread wheat (AABBDD). Due to this event being rare, recent, and having occurred in a restricted Ae. tauschii sub-population close to the Caspian Sea (Wang et al. 2013), little D subgenome variation was captured, and there has been comparatively little time for genetic variation to subsequently accumulate via spontaneous mutation. The effect of this is evident in genetic analyses of bread wheat varieties from across the world (e.g. Wang et al. 2014; Walkowiak et al. 2020; Mellers et al. 2020), where D subgenome variation within genes is typically one-third to one-tenth of that seen on the A and B subgenomes. Consistent throughout the wheat subgenomes however is that gene density and gene variation are lower across the centromeric and adjacent pericentromeric chromosomal regions than in the remaining more distal chromosomal positions (IWGSC 2018). These centromeric and pericentromeric regions are associated with higher frequency of transposable elements (IWGSC 2018), higher levels of epigenetic modifications to DNA and histones associated with heterochromatin (tightly packed DNA), and lower genetic recombination (Gardner et al. 2016; Gardiner et al. 2019), which together are thought to result in the restricted rates of genome evolution observed in these regions (Akhunov et al. 2003). Against this genomic backdrop, in the ~ 9000 years since the speciation of bread wheat has been accumulating natural mutations which have either been retained or lost along the way due to a combination of selection, drift and geneflow. Such shifts in variation have underpinned the many generations of ‘on-farm’ selection that occurred from Neolithic times up until the advent of industrial breeding approaches at the end of the nineteenth century. Accordingly, wheat genetic variation was modulated across this time period by the interplay between human selection, be it conscious (such as selection for larger grains) or unconscious (such as selection for photoperiod insensitive lines; Jones and Lister 2022), and environmental factors such as prevailing climate and disease pressures. This ongoing domestication process resulted in the numerous locally adapted ‘landraces’ that were grown across the world’s wheat growing regions up until the end of the 1800s. Early breeders exploited these sources of genetic diversity by systematically selecting and evaluating such landraces, as well as the crosses made between them. The outcomes of this history are still evident in modern wheat varieties, as these first breeding programmes commonly exploited the landraces that were locally adapted to their regions at the time. Evidence of this history can be seen in modern day wheat. For example, genetic marker analysis of wheat from around the world shows clustering of Chinese landraces and cultivars in genetic diversity space (Cavanagh et al. 2013), while in an analysis of 180 UK varieties released since the year 2000, almost 90% include genetic contributions from the old Ukrainian landrace OSTKA-GALICYJSKA and the Mediterranean landrace from which the early UK variety SQUAREHEAD was developed (Fradgley et al. 2019). Over the years, there have been concerns that the industrial breeding era has resulted in genetic bottlenecks in numerous crops, and that this has restricted genetic diversity in modern wheat. While there are many approaches to measure genetic diversity loss (reviewed by Khoury et al. 2021), for wheat it is clear that more genetic diversity was present in the landraces versus pure-line bred cultivars (e.g. Winfield et al. 2018). The assumption of loss of diversity when within the modern breeding period is not necessarily so apparent, with changes in diversity depending on multiple factors, including the time period and region studied. One factor that has been noted is a reduction in genetic diversity at and soon after the introduction of the ‘Green Revolution’ semi-dwarfing genes across all international breeding programmes from the 1960s onwards (see Chap. 11). However, recent studies of on-farm wheat diversity indicate that at a national level, growers may now actually deploy a much more diverse portfolio of cultivars than was used 100 years ago. For example, in the USA the number of major commercially grown wheat cultivars has increased progressively, increasing fivefold from 1919 (33 cultivars) to 2019 (186 cultivars) with pedigree-based diversity measures of 1353 commercial USA varieties grown across this period indicating this increase in cultivar diversity is likely linked to increased genetic diversity (Chai et al. 2022). In the UK, combining measures of relatedness based on shared parentage (kinship), weighted by the proportional yearly acreage of cultivars over the last 30 years, found an increasing trend in the resulting landscape diversity index (Fradgley 2022). While the dominance of a very low number of varieties across national cropping landscapes is not as common as it once was (such as the use of cv. CAPPELLE-DESPREZ across more than 50% of the UK cropping area in the 1960s; Srinivasan et al. 2003), this is not necessarily the case throughout the wheat growing regions of the world. For example, between 2005 and 2010 the cultivar WYALKATCHEM represented more than 30% of the Australian wheat area sown, while more recently cv. MACE represented over 65% of the wheat cropping area in both 2015 and 2016 (Phan et al. 2020). Notably, these recent examples of low Australian landscape scale cultivar diversity are set against a wider background of a reduction in Australian wheat genetic diversity post Green Revolution (Joukhadar et al. 2017) and highlight the potential vulnerability of such landscape scale cultivar predominance to changes in pest and environmental pressures.

Evolutionary history of hexaploid bread wheat (Triticum aestivum) from its diploid and tetraploid donors progenitors. The unknown or extinct wheat B subgenome donor is a derivative of the S-genome species of the section Sitopsis, which includes diploid Ae. speltoides (diploid SS genome), Ae. bicornis (SbSb), Ae. longissima (SlSl), Ae. searsii (SsSs) and Ae. sharonensis (SshSsh). MYA = millions of years ago
15.2.1 Systematic Broadening of the Wheat Genepool as Wild Wheats Are Deployed
A longstanding concern is that breeding results in loss of genetic diversity—however, as noted above this assumption is not a given. A good example in cereals is the maize long-term selection experiment, where continuous genetic gains within a closed population in response to selection for seed protein and oil content were observed across the 100-year programme, with no significant loss in genetic diversity (Dudley 2007). Presumably, this was achieved via continued selection for genetic loci of small additive effect, as well as the fixing of epistatic interactions (i.e. instances where the allele of one gene hides or masks the phenotype of another gene) as additive effects. It is thus feasible to optimise existing variation present in wheat cultivars into new combinations, and to bring in additional genetic and functional diversity from systematic introgression and analysis of chromosomal regions originating from landraces and species related to wheat. When present in otherwise elite wheat genetic backgrounds, the chromosomal segments present in such ‘wilder wheats’ can often provide agronomic performance gains, despite the possible negative impacts of such chromosomal tracts (due, for example, to linkage drag or local effects on genetic recombination). Reminiscent of the activities at the start of the industrial breeding age, initiatives across the world are once again systematically screening variation captured in wheat landraces and are now supported by modern genetics, genomics, experimental population designs and analysis approaches. For example, the Watkins bread wheat landrace collection of 826 accessions from 32 countries has been genotyped using 41 microsatellite markers (Wingen et al. 2014), and selected accessions from across the genetic diversity space crossed to an elite spring cultivar to create a series of bi-parental genetic mapping populations (Wingen et al. 2017), termed a nested association mapping (NAM) panel. The benefits afforded by ‘wilder wheats’ created via introgressions from wheat relatives are illustrated by the UK cultivar ROBIGUS. Released in the UK in 2020, ROBIGUS delivered high yields and contained particularly novel genetics derived from a wheat wild relative (Gardner et al. 2016) and has been frequently used in the pedigrees of subsequent UK varieties (Fradgley et al. 2019)—without associated loss of wheat cultivar genetic diversity at landscape scale (Fradgley 2022). Genomic analyses now show that the presence of introgressions from wheat relatives is relatively common (e.g. Cheng et al. 2019; Keilwagen et al. 2022; Pont et al. 2019; Przewieslik-Allen et al. 2021; Scott et al. 2020a). Indeed, introgressions often underlie genomic regions conferring agronomically important traits—particularly disease resistance (Aktar-Uz-Zaman et al. 2017). For example, resistance to the wheat fungal disease yellow rust conferred by Yr34 originated from a region of chromosome 5A introgressed over 200 years ago from einkorn wheat (T. monococcum L. ssp. monococcum; Chen et al. 2021), and still confers field resistance in the US (Chen et al. 2021) and UK (Bouvet et al. 2022b). The long breeding history of use and utility of introgression from wheat relatives is exemplified by the extensive use since the late 1980s of synthetic hexaploid wheats in the international wheat breeding programme run by the International Maize and Wheat Improvement Center (CIMMYT) (Das et al. 2016; see also Chap. 11). Synthetic hexaploid wheats address the lack of genetic diversity on the wheat D subgenome by recreating the ancient hybridization event between tetraploid wheat and Ae. tauschii. This is undertaken via inter-specific crosses followed either by embryo rescue, chromosome doubling (Li et al. 2018a, b) or use of specific cytogenetic stocks (Othmeni et al. 2022). While more than 1200 synthetic wheats have been generated by CIMMYT, historically these have sampled a relatively narrow range of Ae. tauschii diversity from the eastern Fertile Crescent. Systematic broadening of the diversity sampled in synthetic wheats is now being undertaken at pre-breeding initiatives at NIAB in the UK, where D subgenome Ae. tauschii genetic diversity from across its natural eco-geographic range is being captured in new synthetics and backcrossed into elite cultivars (Gaurav et al. 2022). While this and other initiatives (e.g. Zhou et al. 2021) are providing new sources of D subgenome genetic variation for breeding, similar approaches are systematically bringing in additional diversity from wheat A and B subgenome donors via the creation of inter-specific hybrids and subsequent backcrossing. For example, the generation of backcross-derived progenies from crosses between 59 diverse accessions of tetraploid T. turgidum ssp. durum with elite spring wheat cv. PARAGON (see also Chap. 8). Introgressions into elite wheat varieties from more distantly related diploid and polyploid grass species are also being generated, including Ambylopyrum muticum (TT genome, Coombes et al. 2022) and Thinopyrum species (Li and Wang 2009; Grewal et al. 2018; Li et al. 2018a, b; Cseh et al. 2019; Baker et al. 2020). The utility of genetic loci originating from the tertiary wheat genepool has begun to lead in the identification of the underlying genes and genetic variants; for example, the wheat Fhb7 locus conferring resistance to the fungal disease Fusarium head blight, and which originated from a Th. elongatum introgression, has been shown to encode an amino acid transferase that detoxifies toxins produced by the infecting fungus (Wang et al. 2020).
15.3 Current Genome-Wide Genotyping Approaches for Wheat
The history of speciation, domestication and breeding outlined above has shaped the heritable variation present across the wheat genome. At the DNA level, this variation includes changes to singe nucleotides (single nucleotide polymorphisms, SNPs), or via other rearrangements that typically involve DNA double strand break repair such as DNA insertions or deletions (InDels), gene copy number variation (CNV) and larger chromosomal rearrangements such as translocation and/or inversion of larger tracts of DNA. In the 2000s, advances in wheat research such as the sequencing across multiple tissues, developmental stages and cultivars of complimentary DNA (cDNA) transcribed from messenger RNA (mRNA), and subsequently the availability of genome assemblies for cv. CHINESE SPRING (the wheat reference genome; IWGSC 2018) and 15 additional wheat cvs. (Walkowiak et al. 2020; Chap. 14) (Table 15.1) have led to detailed catalogues of both genic and non-genic DNA variation. Due to their abundance and nature, wheat studies over the last 10 years have most commonly assayed genic single nucleotide polymorphisms (SNPs) for use in genetic mapping approaches. Since the publication of the first high-density wheat genotyping array in 2013 capable of assaying ~ 9000 SNPs (Cavanagh et al. 2013), several additional arrays ranging from 3000 to 850,000 features are now available (Table 15.2). While SNP genotyping arrays are relatively simple and cheap to use, one drawback is that only those variants that have been pre-selected to be present on the array can be assayed. Thus, if the SNP identification panel used to design the array does not contain adequate sampling of the variants in the target genepool, useful information on the variation present in a target set of germplasm cannot be adequately assessed. This is a common issue for example in synthetic hexaploid wheat and its derived germplasm, where much of the novel D subgenome variation captured in this germplasm may not be assayed. More recently, reductions in costs have meant that sequencing-based genotyping approaches have become increasingly used in wheat. These include complexity reduction approaches such as genotyping by sequencing (GbyS) (Poland et al. 2012), Diversity Array Technology sequencing (DArTseq™; Sansaloni et al. 2011) and exome and/or promotor capture followed by Illumina short-read (i.e. ~ 150 bp) sequencing (Table 15.2). More recently, whole genome low-coverage sequencing is beginning to be used for genotyping in wheat (Table 15.2) and is considered in more detail in Box 1. Natural variation in the form of InDels and CNV are also relatively abundant in the wheat genome (e.g. Pont et al. 2019; Walkowiak et al. 2020; Wang et al. 2022), and despite the relatively limited number of functionally characterised wheat genes to date (Chap. 9), such variation has been shown to be a relatively common source of functional variation. For example, just within the flowering time pathway, deletions across putative cis-regulatory sites caused by double-stranded DNA break repair via nonhomologous recombination have been shown to result in at least seven functional alleles of the VERNALIZATION1 (VRN-1) flowering time gene homoeologues in hexaploid and diploid wheat (Cockram et al. 2007), while CNV at the PHOTOPERIOD-1 (PPD-1) homoeologues determine flowering time in tetraploid and hexaploid wheat (Díaz et al. 2012; Würschum et al. 2019; see also Chap. 11).
PPD-1) homoeologues determine flowering time in tetraploid and hexaploid wheat (Díaz et al. 2012; Würschum et al. 2019; see also Chap. 11).
Box 1: Wheat genotyping via skim sequencing
As genotyping via genome skim sequencing is typically undertaken at significantly less than 1-times genome-wide sequence coverage per line assayed (termed 1 × ), multiple reads at any given chromosomal location are not expected for any single line. Therefore, this approach is suited for experimental populations with defined founders, such that confidence in the DNA variants identified from skim sequence in any one line is achieved via reads obtained from additional lines in the population that carry the same variant. For example, if there are 200 lines in a bi-parental population, with each line sequenced to 0.3 × coverage, we would expect on average 60 × coverage of any single locus, and therefore 30 × coverage of each allele at any bi-allelic locus, i.e. (200 × 0.3)/2. Thus, by cataloguing and the SNPs present at good coverage in the population as a whole, the presence of any of these SNPs identified via a single sequencing read in any given line can be called with good confidence. Pre-determining the sequence variants present in the population founders, for example by exome capture or whole genome assembly, may help the process of variant calling and the imputation of variants that are not directly sequenced in any given line. For example, Scott et al. (2020a) sequenced the 16 founders of a wheat multifounder population via exome + promotor capture identifying 1.13 million SNPs across the 110,790 genes targeted by the capture probes. They then skim sequenced the 501 derived recombinant inbred lines (RILs) at 0.3 × coverage, which directly identified ~ 28% of these SNPs (i.e. 1.13 million SNPs × 0.3 = 339,000 SNPs). SNP imputation in the RILs was then undertaken using the software STICH (Davies et al. 2016), resulting in 94% of the 1.13 million founder SNPs to be called and founder haplotype dosage at each chromosomal location to be assigned for all RILs. Down-sampling the 0.3 × read coverage showed RILs could be accurately inferred from sequence coverage as low as 0.076 × per RIL. Notably, at sequence coverage of 0.076 × and above, imputation accuracy was not dependent on whether or not founder haplotypes were included as a reference panel. This means that accurate RIL haplotype mosaics in the RILs could be achieved without the need to generate data on the 16 founders. In summary, imputation from low-coverage whole genome sequencing of experimental populations represents a relatively straightforward and cost-effective genotyping strategy for bi-parental and multifounder experimental wheat populations and does not suffer from the inherent bias of SNP array genotyping approaches that require the variants targeted to be pre-identified.
15.4 Genetic Mapping Resolution: Population Size, Genetic Recombination and Effect Size
Forward genetic mapping relies largely on the recombination fraction between a QTL and the genetic markers that have been genotyped in the population, and the heritability of the target trait. These considerations are reviewed in more detail elsewhere (e.g. Cockram and Mackay 2018), but in general greater genetic mapping resolution can be attained by increasing population size and/or undertaking additional rounds of crossing. Larger populations also have the benefit of providing greater QTL detection power. Important to consider is the heritability of the target trait and the effect size of the QTL detected. The more heritable a trait is, and the larger its effect size, the easier it is to detect and precisely locate. Indeed, most wheat QTL resolved to the underlying gene level are for highly penetrant major genes, such as gene-for-gene disease resistance loci (e.g. for a recent list of cloned wheat rust resistance genes, see Bouvet et al. 2022b), awn presence/absence (Huang et al. 2020), vernalization response (first undertaken in T. monococcum: Yan et al. 2003; Yan et al. 2004), plant height (Tian et al. 2022) and grain quality (Uauy et al. 2006). If trait heritability is low, phenotypic replication can increase line mean heritability and has been used to refine and update the genetic interval of a locus on chromosome 5A controlling ~ 10% variation for wheat grain size (Brinton 2017; Brinton et al. 2017). Aside from such highly penetrant genetic loci, the genetic architecture of most target traits in wheat is highly quantitative in nature. For example, the mean QTL effect size for grain size traits in wheat is less than 10%, compared to more than 20% in the diploid cereal rice, and is likely due to the buffering effect of homoeologues of overlapping function in hexaploid wheat (Brinton and Uauy 2019).
15.5 Population Types
The identification of functional gene variants via genetic mapping relies on the capture of sufficient genetic diversity and genetic recombination. Fundamentally, two broad experimental population types are employed by researchers interested in identifying genetic loci controlling traits of interest. Both exploit genetic variation, and the reshuffling of this variation via genetic recombination, in order to associate markers or groups of markers (haplotypes, see also Chap. 9) with target traits.
15.5.1 Experimental Populations
Experimental populations are derived from crossing two or more parents to produce progeny in which genetic loci can be identified by the strength of the associations between genetic markers and traits of interest. Examples of some commonly used experimental populations are listed below and are illustrated in Fig. 15.2.

Illustration of experimental population and association mapping panel designs. Number of founders illustrated in each panel is indicated in brackets. Dashed lines indicate inbreeding (via single seed descent or doubled haploid approaches) to produce multiple inbred lines. AIC = advanced intercross, two rounds of intercrossing illustrated, prior to the production of inbred lines. NAM = nested association mapping. NCII = North Carolina II model. MAGIC = multifounder advanced generation intercross. AM = association mapping population
15.5.1.1 Bi-parental
Bi-parental populations are most commonly used in wheat forward genetics research and are constructed by first crossing two parents to generate first filial (F1) derived progeny lines. Inbred progeny are generated either by single seed descent (whereby individual F2 lines are selfed over three or more generations to achieve acceptable levels of homozygosity genome-wide) or via doubled haploid approaches (where haploid F1-derived gametes undergo chromosome doubling, resulting in completely inbred progeny in a single generation) (Fig. 15.2). Despite DH lines typically taking less time to create compared to RILs, DH populations capture less genetic recombination. This is because additional genetic recombination events can occur between regions of heterozygosity from the F2 generation (25% heterozygous) until effective fixing at around the F6 stage (1.6% heterozygous) or beyond, and which on average is equivalent to one additional round of crossing. Bi-parental populations are now beginning to be constructed from wheat cultivars with genome assemblies, such as the CHINESE SPRING × PARAGON population (Wingen et al. 2017).
15.5.1.2 Advanced Intercross
Even when bi-parental populations are created via single seed descent, the amount of genetic recombination captured can be relatively low. One way to increase the number of genetic recombinations is to continue random intercrossing of the F2 for one or more generations before the production of inbred lines (Fig. 15.2). Such advanced intercross (AIC) populations (Darvasi and Soller 1995) designs provide greater precision compared to standard bi-parental populations of the same size. For example, Darvasi and Soller (1995) estimated that eight rounds of random intermating would reduce a QTL interval from 20 to 3.7 cM. While AIC have been used in species such as Arabidopsis (Fitz et al. 2014) and maize (Balint-Kurti et al. 2008), they have yet to be implemented in wheat—likely due to the time required to undertake additional rounds of crossing. However, the advent of ‘speed breeding’ approaches, that allow the generation time of both spring (Watson et al. 2018) and winter (Cha et al. 2022) wheat varieties to be reduced, means that for primary QTL screens, AIC approaches in wheat should become a more attractive prospect.
15.5.1.3 Near Isogenic Line Pairs, Introgression Lines and Chromosome Segment Substitution Lines
A near isogenic line (NIL) captures a relatively small chromosomal region from one ‘donor’ parent within the wider genomic context of a second ‘recipient’ parent (Fig. 15.2). NILs are generated via repeated rounds of backcrossing, often with the use of genetic markers to select for donor at the target chromosomal region, and for the recipient across the remainder of the genome. NILs are commonly used to target specific QTL of interest, allowing the effect of the contrasting alleles captured in the NIL pair to be evaluated using a single pair of lines, rather than a larger population in which additional genetic loci affecting the target trait may be segregating. Following this approach, individual genetic loci controlling a target trait can be investigated in detail, and the underlying physiology and pleiotropic effects on related traits can be assessed. Further, a NIL pair can be crossed to generate further genetic recombination and so further refine the genetic interval. For example, contrasting alleles at a major effect genetic locus for wheat grain weight identified in a bi-parental population of 192 inbred lines was subsequently assessed via phenotypic evaluation of BC2- and BC4-derived NILs, finding the ~ 7% increase in grain weight was (i) mediated predominantly by increased grain length, (ii) the maternal pericarp cell length was longer in the NIL carrying the high grain weight allele, and that (ii) increased grain length was detectable 12 days after fertilisation (Brinton et al. 2017). Additionally, the NILs were used to further refine the genetic interval to 4.3 cM (Brinton et al. 2017), with further analysis indicating that two genetic loci may be present at the locus (Brinton 2017). A series of NILs that capture chromosomal segments from wild and domesticated wheat relatives is termed introgression lines. Recent work in the UK has generated such germplasm resources for a range of wheat relatives. These include diploid Ae. caudata (CC genome. Grewal et al. 2020), Am. muticum (TT. Coombes et al. 2022), Th. bessarabicum (JJ. Grewal et al. 2018), and T. uratu (AuAu. Grewal et al. 2021), tetraploid T. timopheevii (AtAtGG. Devi et al. 2019), hexaploid Th. intermedium (JJ JvsJvs StSt. Cseh et al. 2019) and decaploid Th. elongatum (EbEb EbEb EbEb EStESt EStESt. Baker et al. 2020), with all introgression lines generated using the recipient wheat cv. PARAGON. When a series of NILs is designed to collectively capture the entire donor background, the resulting resource is termed a chromosome segment substitution line (CSSL) population. In wheat, CSSLs populations that capture novel A, B and D subgenome diversity from wheat relatives have recently been developed using (i) a synthetic hexaploid wheat line (Horsnell et al. 2022) and (ii) a tetraploid T. turgidum ssp. dicoccoides accession (TTD-140). Not only do CSSL populations serve as useful sources of novel variation, they can also be used directly for genetic mapping, as recently illustrated in wheat by Horsnell et al. (2022).
15.5.1.4 Multifounder Populations: NAM
While bi-parental populations and derived NILs had long been the mainstay of forward genetic approaches, multifounder populations have recently become commonplace in plant research (reviewed by Scott et al. 2020b). Multi-parent mapping populations capture more variation than bi-parental populations and increase precision via joint linkage and association analysis. Nested association mapping (NAM) populations represent a series of bi-parental populations (termed ‘families’), each of which has the same parent in common (Fig. 15.2). The first NAM population was made in maize (Zea mays L.) by crossing 25 diverse inbred lines with the inbred line B73 (termed here the ‘linking’ founder)—one of the most widely used lines in the history of maize breeding, and the line used for the maize reference genome (Yu et al 2008). Since then, the maize NAM parents have become extensively characterised, including provision of their genome assemblies (Gage et al. 2020). The genetic resolution obtained from NAM populations largely depends on the number of alleles present in the founders and the amount of genetic recombination captured in the progeny. The rarest alleles in any NAM population will be present in half of the progeny from the corresponding family. Therefore, in a NAM with 25 families and 200 progeny per family, rare alleles are expected to be present in 100 of the total 5000 progeny lines, i.e. a frequency of 2%. For NAM design, increasing the number of founders at the expense of family size should be preferable, as the decay of parental linkage disequilibrium for a given allele would likely, on average, be shared among more parents (Gage et al. 2020). NAM populations have now been made in many crop species and can be genetically analysed using association mapping approaches. At least part of the attraction of NAM design is that their composition (a series of bi-parental populations with a common parent) makes them more conceptually familiar to researchers experienced with bi-parental populations. Indeed, once a genetic locus has been identified in a NAM, it is straightforward to continue further analysis using one or more of the relevant constituent bi-parental populations. To date, several NAM populations have been created in wheat (Table 15.3; Fig. 15.3). The founders used include elite cultivars (e.g. Bajgain et al. 2016), genetically diverse landraces (Wingen et al. 2017), as well as germplasm that captures backcrossed chromosomal segments from wheat relatives via synthetic hexaploid wheat and wheat vs tetraploid durum wheat (T. durum ssp. durum) introgression lines. Further, a recent durum NAM has been constructed by crossing 50 durum landraces to an Ethiopian durum cultivar (Kidane et al. 2019). The largest wheat NAM currently available was constructed using 60 inbred worldwide landraces from the Watkins wheat landrace collection, backcrossed to the spring UK cultivar PARAGON, generating a population of 1192 RILs and a mean of 105 RILs per family (Wingen et al. 2017). Therefore, the rarest allele captured in the Watkins NAM would be expected to be present in 4% of the population—a frequency nominally sufficient for detection via genetic analysis.

Features of existing wheat nested association mapping (NAM) populations, comparing mean NAM progeny per component bi-parental population (x-axis) with the number of NAM founders (y-axis) and the size of the resulting population (proportional to the size of the circle)
15.5.1.5 Multifounder Populations: North Carolina II Model
A notable limitation of NAM populations is that while multiple founders are employed, a single ‘linking’ parent is used with which to cross to. The North Carolina II (NCII) design of Comstock and Robinson (1952) is conceptually an extension of NAM, whereby two or more ‘linking’ parents are used such that every progeny family has half-sib relationships both through a common mother and through a common father (Fig. 15.2). Similarly, any combination of populations with founder links between them can be analysed together to undertake genetic analysis and to increase power and precision by increasing sample size (Cockram and Mackay 2018). However, such populations are more commonly used to confer detection of QTL in different genetic backgrounds and on the analysis of epistasis.
15.5.1.6 Multifounder Populations: MAGIC
While NAM and NCII populations capture more diversity than bi-parentals, they capture no additional genetic recombination than bi-parental populations of the same size. Since its pioneering use in mouse in 2002 (The Complex Trait Consortium 2002), the multi-parent advanced generation intercross (MAGIC) design has been applied to many crop species (Scott et al. 2020b). To aid crossing design, MAGIC populations typically use 4, 8 or 16 founders. However, unlike NAM or NCII populations, all MAGIC founders are intercrossed over multiple rounds of crossing to produce progeny that capture equal proportions of each founder genome (Fig. 15.2). Thus, MAGIC combines the benefits of increased genetic diversity afforded by NAM and NCII, with increased amounts of genetic recombination afforded by AIC, while minimising population structure via controlled crossing. In contrast to bi-parental populations, which are typically constructed to target a single target trait and are relatively quick to generate, MAGIC populations aim to capture and recombined multiple alleles across the genome and therefore take much longer to create. However, once complete, MAGIC, as well as other multi-parent populations, are well suited as community resources. In wheat, six MAGIC populations have been published, the first of which was the Australian spring wheat 4-parent MAGIC (Huang et al. 2012). Since then, four additional MAGIC populations have been created: 8-parent populations from Australia (Shah et al. 2019), the UK (Mackay et al. 2014) and Germany (Sannemann et al. 2018; Stadlmeier et al. 2018), as well as a 16-parent European wheat MAGIC (Scott et al. 2020a) (Fig. 15.4). Additionally, a MAGIC-like wheat population made between one male-sterile line crossed and backcrossed with 59 European/worldwide lines, followed by 12 generations of random intermating, has been generated (Thépot et al. 2014). To date, the 8-founder NIAB Elite MAGIC population likely has the most publicly available resources available, including the population and associated 90 k array SNP data (Mackay et al. 2014) and genetic map (Gardner et al. 2016), genome assemblies for two of the founders (Walkowiak et al. 2020), and phenotypic and genetic data for numerous traits including disease (Bouvet et al. 2022a, c; Corsi et al. 2020; Lin et al. 2020a; Riaz et al. 2020) flowering time (Wittern et al. 2022), canopy architecture (Zanella et al. 2022), ear architecture (Dixon et al. 2018), end-use quality and mineral content (Fradgley et al. 2022a). Additionally, BLAST access to the genome assembles for the remaining six founders is currently available ahead of publication (https://www.cropdiversity.ac.uk/8magic-blast/) and release of whole genome skim sequencing data for the RILs is imminent (J Cockram personal communication).

Crossing diagram illustrating the founder selection and pedigree of the wheat 16-parent ‘NIAB Diverse MAGIC’ population. The red and blue lines each track the pedigree of a single recombinant inbred line (RIL) through the pedigree
15.5.2 Founder Selection
Founder choice in any structured population is one of the first decisions addressed and depends to some degree on population type. For a bi-parental population, founders that contrast for a specific trait of interest are typically selected. In some cases, selection criteria will also include selection for specific traits that may otherwise confound the target phenotype. For example, founders with similar ear emergence date may be selected to avoid pleiotropic effects on diseases such as Fusarium head blight that affect the wheat ear. However, the differential presence of alleles of contrasting effect between founders may mean that while the parents may have been selected for similar phenotype, segregation for the phenotype may still be observed in the progeny. For NAM and MAGIC populations, founders should generally be selected to maximise genetic diversity, particularly in those designs that include larger founder numbers. For NAM populations, the selection of the ‘linking’ founder is notably important as each progeny line will sample 50% of its genome, and its genome will be highly represented in the population. ‘Linking’ founders typically represent a line which has been particularly well characterised, or is common in the wheat pedigree within the target geographical region. For example, the cultivar PARAGON has been selected as the ‘linking’ founder in three wheat NAM populations: Watkins-60 (Wingen et al. 2017), NIAB SHW and NIAB TetHex (data repository at https://niab.github.io/niab-dfw-wp3/). PARAGON is a spring UK variety released in 1988 which has a sequenced genome (Walkowiak et al. 2020), RNA sequence (RNA-seq) data from multiple tissues and a gamma-irradiated series of deletion lines (available via https://www.jic.ac.uk/research-impact/germplasm-resource-unit/). Similar considerations apply to the selection of linking founders in NCII population designs, although as two or more such founders are used, more flexibility is afforded.
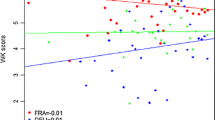
If the aim of the population is to generate data under field conditions, founders should be suited for growth in the environments under which they will be phenotyped. When constructing populations using elite varieties, this should be relatively straightforward. For example, in the NIAB Diverse MAGIC population, the 16 founders were selected to sample maximum genetic diversity across a wider collection of 94 European winter wheat cultivars released over a 70 year period, for assessment under UK field conditions (Scott et al. 2020a). However, for populations that capture variation from landrace or species related to wheat, especially if these donors originate from geographic areas distant to the target environment, adaptability of the resulting populations to local field environments could be more problematic. In bi-parental or NAM populations, one way to address this is to generate populations from backcross-1 (BC1) generation (where each progeny line contains on average 25% of the non-recurrent founder genome) or beyond, rather than from the F1 which is expected to contain 50% contribution from each founder. This approach is logistically harder, as it involves an additional round of crossing and requires more progeny than an F1-derived population to effectively sample non-recurrent founder genome. However, if the aim is to generate phenotypic data under field conditions, such approaches may be beneficial. For MAGIC designs, as each progeny line represents a balanced genomic mosaic of all founders, the inclusion of one, or possibly more, ‘wilder’ founder genomes is slightly less problematic. For example, in an 8-founder MAGIC which includes one ‘wilder’ founder, each RIL would be expected to contain a 1/8th genomic contribution from the ‘wilder’ founder. While no such MAGIC populations have been constructed to date in wheat, the most diverse is the INRA MAGIC-like population developed using one male-sterile line (cv. PROBUS) crossed and backcrossed with 59 European/worldwide lines before 12 generation of random intermating to generate 1000 lines (Thépot et al. 2014). Finally, for all population designs, it may be useful to consider the size and extent of any genomic rearrangements (e.g. the chromosome 5AL/7AL translocation Walkowiak et al. 2020) or chromosomal introgressions from wheat relatives, as their presence is likely to disrupt local genetic recombination rates. While such regions may specifically be sought, for example the Ae. tauschii (D) and T. durum ssp. durum (AB) genomic contributions captured in the NIAB SHW NAM, it is possible that one or more founders are unintentionally selected that contain such features. For example, in the 16-founder NIAB Diverse MAGIC population, cv. MARIS FUNDIN carries a large introgression of 540 Mb from T. timopheevi on chromosome 2B which is substantially over-represented in the MAGIC progeny (Fig. 15.5) (Scott et al. 2020a). Segregation distortion due to introgressions was also identified in the 8-founder NIAB Elite MAGIC, for example due to the chromosome 1B/1R wheat/rye introgression in cvs. BROMPTON and RIALTO and the presence of an introgression on the long arm of chromosome 4A in cv. ROBIGUS (Gardner et al. 2016).

~ 540 Mb chromosome 2B introgression from T. timopheevi present in the NIAB Diverse MAGIC founder MARIS FUNDIN, as identified by analysis of exome-promotor capture sequence data of the 16 founders. The introgression is visualised here by the increase in non-reference (relative to chromosome 2B IWGSC RefSeq v1.0, cv. CHINESE SPRING) SNP variants (top) and as reduced sequence coverage (bottom) in MARIS FUNDIN, compared to the remaining 15 founders. Scott et al. (2020a) find the introgression to be substantially over-represented in the MAGIC progeny
15.5.3 Association Mapping Panels
The experimental populations described above take time to construct. However, it is possible to exploit the genetic variation and historical genetic recombination captured in existing collections of wheat varieties, landraces or accessions (Fig. 15.2). Such association mapping approaches aim to locate QTL based on the strength of the association between genetic markers and the target trait(s) and rely on the decay of linkage disequilibrium between markers and QTL over genetic distance (Cockram and Mackay 2018). Genetic analysis of association mapping panels can be conducted using markers from candidate genes, or from across the genome using a whole genome association scan (GWAS) approach. Most commonly, single markers are regressed against the target trait. However, power can be increased by constructing haplotypes from the genotypic allele calls of two or more genetic variants that are closely physically or genetically linked within a defined region (haploblock). Use of haplotypes in GWAS can improve the estimation of allelic effects and increase statistical significance and is increasingly used in wheat. For example, linkage disequilibrium approach to defining haploblocks in a panel of 6333 wheat lines genotyped with 14,027 GbyS genetic markers resulted in the identification of 537 genome-wide haploblocks for downstream GWAS of grain yield (Sehgal et al. 2020). Alleles present at a frequency of less than 5% within the panel will typically not be detected, even if these alleles have relatively high effect sizes and/or the causative polymorphism is assayed. In human genetics, approaches that help identify rare alleles in GWAS are increasingly being used (reviewed by Lee et al. 2014), such as aggregation tests that evaluate cumulative effects of multiple genetic variants in a gene or region. The ability to generate experimental populations in plants means that such approaches are not as necessary to explore.
Unlike the case in most experimental populations in which allele frequency is relatively equally distributed among the progeny, association mapping panels are often characterised by notable levels of population substructure or subdivision. This is due to the differences in the shared ancestry of the lines over time, due to non-random mating. In cereal crops, population structure commonly arises from (i) physical separation, i.e. (geographic location), (ii) the contrasting germplasm preferences within different breeding companies, (iii) seasonal growth habit (i.e. spring or winter-sown) and (iv) traits underlying end-use quality (such as malting or feed in barley, or bread making versus in wheat) (Cockram et al. 2010; White et al. 2022) and yield (Sharma et al. 2022). For example, while relatively few major genetic determinants control the spring versus winter phenotype (Bentley et al. 2013), the common practice that spring cultivars are typically bred from other spring lines, while winters are bred from winters means that any genetic variants present at notably different frequencies between these two germplasm pools continue to show skews in their frequency in progeny lines. Thus here, if a favourable allele controlling a trait of interest happened to segregate predominantly in the spring pool, then the population structure inherent within spring varieties may lead to false-positive genotype-trait associations (termed Type-I errors) that are not due to close linkage of markers with the underlying QTL. It is possible to control statistically for population structure (Q) by using genetic markers to determine a Q-matrix of population membership estimates for each accession in the panel. Q-matrices can be determined using programmes such as STRUCTURE (Pritchard et al. 2000) or via principal component analysis (Zhao et al. 2007). Additional correction for more recent similarities due to close kinship (K) can also be included and can be determined using genetic markers. Indeed, approaches such as the Q + K mixed model (Yu et al. 2006) that account for multiple levels of relatedness between individuals have been shown to control well for false-positive as well as false-negative (Type-II error) associations and often lead to higher power than correction via Q or K alone (Yu et al. 2006). However, accounting for population structure/kinship sacrifices some level of experimental power to detect those genetic loci that are correlated with the adjustments made. Nevertheless, power and precision to detect genetic loci in association mapping panels can be high, compared to experimental populations of the same size. While improved power can be achieved by increasing the number of individuals in the panel, the inclusion of additional accessions may increase population substructure and/or kinship. Similarly, linkage disequilibrium may decay quite slowly in with genetic distance in cultivars (due to close kinship among all lines), which will reduce the precision to detect QTL (Cockram and Mackay 2018) but will increase power. Conversely, linkage disequilibrium in panel’s landraces is typically higher, enabling greater genetic mapping precision. Genotyped wheat landraces collections are now available that sample diversity with single countries (e.g. China, Zhou et al. 2017) or from around the world—such as the Watkins (Wingen et al. 2014) and Vavilov collections (Riaz et al. 2018). These are beginning to be used for GWAS of agronomic traits, such as disease resistance (tan spot, Halder et al. 2019; leaf rust. Riaz et al. 2018, stripe rust, Jambuthenne et al. 2022) and pre-harvest sprouting (Zhou et al. 2017). Given the multiple variables affecting GWAS in association mapping panels, it is useful to determine the efficacy of experimental design by undertaking power calculations, especially if population size is relatively small (e.g. White et al. 2022).
15.6 Reverse Genetics Germplasm Platforms
Functional validation of genes genetically mapped using experimental or association mapping populations can be undertaken using reverse genetics approaches. Transgenic methods aim to alter gene expression or function, typically via gene overexpression, gene silencing or gene editing (reviewed in wheat by Adamski et al. 2020). Alternatively, non-transgenic reverse transgenics approaches are available that exploit genetic variation induced by mutagenizing agents. In wheat, the most commonly used are Targeting Induced Local Lesions in Genomes (TILLING) populations, created by using an inbred donor line (termed the M0 generation) and applying the chemical agent ethyl methanesulphonate (EMS). The resulting EMS treated seed is termed the M1 generation, which can be subsequently selfed over several generations to generate a population of TILLING lines in which the EMS-generated mutations become progressively fixed in homozygous state. Bespoke experiment-specific TILLING populations are frequently used to determine genes underlying traits controlled by single major effect genes, such as gene-for-gene disease resistance. In such cases, a wheat line for which resistance to the target disease is controlled by a single major effect locus is mutated, and susceptible TILLING lines identified phenotypically. Assuming the underlying gene can be sequenced, relatively low numbers of TLLING lines with independent mutations at the target locus are generally sufficient to give a high statistical probability of identifying the causative gene. For example, Sánchez-Martín et al. (2016) estimated that the probability that the 12 kb gene containing contig of their target wheat gene (Pm2 conferring resistance to powdery mildew) being mutated across all 12 identified powdery mildew susceptible TILLING mutants was 1 in 300,000,000,000. Several approaches to applying DNA sequencing to such gene identification approaches have been published: the first uses exome capture of pre-determined candidate gene families (termed resistance-gene enrichment sequencing, RenSeq, when applied to NRL disease resistance gene families; Jupe et al. 2013). The second, termed MutChromSeq, involves flow sorting and direct sequencing of the target chromosome in each of the phenotypically identified TILLING lines (Sánchez-Martín et al. 2016). In addition to such experiment-specific TILLING resources, exome capture followed by DNA sequencing of large numbers of TILLING lines generated from the spring bread wheat cv. CADENZA (1200 lines) and the tetraploid wheat cv. KRONOS (1535 lines) have been made publicly available (Krasileva et al. 2016). The resulting TILLING mutations have been aligned against the bread wheat reference genome of cv. CHINESE SPRING (RefSeq v1.1; IWGSC 2018) and searchable via the Ensembl plants (Cunningham et al. 2022) genome browser. The effects of mutations on protein sequence have been predicted in relation to CHINESE SPRING gene models, with deleterious mutations determined to be present in around 90% of the captured genes. The ability to identify and prioritise TILLING mutants in silico means these resources serve as useful genome-wide resources for gene functional validation in wheat. Considerations for the identification and validation of wheat TILLING mutants in the CADENZA and KRONOS populations are listed in more detail by Adamski et al. (2020) and include the need to combine TILLING mutants in multiple homoeologues to overcome possible functional redundancy as well as the need to undertake sufficient rounds of backcrossing to remove background mutations. Examples of their use for gene functional characterisation include (i) wheat candidate genes orthologous to map-based cloned gene from model species (e.g. TaGRAIN WIDTH2, Simmonds et al. 2016), (ii) wheat genes identified via forward phenotypic screening followed by bulk segregant analysis of backcross derived progeny between mutant line and wild-type (e.g. HOMEOBOX DOMAIN-2, Dixon et al. 2022) and (iii) candidate genes underlying wheat genetic loci previously refined by fine-mapping (e.g. WHEAT ORTHOLOG OF APO1, Kuzay et al. 2022; EARLY FLOWERING 3, Wittern et al. 2022). While the ability to screen in silico the cv. CADENZA and KRONOS TILLING populations provide proven community resources for gene functional characterisation, they can only be used for those genes present in the two founding cultivars used. The availability of annotated genome assemblies for multiple wheat varieties now provides the underpinning knowledge from which it may in future be possible to develop additional sequenced TILLING resources that target genes not captured in cv. CADENZA and KRONOS.
15.7 The Future of Genetic Recombination
Genetic recombination in wheat is enriched in the telomeric regions and becomes progressively less frequent towards the pericentromeric and centromeric regions, with 80% of recombination events occurring in less than a quarter of the genome (e.g. Gardner et al. 2016; IWGSC 2018). As genetic mapping relies on the occurrence of recombination, being able to increase recombination at chromosomal regions of interest would help both genetic mapping precision, and the ability to recombine different haplotypes in breeding. Analysis of crossover events in RIL populations has identified QTL for genetic recombination frequency, such as a locus on chromosome 6A in the CHINESE SPRING × PARAGON population controlling around 6% of the variation (Gardiner et al. 2019). Further, recent work shows that recombination events in wheat pericentric regions can be increased in some chromosomes by increasing temperature during meiosis (Coulton et al. 2020), although this does come with reduced fertility (Draeger and Moore 2017). Transgenic approaches for altering genetic recombination rates and locations are also now being investigated. For example, transient virus induced gene silencing (VIGS) of wheat candidate genes homologous to genes in other species shown to control genetic recombination shows it is possible to alter the distribution of recombination along chromosomes (Raz et al. 2021). VIGS silencing of the durum wheat homologue of the anti-cross over gene XRCC2 (a paralogue of RAD51) in F1 plants ahead of meiosis resulted in increased genetic recombination across much of the pericentromeric region of chromosome 4B, as well the more distal pericentromeric regions of chromosome 5B (Raz et al. 2021). Such results indicate that it should be possible to increase genetic recombination in at least some of the pericentromeric landscape of wheat. The maturation of gene editing methodologies may soon enable the targeting of cross-overs and genetic recombination to more specific genomic locations.
15.8 Conclusions
In parallel to the efforts to provide wheat genomic and genotyping tools, the wheat community has generated extensive resources to support genetic locus and gene characterisation via forward and reverse genetics approaches. For highly penetrant wheat genetic loci originating from natural variants or via induced mutation, and where phenotype effectively acts as a genetic marker, various routes have been used to identify the underlying genetic loci, including fine-mapping in bi-parental derived germplasm, as well as reverse genetics approaches such as RenSeq and MutChromSeq where the identification of multiple independent alleles rather that genetic recombination is required. For genetic loci of a more quantitative nature, to date it is those which account for an unusually high proportion of genetic variation that have been fine-mapped or map-based cloned, using bi-parental populations and also more recently via multifounder populations. The vast majority of remaining heritable variation in the wheat genepool is much more quantitative in nature, typically accounting for 3–5% of the phenotypic variation. For such loci, including those located in genomic regions with very low genetic recombination, identification of the underlying genes and variants via forward mapping approaches will continue to pose a challenge. However, genetic mapping approaches will allow their alleles and linked haplotypes to be determined, and increasingly, for the epistatic non-additive interaction effects of these loci to be characterised. For wheat breeding, advances in our knowledge of genetic loci and gene function will best be exploited within a quantitative genetics framework (Mackay et al. 2021). Trait improvement in the context of breeding over the next decade will likely focus on integration of multi-trait ensemble phenotypic weighting approaches (e.g. Fradgley et al. 2022b) combined with improved genomic selection methodologies and field-based phenotyping at increasing throughput and precision. The next decade will likely also see the maturation of approaches to engineer increased genetic recombination, and to design via gene editing new alleles with improved function. Finally, computer vision, artificial intelligence and machine learning and approaches are now maturing to the point at which they can more readily be applied to complex challenges such as crop phenotyping and plant breeding. Such approaches need to be efficiently combined to underpin future breeding for improved crop performance, quality and resilience.
References
Adamski NM, Borrill P, Brinton, Harrington SA, Marchal C et al (2020) A roadmap for gene functional characterisation in crops with large genomes: lessons from polyploid wheat. eLife, 9:e55646
Akhunov ED, Goodyear AW, Geng S, Qi L-L, Echalier B et al (2003) The organization and rate of evolution of wheat genomes are correlated with recombination rates along chromosome arms. Genome Res 13:753–763
Aktar-Uz-Zaman M, Tuhina-Khatun M, Hanafi MM, Sahebi M (2017) Genetic analysis of rust resistance genes in global wheat cultivars: an overview. Biotechnol Biotechnol Equip 31:431–445
Allen AM, Winfield M, Burridge AJ, Rownie RC, Benbow HR, Barker GLA, Wilkinson PA, Coghill J, Waterfall C, Davassi A, Scopes G, Pirani A, Webster T, Brew F, Bloor C, Griffiths S, Bentley AR, Alda M, Jack P, Phillips AL, Edwards KJ (2017) Characterization of a wheat breeders’ array suitable for high-throughput SNP genotyping of global accessions of hexaploid bread wheat (Triticum aestivum). Plant Biotechnol J 15:390–401
Avni R, Nave M, Barad O, Baruch K, Twardziok SO et al (2017) Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 357:93–97
Bajgain P, Rouse MN, Tsilo TJ, Macharia GK, Bhavani S et al (2016) Nested association mapping of stem rust resistance in wheat using genotyping by sequencing. PLoS ONE 11:1–22
Baker L, Grewal S, Yang C-y, Hubbart-Edwards S, Scholefield D, Ashling S, Burridge AJ, Przewieslik-Allen AM, Wilkinson PA, King IP, King J (2020) Exploiting the genome of Thinopyrum elongatum to expand the gene pool of hexaploid wheat. Theor Appl Genet 133:2213–2226
Balint-Kurti PJ, Wisser R, Zwonitzer JC (2008) Use of an advanced intercross line population for precise mapping of quantitative trait loci for gray leaf spot resistance in maize. Crop Science, 48: 1696–1704.
Bentley AR, Jensen EF, Mackay IJ, Hönicka H. Fladung M, Hori K, Yano M, Mullet JE, Armstead IP, Hayes C, Thorogood D, Lovatt A, Morris R, Pullen N, Mutasa-Göttgens E, Cockram J (2013). Flowering time. In: Cole C (ed) Genomics and breeding for climate-resilient crops, vol 2. Springer, Berline, pp 1–67
Bouvet L, Percival-Alwyn L, Berry S, Fenwick P, Holdgate S, Mackay IJ, Cockram J (2022a) Genetic resistance to yellow rust infection of the wheat ear is controlled by genes controlling foliar resistance and flowering time. Crop Sci 62:1758–1770
Bouvet L, Holdgate S, James L, Thomas J, Mackay IJ, Cockram J (2022b) The evolving battle between yellow rust and wheat: implications for global food security. Theor Appl Genet 135:741–753
Bouvet L, Percival-Alwyn L, Berry S, Fenwick P, Mantello CC, Sharma R, Holdgate IJ, Mackay IJ, Cockram J (2022c) Wheat genetic loci conferring resistance to stripe rust in the face of genetically diverse races of the fungus Puccinia striiformis f. sp. tritici. Theor Appl Genet 135:301–319
Brinton J (2017) Deciphering the molecular mechanisms controlling grain length and width in polyploid wheat. PhD thesis, submitted to the University of East Anglia, Norwich, UK
Brinton J, Simmonds J, Minter F, Leverington-Waite M, Snape J, Uauy C (2017) Increased pericarp cell length underlies a major quantitative trait locus for grain weight in hexaploid wheat. New Phytol 215:1026–1038
Brinton J, Uauy C (2019) A reductionist approach to dissecting grain weight and yield in wheat. J Integr Plant Biol 61:337–358
Cavanagh CR, Chao S, Wang S, Huang BE, Stephen S et al (2013) Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc Natl Acad Sci USA 110:8057–8062
Cha J-K, O’Connor K, Alahmad S, Lee J-H, Dinglasan E, Park H, Lee S-M, Hirsz D, Kwon S-W, Kwon Y, Kim K-M, Ko J-M, Hickey LT, Shin D, Dixon LE (2022) Speed vernalization to accelerate generation advance in winter cereal crops. Mol Plant 15:1300–1309
Chai Y, Pardey PG, Silverstein AT (2022) Scientific selection: a century of increasing crop varietal diversity in UK wheat. Proc Natl Acad Sci USA 119:e2210773119
Chen S, Hegarty J, Shen T, Hua L, Li H, Luo J, Li H, Bai S, Zhang C, Dubcovsky J (2021) Stripe rust resistance gene Yr34 (synonym Yr48) is located within a distal translocation of Triticum monococcum chromosome 5AmL into common wheat. Theor Appl Genet 134:2197–2211
Cheng H, Liu J, Wen J, Nie X, Xu L, Chen N, Li Z, Wang Q, Zheng Z, Li M, Cui L, Liu Z, Bia J, Wang Z, Xu S, Yang Q, Appels R, Han D, Song W, Sun Q, Jiang Y (2019). Frequent intra- and inter-species introgression shapes the landscape of genetic variation in bread wheat. Genome Biology 20:136
Clavijo BJ, Venturini L, Schudoma C, Accinelli GG, Kaithakottil G et al (2017) An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res 27:885–896
Cockram J, Mackay I, O’Sullivan DM (2007) The role of double-stranded break repair in the creation of phenotypic diversity in cereal VRN1 loci. Genetics 177:2535–2539
Cockram J, White J, Zuluaga DL, Smith D, Comadran J et al (2010) Genome-wide association mapping to candidate polymorphism resolution in the un-sequenced barley genome. Proc Natl Acad Sci USA 107:21611–21616
Cockram J, Mackay I (2018) Genetic mapping populations for conducting high-resolution trait mapping in plants. In: Varshney R, Pandey M, Chitikineni A (eds) Plant genetics and molecular biology. Advances in biochemical engineering/biotechnology, vol 164. Springer, Cham
Comstock RH, Robinson HF (1952) In: Gowen JW (ed) Heterosis. Iowa State College Press, Ames, IA., pp 495–516
Coombes B, Fellers JP, Grewal S, Rusholme-Pilcher R, Hubbart-Edwards S, Yang CY, Joynson R, King IP, King J, Hall A (2022) Whole genome sequencing uncovers the structural and transcriptomic landscape of hexaploid wheat/Ambylopyrum muticum introgression lines. Plant Biotechnol J. https://doi.org/10.1111/pbi.13859
Corsi B, Percival-Alwyn L, Downie RC, Venturini L, Iagallo EM, Mantello CC, McCormick-Barnes C, See PT, Oliver RP, Moffat CS, Cockram J (2020) Genetic analysis of wheat sensitivity to the ToxB fungal effector from Pyrenophora tritici-repentis, the causal agent of tan spot. Theor Appl Genet 133:935–950
Corsi B, Obinu L, Zanella CM, Cutrupi S, Day R et al (2021) Analysis of a German multi-parental population identifies eight genetic loci controlling two or more yield components in wheat, including the genetic loci Rht24, WAPO-A1 and WAPO-B1 and multi-trait QTL on chromosomes 5A and 6A. Theor Appl Genet 134:1435–1454
Coulton A, Burrdidge A, Edwards KJ (2020) Examining the effects of temperature on recombination in wheat. Front Plant Sci 11:230
Cseh A, Yang C-Y, Hubbart-Edwards S, Scholefield D, Ashling S, Burridge AJ, Wilkinson PA, King IP, King J, Grewal S (2019) Development and validation of Thinopyrum intermedium–exome based SNP marker set for identification of the St, Jr and Jvs genomes in a wheat background. Theor Appl Genet 132:1555–1570
Cui F, Zhang N, Fan X-I, Zhang Wm Zhao C-H, Yang L-J, Pan R-Q, Chen M, Han J, Zhao X-Q, Ji J, Tong Y-P, Zhange H-X, Jia J-Z, Zhao G-Y, Li J-M (2017) Utilization of a Wheat660k SNP array-derived high-density genetic map for high-resolution mapping of a major QTL for kernel number. Sci Rep 7:3788
Cunningham F, Allen JE, Allen A, Alvarez-Jarreta J, Amode MR et al (2022) Ensembl 2022. Nucleic Acids Res 50:D988–D995
Das MK, Bai G, Mujeeb-Kazi A, Rajaram S (2016) Genetic diversity among synthetic hexaploid wheat accessions (Triticum aestivum) with resistance to several fungal diseases. Genet Resour Crop Evol 63:1285–1296
Darvasi A, Soller M (1995) Advanced intercross lines, and experimental population for fine genetic mapping. Genetics 141:1199–1207
Davies RW, Flint J, Myers S, Mott R (2016) Rapid genotype imputation from sequence without reference panels. Nat Genet 48:965–969
Devi U, Grewal S, Yang C-y, Hubbart-Edwards S, Scholefield D, Ashling S et al (2019) Development and characterisation of interspecific hybrid lines with genome-wide introgressions from Triticum timopheevii in a hexaploid wheat background. BMC Plant Biol 19:183. https://doi.org/10.1186/s12870-019-1785-z
Díaz A, Zikhali M, Turner AS, Isaac P, Laurie DA (2012) Copy number variation affecting the Photoperiod-B1 and Vernalization-A1 genes is associated with altered flowering time in wheat (Triticum aestivum). PLoS ONE 7:e33234
Dixon LE, Greenwood JR, Bencivenga S, Zhang P, Cockram J, Mellers G et al (2018) TEOSINTE BRANCHED 1 regulates inflorescence architecture and development in bread wheat (Triticum aestivum L.). Plant Cell 30:563–581
Dixon LE, Pasquarello M, Badgami R, Levin KA, Poschet G, Ng PQ, Orford S, Chayut N, Adamski NM, Brinton J, Simmonds J, Steuernagel B, Searle IR, Uauy C, Boden SA (2022). MicroRNA-resistant alleles of HOMEOBOX DOMAIN-2 modify inflorescence branching and increase grain protein content of wheat. Science Advances 8:eabn5907
Downie RC, Bouvet L, Furuki E, Gosman N, Gardner KA, Mackay IJ, Mantello CC, Mellers G, Phan H, Rose GA, Tan K-C, Oliver R, Cockram J (2018) Assessing European wheat sensitivities to Parastagonospora nodorum necrotrophic effectors and fine-mapping of the Sn3-B1 locus conferring sensitivity to the effector SnTox3. Front Plant Sci 9:881
Draeger T, Moore G (2017) Short periods of high temperature during meiosis prevent normal meiotic progression and reduce grain number in hexaploid wheat (Triticum aestivum L.). Theor Appl Genet 130:1785–1800
Dudley JW (2007) From means to QTL: the Illinois long-term selection experiment as a case study in quantitative genetics. Crop Sci 47:S20–S31
Fitz Gerald JN, Carlson AL, Smith E, Maloof JN, Weigel D, Chory J, Borevitz JO, Swanson RJ (2014) New Arabidopsis advanced intercross recombinant inbred lines reveal female control of nonrandom mating. Plant Physiol 165:175–185
Fradgley N (2022) Working towards a diverse UK landscape. In: NIAB landmark, issue 51, Winter 2022/2023, pp 6–7
Fradgley N, Gardner KA, Cockram J, Elderfield J, Hickey JM, Howell P, Jackson R, Mackay I (2019) A large-scale pedigree resource of wheat reveals evidence for adaptation and selection by breeders. PLoS Biol 17:e3000071
Fradgley NS, Gardner KA, Kerton M, Swarbreck SM, Bentley AR (2022a) Trade-offs in the genetic control of functional and nutritional quality traits in UK winter wheat. Heredity 128:420–433
Fradgley NS, Gardner KA, Bentley AR, Howell P, Mackay IJ, Scott MF, Mott R, Cockram J (2022b). Multi-trait ensemble genomic prediction and simulations of recurrent selection highlight importance of complex trait genetic architecture in long-term genetic gains in wheat. bioRxiv, https://doi.org/10.1101/2022.11.08.515457
Gage JL, Monier B, Giri A, Buckler ES (2020) Ten years of the maize nested association mapping population: impact, limitations and future directions. Plant Cell 32:2083–2093
Gardiner L-J, Wingen LU, Bailey P, Joynson R, Brabbs T, Wright J, Higgins JD, Hall N, Griffiths S, Clavijo BJ, Hall A (2019) Analysis of the recombination landscape of hexaploid bread wheat reveals genes controlling recombination and gene conversion frequency. Genome Biol 20:69
Gardner KA, Wittern LM, Mackay IJ (2016) A highly recombined, high-density, eight founder wheat MAGIC map reveals extensive segregation distortion and genomic locations of introgression segments. Plant Biotechnol J 14:1406–1417
Gaurav K, Arora S, Silva P, Sanchez-Martín J, Horsnell R et al (2022) Population genomic analysis of Aegilops tauschii identifies targets for bread wheat improvement. Nat Biotechnol 40:422–431
Geyer M, Mohler V, Hartl L (2022) Genetics of the inverse relationship between grain yield and grain protein content in common wheat. Plants 11:2146
Grewal S, Yang C, Edwards S, Scholefield D, Ashling S, Burridge AJ, King IP, King J (2018) Characterisation of Thinopyrum bessarabicum chromosomes through genome-wide introgressions into wheat. Theor Appl Genet 131:389–406
Grewal S, Hubbart-Edwards S, Yang C, Devi U, Baker L, Heath J, Ashling S, Scholefield D, Howells C, Yards J, Isaac P, King IP, King J (2020) Rapid identification of homozygosity and site of wild relative introgressions in wheat through chromosome-specific KASP genotyping arrays
Grewal S, Guwela V, Newell C, Yang C-y, Ashling S, Scholefield D, Hubbart-Edwards S, Burridge A, Stride A, King IP, King J (2021) Generation of doubled haploid wheat—Triticum urartu introgression lines and their characterisation using chromosome-specific KASP markers. Front Plant Sci 12:643636
Halder J, Zhang J, Ali S, Sidhu JS, Gill HS, Talukder SK, Kleinjan J, Turnipseed B, Sehgal SK (2019) Mining the genomic characterization of resistance to tan spot, Stagonospora nodorum blotch (SNB), and Fusarium head blight in Watkins core collection of wheat landraces. BMC Plant Biol 19:480
Horsnell R, Leight FJ, Wright TIC, Burrudge A, Ligeza A, Przewieslik-Allen MA, Howell P, Uauy C, Edwards KJ, Bentley AR (2022) A wheat chromosome segment substitution line series supports characterisation and use of progenitor genetic variation. Plant Genome e20288. https://doi.org/10.1002/tpg2.20288
Huang BE, George AW, Forrest KL, Kilian A, Hayden MJ, Morell MK, Cavanagh CR (2012) A multiparent advanced generation inter-cross population for genetic analysis in wheat. Plant Biotechnol J 10:826–839
Huang D, Zheng Q, Melchkart T, Bekkaoui Y, Konkin DJF, Kagale S, Martucci M, You FM, Clarke M, Adamski NM, Chinoy C, Steed A, McCartney CA, Cutler AJ, Nicholson P, Feurtado JA (2020) Dominant inhibition of awn development by a putative zinc-finger transcriptional repressor expressed at the B1 locus in wheat. New Phytol 225:340–355
International Wheat Genome Sequencing Consortium (IWGSC), Appels R, Eversole K, Stein N, Feuillet C, Keller B, Rogers J, Pozniak C et al (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361:eaar7191
Jambuthenne DT, Riaz A, Athiyannan N, Alahmad S, Ng WL, Ziems L, Afanasenko O, Periyannan SK, Aitken E, Platz G, Godwin I, Voss-Fels KP, Dinglasan E, Hickey LT (2022) Mining the Vavilov wheat diversity panel for new sources of adult plant resistance to stripe rust. Theor Appl Genet 135:1355–1373
Jones MK, Lister D (2022) The domestication of the seasons: the exploitation of variations in crop seasonality responses by later prehistoric farmers. Front Ecol Evol 10:907536
Jones H, Norris C, Cockram J, Lee D (2013) Variety protection and plant breeders’ rights in the ‘DNA era.’ In: Lübberstedt T, Varshney R (eds) Diagnostics in plant breeding. Springer, Dordrecht, pp 369–402
Jordan KW, Wang S, Lun Y, Gardiner L-J, MacLachlan R, Hucl P et al (2015) A haplotype map of allohexaploid wheat reveals distinct patterns of selection on homoeologous genomes. Genome Biol 16:48
Jordan KW, Wang S, He F, Chao S, Lun Y et al (2018) The genetic architecture of genome-wide recombination rate variation in allopolyploid wheat revealed by nested association mapping. Plant J 95:1039–1054
Joukhadar R, Daetwyler HD, Bansal UK, Gendall AR, Hayden MJ (2017) Genetic diversity, population structure and ancestral origin of Australian wheat. Front Plant Sci 8:2115
Jupe F, Witek K, Verweij W, Sliwka J, Pritchard L et al (2013) Resistance gene enrichment sequencing (RenSeq) enables reannotation of the NB-LRR gene family from sequenced plant genomes and rapid mapping of resistance loci in segregating populations. Plant J 76:530–544
Keilwagen J, Lehnert H, Berner T, Badaeva E, Himmelbach A, Brner A, Kilian B (2022) Detecting major introgressions in wheat and their putative origins using coverage analysis. Sci Rep 12:1908
Khokhar JD, King J, King IP, Young SD, Foulkes MJ et al (2020) Novel sources of variation in grain zinc (Zn) concentration in bread wheat germplasm derived from Watkins landraces. PLoS ONE 15:e0229107
Khoury CK, Brush S, Costich DE, Curry HA, de Haan S, Engels JMM, Guarino L, Hoban S, Nercer KL, Miller AJ, Nabhan GP, Perales HR, Richards C, Riggins C, Thormann I (2021) Crop genetic erosion: understanding and responding to loss of crop diversity. New Phytol 233:84–118
Kidane YG, Gesesse CA, Hailemariam BN, Desta EA, Mengistu DK, Fadda C, Pè ME, Dell’Acqua M (2019) A large nested association mapping population for breeding and quantitative trait locus mapping in Ethiopian durum wheat. Plant Biotechnol J 17:1380–1393
King J, Grewal S, Othmeni M, Coombes B, Yang C-y, Walter N, Ashling S, Scholefield D, Walker J, Hunnart-Edwards S, Hall A, King IP (2022) Introgression of the Triticum timopeevii genome into wheat detected by chromosome-specific Kopetitive Allelle Specific PCR markers. Front Plant Sci 13:919519
Krasileva KV, Vasquez-Gross HA, Howell T, Bailey P, Paraiso F et al (2016) Uncovering hidden variation in polyploid wheat. Proc Natl Acad Sci USA 114:E913–E921
Kuzay S, Lin H, Li C, Chen S, Woods DP, Zhang J, Lan T, von Korff M, Dubcovsky J (2022) WAPO-A1 is the coausal gene of the 7AL QTL for spikelet number per spike in wheat. PLoS Genet 18:e1009747
Lee S, Abecasis GR, Boehnke M, Lin X (2014) Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet 95:5–23
Levy AA, Feldman M (2022) Evolution and origin of bread wheat. Plant Cell 34:2549–2567
Li H, Wang X (2009) Thinopyrum ponticum and Th. intermedium: the promising source of resistance to fungal and viral diseases of wheat. J Genet Genomics 36:557–565
Li A, Liu D, Yang W, Kishii M, Mao L (2018a) Synthetic hexaploid wheat: yesterday, today, and tomorrow. Engineering 4:552–558
Li DY, Long D, Li TH, Wu YL, Wang Y, Zeng J, Xu LL, Fan X, Sha LN, Zhang HQ, Zhou YH, Kang HY (2018b) Cytogenetics and stripe rust resistance of wheat–Thinopyrum elongatum hybrid derivatives. Mol Cytogenet 11:16
Lin M, Corsi B, Ficke A, Tan K-C, Cockram J, Lillemo M (2020a) Genetic mapping using a wheat multi-founder population reveals a locus on chromosome 2A controlling resistance to both leaf and glume blotch caused by the necrotrophic fungal pathogen Parastagonospora nodorum. Theor Appl Genet 133:785–808
Lin M, Stadlmeier M, Mohler V, Tan KC, Ficke A, Cockram J, Lillemo M (2020b) Identification and cross-validation of genetic loci conferring resistance to Septoria nodorum blotch using a German multi-founder winter wheat population. Theor Appl Genet 134:125–142
Lisker A, Maurer A, Schmutzer T, Kazman E, Cöster H, Holzapfel J, Ebmeyer E, Alqudah AM, Sannemann W, Pillen K (2022) A haplotype-based GWAS identified trait-improving QTL alleles controlling agronomic traits under contrasting nitrogen fertilization treatments in the MAGIC wheat population WM-800. Plants 11:3508
Luo MC, Gu YQ, Puiu D, Wang H, Twardziok SO et al (2017) Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 551:498–502
Mackay IJ, Horwell A, Garner J, White J, McKee J, Philpott H (2011) Reanalyses of the historical series of UK variety trials to quantify the contributions of genetic and environmental factors to trends and variability in yield over time. Theor Appl Genet 122:225–238
Mackay I, Bansept-Basler P, Barber T, Bentley AR, Cockram J et al (2014) An eight-parent multiparent advanced generation intercross population for winter-sown wheat: creation, properties and validation. G3: Genes Genomes Genetics 4:1603–1610
Mackay IJ, Cockram H, Howell P, Powell W (2021) Understanding the classics: the unifying concepts of transgressive segregation, inbreeding depression and heterosis and their central relevance for crop breeding. Plant Biotechnol J 19:26–34
Mellers G, Aguilera JG, Bird N, Bonato ALV, Bonow S et al (2020) Genetic characterization of a wheat association mapping panel relevant to Brazilian breeding using a high-density single nucleotide polymorphism array. G3: Genes, Genomes, Genetics 10:2229
Naish M, Alonge M, Wlodzimierz P, Tock AJ, Abramson BW et al (2021) The genetic and epigenetic landscape of the Arabidopsis centromeres. Science 374:eabi7489
Othmeni M, Grewal S, Walker J, Yang C-Y, King IP, King J (2022) Assessing the potential of using the Langdon 5D(5B) substitution line for the introgression of Aegilops tauschii into durum wheat. Front Plant Sci 13:927728
Phan HTT, Jones DAB, Rybak K, Dodhia KN, Lopez-Ruiz FJ, Valade R, Gout L, Lebrun M-H, Brunner PC, Oliver RP, Tan K-C (2020) Low amplitude boom-and-bust cycles define the Septoria Nodorum Blotch interaction. Front Plant Sci 10:1785
Poland JA, Brown PJ, Sorrells ME, Jannink JL (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 7:e32253
Pont C, Leroy T, Seidel M, Tondelli A, Duchemin W et al (2019) Tracing the ancestry of modern bread wheats. Nat Genet 51:905–911
Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000) Association mapping in structured populations. Am J Hum Genet 67:170–181
Przewieslik-Allen AM, Wilkinson PA, Burridge A, Winfield MO, Dai X et al (2021) The role of gene flow and chromosomal instability in shaping the bread wheat genome. Nature Plants 7:172–183
Raz A, Dahan-Meir T, Melamed-Bessudo C, Leshkowitz D, Levy AA (2021) Redistribution of meiotic crossovers along wheat chromosomes by virus-induced gene silencing. Front Plant Sci 11:635139
Riaz A, Hathorn A, Dinglasan E, Ziems L, Richard C, Singh D, Mitrofanova O, Afanasenko O, Aitken E, Godwin I, Hickey L (2017) Into the vault of the Vavilov wheats: old diversity for new alleles. Genet Resour Crop Evol 64:531–544
Riaz A, Athiyannan N, Periyannan SK, Afanasenko O, Mitrofanova OP, Platz GJ, Aitken EAB, Snowdon RJ, Lagudah ES, Hickey LT, Voss-Fels KP (2018) Unlocking new alleles for leaf rust resistance in the Vavilov wheat collection. Theor Appl Genet 131:127–144
Riaz A, KockAppelgren P, Hehir J, Kang J, Meade F, Cockram J, Milbourne D, Spink J, Mullins W, Byrne S (2020) Genetic analysis using a multi-parent wheat population identifies novel sources of septoria tritici blotch resistance. MDPI Genes 11:887
Rimbert H, Barrier B, Navarro J, Kitt J, Choulet F et al (2018) High throughput SNP discovery and genotyping in hexaploid wheat. PLoS ONE 13:e0186329
Sansaloni C, Petroli C, Jaccoud D, Carling J, Detering F, Grattapaglia D, Kilian A (2011) Diversity arrays technology (DArT) and next-generation sequencing combined: genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc 5:P54
Sansaloni C, Franco J, Santos B, Percival-Alwyn L, Singh S et al (2020) Diversity analysis of 80,000 wheat accessions reveals consequences and opportunities of selection footprints. Nat Commun 11:4572
Sánchez-Martín J, Steueragel B, Ghosh S, Herren G, Hurni S et al (2016) Rapid gene isolation in barley and wheat by mutant chromosome sequencing. Genome Biol 17:221
Sannemann W, Lisker A, Maurer A, Léon J, Kazman E, Cöster H et al (2018) Adaptive selection of founder segments and epistatic control of plant height in the MAGIC winter wheat population WM-800. BMC Genomics 19:1–16
Scott M, Fradgley N, Bentley AR, Brabbs T, Corke F, Gardner KA, Horsnell R, Howell P, Ladejobi F, Mackay I, Mott R, Cockram J (2020a) Limited haplotype diversity underlies polygenic trait architecture across 70 years of plant breeding. Genome Biol 22:137
Scott MF, Ladejobi O, Amer S, Bentley AR, Biernaskie J et al (2020b) Multi-parent populations in crops: a toolbox integrating genomics and genetic mapping with pre-breeding. Heredity 125:396–416
Sehgal D, Mondal S, Crespo-Herrera L, Velu G, Juliana P, Huerta-Esoino J, Shrestha S, Poland J, Singh R, Dreisigacker S (2020) Haplotype-based, genome-wide association study reveals stable genomic regions for grain yield in CIMMYT spring bread wheat. Front Genet 11:589490
Senapati N, Semenov MA, Halford NG, Hawkesford MJ, Asseng S et al (2022) Global wheat production could benefit from closing the genetic yield gap. Nature Food 3:532–541
Shah R, Huang BE, Whan A, Newberry M, Verbyla K et al (2019) The complex genetic architecture of recombination and structural variation in wheat uncovered using a large 8-founder MAGIC population. bioRxiv, p 594317. https://doi.org/10.1101/594317
Sharma R, Cockram J, Gardner KA, Russell J, Ramsay L, Thomas WTB, O’Sullivan DM, Powell W, Mackay IJ (2022) Trends of genetic changes uncovered by Env- and Eigen-GWAS in wheat and barley. Theor Appl Genet 135:667–678
Simmonds NW (1995) The relation between yield and protein in cereal grain. J Sci Food Agric 67:309–315
Simmonds J, Scott P, Brinton J, Mestre TC, Bush M, del Blanco A, Dubcovsky J, Uauy C (2016) A splice acceptor site mutation in TaGW2-A1 increases thousand grain weight in tetraploid and hexaploid wheat through wider and longer grains. Theor Appl Genet 129:1099–1112
Srinivasan CS, Thirtle C, Palladino P (2003) Winter wheat in England and Wales, 1924–1995: what do indices of genetic diversity reveal? Plant Genetic Resources 1:43–57
Stadlmeier M, Hartl L, Mohler V (2018) Usefulness of a multiparent advanced generation intercross population with a greatly reduced mating design for genetic studies in winter wheat. Front Plant Sci 871:1–12
Stadlmeier M, Nistrup Jørgensen L, Fejer Justesen A, Corsi B, Cockram J, Hartl L, Mohler V (2019) Genetic dissection of resistance to the three fungal plant pathogens B. graminis, Z. tritici, and P. tritci-repentis in the background of a multiparental winter wheat population. G3 9:1745–1757
Steed A, King J, Herwal S, Yang C-Y, Clarke M, Devi U, King IP, Nicholson P (2022) Identification of Fusarium head blight resistance in Triticum timopheevii accessions and characterization of wheat-T. timopheevii introgression lines for enhanced resistance. Frontiers in Plant Science 13:943211
The Complex Trait Consortium (2002) The collaborative cross, a community resource for the genetic analysis of complex traits. Nature Genetics 36:1133–1137
Thépot S, Restoux G, Goldringer I, Hospital F, Gouache D et al (2014) Efficiently tracking selection in a multiparental population: the case of earliness in wheat. Genetics 199:609–623
Tian X, Xia X, Xu D, Liu Y, Xie L, Hassan MA, Song J, Li F, Wang D, Zhang Y, Hao Y, Li G, Chu C, He Z, Cao S (2022) Rht24b, and ancient variation of TaGA2ox-A9, reduces plant height without yield penalty in wheat. New Phytol 233:738–750
Uauy C, Distelfeld A, Fahima T, Blechl A, Dubcovsky J (2006) A NAC gene regulating senescence improves grain protein, zinc, and iron content in wheat. Science 314:1298–1301
Walkowiak S, Gao L, Monat C, Haberer G, Kassa MT et al (2020) Multiple wheat genomes reveal global variation in modern breeding. Nature 588:277–283
Wang J, Luo M-C, Chen Z, You FM, Wei Y et al (2013) Aegilops tauschii single nucleotide polymorphisms shed light on the origins of wheat D-genome genetic diversity and pinpoint the geographic origin of hexaploid wheat. New Phytol 198:925–937
Wang S, Wong D, Forrest K, Allen A, Chao S et al (2014) Characterization of polyploid wheat genomic diversity using a high-density 90 000 single nucleotide polymorphism array. Plant Biotechnol J 12:787–796
Wang H, Sun S, Ge W, Zhao L, Hou B et al (2020) Horizontal gene transfer of Fhb7 from fungus underlies Fusarium head blight resistance in wheat. Science 368:6493
Wang Z, Deng Z, Kong X, Wang F, Guan J, Cui D, Sun G, Liao R, Fu M, Che Y, Hao C, Geng S, Zhang X, Zhou P, Mao L, Liu S, Li A (2022) InDels identification and association analysis with spike and awn length in Chinese wheat mini-core collection. Int J Mol Sci 23:5587
Watson A, Ghosh S, Williams MJ, Cuddy WS, Simmonds J et al (2018) Speed breeding is a powerful tool to accelerate crop research and breeding. Nature Plants 4:23–29
White J, Sharma R, Balding D, Cockram J, Mackay IJ (2022) Genome-wide association mapping of Hagberg falling number, protein content, test weight, and grain yield in U.K. wheat. Crop Sci 62:965–981
Winfield MO, Allen AM, Burridge AJ, Barker GLA, Benbow HR et al (2016) High-density SNP genotyping array for hexaploid wheat and its secondary and tertiary gene pool. Plant Biotechnol J 14:1195–1206
Winfield MO, Allen AM, Wilkinson PA, Burridge AJ, Baker GLA, Coghill J, Waterfall C, Wingen LU, Griffiths S, Edwards KJ (2018) High-density genotyping of the A. E. Watkins collection of hexaploid landraces identifies a large molecular diversity compared to elite bread wheat. Plant Biotechnology Journal 16:165–175
Wingen LU, Orford S, Goram R, Leverington-Waite M, Bilham L, Patsiou TS, Ambrose M, Dicks J, Griffiths S (2014) Establishing the A. E. Watkins landrace cultivar collection as a resource for systematic gene discovery in bread wheat. Theor Appl Genet 127:1831–1842
Wingen LU, West C, Leverington-Waite M, Collier S, Orford S, Goram R, Yang C-Y, King J, Allen AM, Burridge A, Edwards KJ, Griffiths S (2017) Wheat landrace genome diversity. Genetics 205:1657–1676
Wittern L, Steed G, Taylory LJ, Ramirez DC, Pingarron-Cardenas G, Gardner K, Greenland A, Hannah MA, Webb AAR (2022). Wheat EARLY FLOWERING3 is a dawn-expressed circadian oscillator component that regulates heading date. bioRxiv. https://doi.org/10.1101/2021.09.03.458922.
Würschum T, Rapp M, Miedaner T, Longin CFH, Leiser WL (2019) Copy number variation of Ppd-B1 is the major determinant of heading time in durum wheat. BMC Genet 20:64
Yan L, Loukoianov A, Tranquilli G, Helguera M, Fahima T, Dubcovsky J (2003) Positional cloning of the wheat vernalization gene VRN1. Proc Natl Acad Sci USA 100:6263–6268
Yan L, Loukoianov A, Blechl A, Tranquilli G, Ramakrishana W, SanMiguel P, Bennetzen JL, Echenique V, Dubcovsky J (2004) The wheat VRN2 gene is a flowering repressor down-regulated by vernalization. Science 303:1640–1644
Yu J, Pressoir G, Briggs WH, Bi IV, Yamasaki M et al (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Yu J, Holland JB, McMullen MD, Buckler ES (2008) Genetic design and statistical power of nested association mapping in maize. Genetics 178:539–551
Zanella CM, Rotondo M, McCormick-Barnes C, Mellers G, Corsi B, Berry S, Ciccone G, Day R, Faralli M, Galle A, Gardner KA, Jacobs J, Ober E, Sanchez del Rio A, Van Rie J, Lawson T, Cockram J(2022) Longer epidermal cells underlie a quantitative source of variation for wheat flag leaf size. New Phytologist, online-first. https://doi.org/10.1111/nph.18676
Zhao K, Aranzana MJ, Kim S, Lister C, Shindo C et al (2007) An Arabidopsis example of association mapping in structured samples. PLoS Genet 3:e4
Zhou Y, Tang H, Cheng M-P, Dankwa KO, Chen Z-X et al (2017) Genome-wide association study for pre-harvest sprouting resistance in a large collection of Chinese wheat landraces. Front Plant Sci 6:401
Zhou Y, Bai S, Li H, Sun G, Zhang D et al (2021) Introgressing the Aegilops tauschii genome into wheat as a basis for cereal improvement. Nature Plants 7:774–786
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Glossary
- 2n = 6x = 42, AABBDD
-
n is the gametic chromosome number, 2n is somatic chromosome number. x is the basic chromosome number, which for wheat is 7. Bread wheat is hexaploid with 6 chromosome sets in its genome (6x), termed the AA, BB and DD subgenomes. Thus, a somatic cell of the hexaploid bread wheat genome has a total of 42 chromosomes, summed across its AA BB and DD subgenomes.
- Advanced intercross (AIC)
-
A bi-parental populations, where F2 progeny are intercrossed over one or more generations before the generation of inbred lines.
- Association mapping
-
A method for genetic mapping of QTL that uses historic linkage disequilibrium to associated phenotype to genetic markers. Also known as ‘linkage disequilibrium mapping’.
- Copy number variation (CNV)
-
Differences in the number of copies of a particular gene or chromosomal region. Where there is a presence or absence of a gene/region, it can also be termed presence/absence variation (PAV).
- Genetic recombination
-
The rearrangement of DNA sequences by the breakage and re-joining of chromosome segments.
- Genome-wide association study (GWAS)
-
A method for genetic mapping, using a collection of varieties, landraces or lines from an experimental population with phenotypic and genome-wide genotypic datasets.
- Haplotype
-
A set of DNA markers located sufficiently closely linked on the same chromosome to be frequently inherited as a single unit.
- Linkage disequilibrium (LD)
-
Non-random association of genetic markers at separate loci located that are typically located on the same chromosome.
- Experimental population
-
A population of lines created by crossing two or more founders.
- Multi-parent advanced generation intercross (MAGIC)
-
Experimental populations typically made by intercrossing 4, 8 or 16 founders over multiple generations so that the outputs of the crossing have contributions from each of the founders. Inbred lines are then derived by single seed descent.
- Nested association mapping (NAM)
-
A collection of two or more bi-parental populations, where all individual bi-parental populations share one founder in common (i.e. a single recurrent parent is used). E.g. Founder-1 × Founder-2, 1 × 3, 1 × 4.
- North Carolina II (NCII) model
-
A collection of three or more bi-parental populations, where any single bi-parental populations shares at least one founder in common with any other population, but where two or more recurrent parents are used. E.g. Founder-1 × Founder-2, 1 × 3, 2 × 3.
- Population substructure
-
Presence of a systematic difference in allele frequencies between groups of accessions, due to non-random mating.
- Single nucleotide polymorphism (SNP)
-
A genomic variant at a single base position in a DNA strand.
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2024 The Author(s)
About this chapter

Cite this chapter
Cockram, J. (2024). Genome-Wide Resources for Genetic Locus Discovery and Gene Functional Analysis in Wheat. In: Appels, R., Eversole, K., Feuillet, C., Gallagher, D. (eds) The Wheat Genome. Compendium of Plant Genomes. Springer, Cham. https://doi.org/10.1007/978-3-031-38294-9_15
Download citation
DOI: https://doi.org/10.1007/978-3-031-38294-9_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-38292-5
Online ISBN: 978-3-031-38294-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)