
Abstract
Mass spectrometry is of paramount importance in many studies of pristine and doped helium droplets. Here, we attempt to review the body of work that has been performed in this field. Special focus is given to experiments conducted by the group of Paul Scheier at the University of Innsbruck. We specifically highlight recent studies of highly charged helium droplets and the successive development of pickup into highly charged and mass selected droplets.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


3.1 Foreword and Introduction
Mass spectrometry (MS) has been an invaluable tool in various fields of scientific research since the beginning of the 20th century. Many ground-breaking advances in atomic and molecular physics, chemistry and biology would have been impossible without this technique—research of helium nanodroplets (HNDs) being no exception. This chapter aims to give the reader an overview of mass spectrometric research utilizing HNDs (HND MS). We shall assume that the reader is familiar with, and therefore we will not discuss the basic principles, techniques and instrumentation of MS. We would, however, like to briefly discuss some important concepts for the interpretation of HND mass spectra that might not be as widely known—the experienced reader familiar with cluster and HND MS is encouraged to skip the following paragraph.
The size distribution of cluster ions as observed in a mass spectrum (i.e. the ion yield as a function of m/z) is influenced by various factors, such as the size distribution of the neutral precursor clusters, ionization cross section, ion transmission efficiency of the ion optics and the MS as well as the detector efficiency. While these factors are often difficult to determine, they can be expected to vary rather smoothly over the mass range. Hence, abrupt intensity variations of neighboring cluster ion peaks are unlikely caused by the experimental setup, but rather the intrinsic cluster properties which are known to be able to vary dramatically with the addition or removal of a single atom or molecule [1]. Cluster ion peaks with anomalously increased intensity compared to neighboring peaks are referred to as “magic number” clusters while those with anomalously decreased intensity are called “antimagic” [2–4], in analogy to the concept in nuclear physics [5–7]. The occurrence of magic cluster ions generally requires both a particularly stable cluster structure (neutral or ionic) and an energetic process inducing fragmentation of the emanating clusters or cluster ions. In principle, the variations could be produced by fragmentation during the dopant cluster formation process, however, HNDs are excellent at stabilizing even weakly bound structures via evaporative cooling. Hence, a smooth neutral cluster size distribution can be expected and fragmentation will typically be caused by excess energy transferred to the nascent cluster ion during the ionization process. This results in the depletion of weakly bound clusters and the corresponding relative enrichment of magic number cluster ions [8]. Many of the observed magic numbers are linked to shell closures [1] of geometric [9–13] or electronic nature, the latter being described by the jellium model [3, 14, 15]. Magic numbers are not only found in homogenous clusters, but also in progressions of heterogenous complexes where ions are solvated by different numbers of ligands such as rare gas atoms or small molecules. Again, these magic numbers can often be interpreted in terms of corresponding structures [16, 17] and may be used in the structure analysis of the solvated ion [18–21], for which rare gas atoms and especially He are desirable due to the minimal influence of the weakly bound ligands on the underlying structure. Nonetheless, the innermost ligands are often bound strongly enough to be heavily localized around the central ion in these complexes, which led to the term “snowballs” for ions complexed with He atoms [22–26].
Furthermore, we introduced a few simplifications in order to avoid repetition and for the sake of an easier reading experience:
-
(1)
While 3He is a unique and fascinating species in its own right, it is almost irrelevant in terms of natural abundance and HND MS. Thus, whenever “helium” or “He” is mentioned in this chapter, it refers to the dominant 4He isotope.
-
(2)
Electron ionization (EI) is by far the most common ionization method in HND MS. Thus, when no specific method of ionization is mentioned, EI was used in the discussed experiment.
-
(3)
Deuterium or deuterated compounds are sometimes used in HND MS instead of or complementary to naturally occurring hydrogen or its compounds, achieving mainly two things. For one, the larger spacing between attached D atoms or molecules can make the interpretation of mass spectra easier. In addition, ambiguities in chemical reactions may be resolved, for example one can determine whether a reaction involving hydrogen proceeds via a deuterated dopant or residual water molecules. Since hydrogen and deuterium atoms are chemically very similar, the results are usually transferrable between the two species. Thus, we simply refer to “hydrogen” or “H” in the corresponding discussion if deuterium or deuterated compounds are used in a complementary way and the results are very similar.
We structured our chapter as follows. First, we will present an overview of mass spectrometric work utilizing HNDs performed by groups all over the world. The second part of our chapter focuses on recent advances in mass spectrometry of pristine and doped HNDs by three experiments of our group. While we gave our best effort to include all work that has been performed in the field of HND MS and give credit to the contributors, this is obviously a difficult task due to limited time, space and the imperfect human nature. We want to apologize to any contributors whose name we did not mention or whose contribution we missed entirely. But without further ado, let’s begin!
3.2 History of HND Mass Spectrometry
The first report of a mass spectrometric study of small helium clusters was made in 1975 by van Deursen and Reuss [27]. The authors attempted to answer the question whether a bound state of He2 exists by observing Hen+ up to n = 13. The experiment produced helium clusters in a supersonic nozzle expansion and used a magnetic sector analyzer instrument (MSA) for ion detection. Five years later, Gspann and Vollmar employed time-of-flight mass spectrometry (ToF-MS) to observe metastable excitations of large neutral or cationic HeN clusters and determined their size to be in the range of N = 106–108 [28]. Gspann also observed the ejection of “charged miniclusters” [29] and confirmed the existence of large anionic HeN (N > 2×106) in 1991 using a similar experiment [30]. In 1983, Stephens and King used a free jet expansion cluster source and a quadrupole mass spectrometer (QMS) to record mass spectra of small Hen+ and make the first report of magic numbers in small He cluster ions, finding n = 7, 10, 14, 23 and 30 to be anomalously abundant [31]. Naturally, these first studies attempted to understand HNDs and thus targeted pristine HNDs as well as small Hen cluster (ions), however, this was about to change.
3.2.1 Pioneering Work by the Toennies Group (Göttingen)
Arguably the single most important, ground-breaking discovery in HND research was made in 1990 when Scheidemann and co-workers demonstrated the HND’s ability to capture various foreign atoms and molecules (Ne, Ar, Kr, H2, O2, H2O, CH4 and SF6) [32, 33]. A HND beam was crossed with beams of foreign atomic or molecular species in various configurations and fragments emerging from the doped droplets upon ionization were analyzed using a MSA. The mass spectra revealed that not only were the foreign gas-phase species captured by the HNDs, but they also coagulated and formed clusters of their own as large as (H2O)18, in or on—this was not clear at the time—the HNDs. These discoveries opened up a plethora of possibilities and applications such as HND isolation spectroscopy, growing dopant clusters and studying chemical reactions inside HNDs. The following years saw an unprecedented growth of the field with numerous research teams around the globe picking up research based around HNDs, manifesting in a huge increase in publication numbers in the field.
The described foreign species pickup experiment came out of one of the first and most successful groups conducting extensive studies on pristine and doped HNDs, their properties, ionization mechanisms and applications, the research group for molecular interactions led by J. P. Toennies at the MPI for Fluid Dynamics (now the MPI for Dynamics and Self-Organization) in Göttingen, Germany.

An early HND MS setup from the Toennies group in Göttingen. This basic combination of a HND source followed by a pickup chamber and a MS (in this case two) has been and still is utilized by groups all over the world. This experiment was designed to record mass spectra of small ionic fragments from doped or pristine HNDs and measure the velocity of the HND beam but could easily be modified to be used for laser depletion spectroscopy. Reproduced with permission from Ref. [34]. © copyright AIP Publishing. All rights reserved

Reproduced with permission from Refs. [31] (a) and [35] (b). © copyright AIP Publishing. All rights reserved
a EI mass spectra of small Hen+ from large droplets produced at T0 = 5K, p0 = 20 bar (circles, top trace), recorded with the setup shown in Fig. 2.1. The second mass spectrum (squares, bottom trace) was recorded using a similar setup but much smaller droplets produced at T0 = 4.2K, p0 = 0.5 bar. The most interesting feature is the dominant magic He4+ in the top mass spectrum which is entirely absent in the bottom one, indicating the difference in droplet size. b Mass spectrum showing Hen+ fragments produced via photoionization of small HNDs (<N> = 2100). The close resemblance of the Hen+ distributions produced via EI and PI indicates similarities in the respective ionization processes.
Despite their pioneering work laying the foundation for applications of HNDs in spectroscopy, chemistry and cluster physics, much of the mass spectrometric work conducted in the Toennies group came rather early and was aimed at understanding the relevant characteristics of and processes in pristine HNDs (Fig. 2.1). Two early studies published by Buchenau and co-workers in 1990/91 characterized HNDs with source stagnation pressures p0 between 8–20 bar and source temperatures T0 ranging from 5–20 K by investigating a number of their basic properties [34, 36]. The experiments yielded time-of-flight (ToF) spectra, mass spectra of small Hen+ (n ≤ 30) cluster ions (Fig. 2.2a) and signal intensities as a function of the EI energy. By analyzing these measurements in combination with the He phase diagram, the authors classified different regimes for the formation of HNDs and the ionization/fragmentation processes leading to the ejection of small Hen+ fragment ions. The photoionization (PI) of pure and doped helium droplets was studied by Fröchtenicht et al. in a comprehensive investigation using a synchrotron radiation source and a linear time-of-flight mass spectrometer (ToF-MS) [35]. The authors studied in detail the ionization processes of pristine and SF6-doped HNDs with a wide range of droplet sizes (<N> ~ 102–107) at photon energies between 15 and 30 eV. The recorded mass spectra of small Hen+ were found to be similar to those utilizing EI, with pronounced magic numbers at n = 7, 10 and 14 as previously observed in EI experiments (Fig. 2.2b) [31]. Ion signals of Hen+ and impurities were also found below the ionization potential of free He atoms (<24.6 eV) and assigned to autoionization and Penning ionization processes at energies corresponding to excitations of free He atoms. On the other hand, no direct photoionization of embedded dopant SF6 molecules was observed. These findings led the authors to conclude that both EI and PI proceed in a fairly similar manner in HNDs after being predominantly initiated by the excitation/ionization of a He atom. A set of studies by Lewerenz and co-workers as well as Bartelt and co-workers expanded on the findings of the pickup studies by growing clusters of the heavier rare gas atoms Ar, Kr and Xe, H2O and SF6 molecules [37] as well as the metals Ag, In and Eu [38]. For the non-metals, the emanating cluster ions displayed a Poisson distribution, which is expected for pickup of gas-phase species and subsequent growth of neutral clusters [22, 39]. This indicates that fragmentation due to EI is largely suppressed by efficient cooling of the nascent cluster ions provided by the surrounding HND [37]. In contrast, the mass spectra of metal cluster ions produced in HNDs are fairly similar to those obtained from free cluster beams. The size distribution of silver cluster ions Agn+ show the well-known odd-even oscillations [40] with a clear enhancement of the n = 3, 5, 7, 9 ions [38]. The indium mass spectrum features strong signals of both In+ and In7+. Whereas the latter can simply be explained by an increased stability of In7+, the explanation for the enhanced monomer signal is a little more complex. Indium clusters between 8–20 atoms feature a similar or slightly higher ionization potential compared to the indium atom, thus fragmentation of these cluster is more likely to proceed by ejection of a charged monomer instead of a neutral one [41]. Additionally, the authors concluded that, in contrast to alkali metal atoms and clusters, the observed metal atoms and clusters reside inside the HNDs and should thus adopt the HNDs’ temperature of ~380 mK [42], which would be about 2 orders of magnitude lower than previously observed for free metal clusters [38].
Another interesting topic tackled by the Toennies group are chemical reactions proceeding inside HNDs. In 1993, Scheidemann et al. utilized a MSA to record mass spectra of SF6 molecules embedded in HNDs [43]. In contrast to EI of a beam of free SF6 molecules, where all possible fragments SFn+ (n = 0–5) and F+, but no intact SF6+ are detected [44–47], the HND mass spectrum essentially yields SF5+ as the only fragment and, seemingly, intact SF6+ ions. The authors concluded that the SF6 molecules reside inside the HND, the dominant ionization mechanism is charge transfer from He+ and the expected rich fragmentation of the nascent SF6+ ions is efficiently quenched by the HND. Later, the apparent observation of SF6+ was attributed to an impurity [48], likely SF5+ complexed with a water molecule. In the concluding remarks the authors hint at the potential application of HNDs of studying chemical reactions via the unique possibility of identifying “frozen” reaction intermediates [43]. In 2004, Farnik and Toennies followed up on this seminal idea with a comprehensive study of ion–molecule reactions, very fittingly describing HNDs as “flying nano-cryo-reactors” [49]. The authors utilized HNDs doped with D2, N2 and CH4 in two successive pickup chambers and a MSA to record mass spectra of ions produced in initial charge transfer reactions with He+ and secondary reactions. By monitoring the product intensities as a function of the pickup chamber pressure(s), reaction pathways were identified. The reaction products of the molecules with He+ differ significantly from the gas-phase equivalents. Similar to the SF6 experiment, dissociative charge transfer reactions are largely suppressed by the HND environment. For CH4, the gas phase reaction produces all possible fragment ions except C+ [50], but in the HND environment only CH3+ and CH4+ are detected, which are minor contributions to the total ion yield in the gas phase reaction. For N2, both N+ and N2+ are produced in the gas phase [50, 51], but again, the dissociation reactions appears to be suppressed in the HND, where N2+ is the sole reaction product. The dominating reaction products produced in the D2–doped HNDs are HemD+ (m < 20), produced in a dissociative reaction of He+ and D2, and D3+ formed in a secondary reaction of D2 with D2+, which is initially formed by non-dissociative charge transfer from He+. Other secondary reaction products included CH5+, (from CH4+ + CH4), but not C2H5+ (from CH3+ + CH4), which are both expected from gas-phase experiments [50, 52], as well as CH4D+ and CH3D2+ (from reactions of CH4+ and CH3+ upon additional doping of D2) and N2D+ (from N2+ + D2 upon additional doping of D2). The authors also observed molecular ions complexed with He, such as HemN2+ (m = 1, 2, 4) and HemCH3+ (m =1, 2), which was unexpected at the time, since until then mostly atomic species were observed complexed with He [49].
3.2.2 Review of more recent research
The basic setup of a HND source, a pick-up region and a mass spectrometer proved to be a simple, but efficient way to investigate pure HNDs, dopant clusters, ionization mechanisms and chemical reactions inside HNDs that was soon applied and adapted by several groups to reach their individual scientific goals. Among the first of these groups were those of K. Janda at UC Irvine and V. Kresin at USC Los Angeles.
3.2.2.1 Janda Group (Irvine)
Janda and co-workers used a HND-pickup-QMS setup to study processes following the ionization of small HNDs (<N> ~ 100-20000) in either pristine [53] or doped (NO, Ne, Ar or Xe) [48, 54–56] condition. The study utilizing pristine HNDs showed that the relative intensity of small Hen+ fragments (up to n ≈ 135) did not vary significantly with the average HND size in the range of 100–15,000 He atoms [53]. The authors concluded that small Hen+ originating from larger HNDs are not produced in a thermal process by evaporating excess He, but rather an impulsive process following the formation of a He2+ core, leading to the ejection of small Hen+ from the HND as the ionic core may drag along n–2 additional He atoms. By comparing the dopant and He2+ ion yields for different average HND sizes, Callicoatt et al. [48, 56] as well as Ruchti et al. [54, 55] determined the probability of charge transfer from He+ to the dopant (cluster). It was clearly shown that the probability is highest for very small HNDs (<N> on the order of a few hundred), but decreases gradually for larger HNDs (<N> on the order of a few thousand). While this trend was universally observed, the exact probabilities ranged from a few per cent and unity, depending on the dopant species and dopant cluster size. The authors also estimated the average number of resonant charge hops by a positive He+ hole before self-trapping and localizing as He2+. A relatively simple model yielded a value of 70 for HNDs doped with NO [56], considerably lower than previous estimates on the order of 104 made by Scheidemann et al. [43]. A refined model taking into account polarization arrives at an even lower number of 3–4 hops before charge localization at He2+ or the dopant (Ar) [48], which is fairly close to the most recent estimate of ~10 hops before self-trapping as He2+ [57]. Three studies of HNDs (<N> typically ~1000–3000 He atoms) doped with Ne, Ar and Xe investigated the production of small fragment ions produced upon EI by varying parameters such as the average HND size, dopant pickup pressure and electron energy [48, 54, 55]. The studies of Ne and Ar revealed qualitatively similar patterns where HNDs doped with single atoms produced RgHen+ (Rg = Ne, Ar) with evidence for a shell closure after n = 12 for ArHen+, but no bare ions. On the other hand, HNDs doped with two or more rare gas atoms primarily produced Rg2+ for the smaller and RgHen+ as well as Rg2Hen+ for the larger droplet sizes studied [48, 54]. In contrast, bare Xe+ is much more likely to be formed, both from droplets containing only a single Xe atom as well as more heavily doped HNDs with up to four Xe atoms, which is attributed to an electronically excited state of Xe+ accessible by charge transfer from He+, which does not exist for Ne+ or Ar+ [55]. While the most likely fragment from HNDs doped with two Xe atoms is Xe2+, analogous to Ne and Ar, small complexes XeHen+ (0 ≤ n ≤ 3) are more readily formed than for ArHen+ and no shell closure is evident at n = 12 (a possible shell closure of XeHen+ after n = 17 indicated in the mass spectrum (Fig. 2.3) is not discussed by the authors). Finally, Xe2Hen+ complexes are notably absent for all experimental conditions, which indicates a weaker caging effect of the relatively small HNDs (<N> up to 3300) for Xe2+ compared to Ne2+ and Ar2+, which could be due to the production highly repulsive Xe2+ states that cannot be cooled by the HND.

Reproduced with permission from Ref. [55]. © copyright Royal Society of Chemistry. All rights reserved
Simplified mass spectrum showing the progression of XeHen+ from EI of Xe-doped HNDs. In contrast to NeHen+ [54] and ArHen+ [48], there is a higher yield of complexes with small n, however, there are no immediately obvious features. A possible magic number character of n = 5 and shell closure after n = 17 (marked with asterisks) are not discussed by the authors.

Reproduced with permission from Refs. [58] (a) and [59] (b). © copyright AIP Publishing (a) and American Physical Society (b). All rights reserved
a Ion yields as a function of electron energy for small He and Li complexes and b three earth alkali atoms as well as xenon grown in HNDs consisting of approximately 2000–5000 and 10000 atoms, respectively. The shape of the curves hint at the location of the neutral dopants: whereas the Xe ion yield closely resembles that of He+ and He2+ and is consistent with an interior location, all other curves show distinct evidence of Penning ionization and thus a surface location.
3.2.2.2 Kresin Group (Los Angeles)
Kresin and co-workers introduced a similar HND-pickup-QMS setup to investigate alkali and alkaline earth metals as well as amino acids. Scheidemann, Vongehr and co-workers were able to show that alkali metal atoms and small clusters (Li and Na) are preferably ionized by excited He* via a Penning process, indicating these species, as He*, are located at or near the surface of HNDs (Fig. 2.4a) [58, 60]. This finding corroborated theoretical predictions made for alkali metal atoms (Li–Cs) [61–63] and dimers (Li2 and Na2) [64]. A few years later, Ren and Kresin were able to show that the alkali metal’s neighbors in the periodic table, alkaline earth metals (Mg–Sr), are also located in surface “dimples” (Fig. 2.4b) [59]—this was previously heavily debated, especially for Mg [65–69]. Kresin’s group also performed studies examining the possibilities of controlling amino acid fragmentation in HNDs [70, 71] as well as proton transfer reactions between amino acids in HNDs [72]. Ren and co-workers showed that by co-doping HNDs with water, fragmentation of embedded glycine and tryptophan (albeit to a lesser degree compared to glycine) molecules could be suppressed significantly, whereas the HND environment alone was unable to significantly reduce fragmentation compared to the gas phase [70]. The authors proposed a “charge-steering” effect (also see [73] which will be discussed later on) where charge transfer from He+ is favored to occur on (H2O)n due to attractive forces caused by the water’s relatively large dipole moment, followed by ionization of the amino acid via proton transfer, much softer than direct charge transfer from He+. Ren and Kresin conducted a follow-up study to support this hypothesis using glycine (complexes) as well as alkanes and alkanethiols [71]. They showed that fragmentation patterns of molecules with similar dipole moments as water were practically unaffected, whereas those with lower dipole moments were efficiently protected from fragmentation by the presence of water. Finally, Bellina and co-workers studied the proton transfer reaction in histidine-tryptophan complexes yielding protonated histidine. Additional mass spectra using methyl-tryptophan and indole instead of tryptophan together with density functional theory (DFT) calculations revealed that protonation occurs from the indole to the imidazole side chain of tryptophan and histidine, respectively, at the site of the N–H∙ ∙ ∙N bond between the two functional groups, which can be viewed as a model system for heterodimers between aromatic amino acids [72] ( (Figs. 2.5 and 2.6).

Reproduced with permission from Ref. [74]. © copyright American Physical Society. All rights reserved
Smoothed mass spectrum of MgN produced via PI of doped HNDs (top). Anomalously abundant ion signals indicate magic number clusters. The authors propose a model where electronic shells are filled by the 2N delocalized electrons of the cluster, as illustrated below the mass spectrum. Unexpected shell closures are explained in terms of level reorganization upon increased filling of some shells.

Reproduced with permission from Ref. [75]. © copyright AIP Publishing. All rights reserved
Mass spectra showing bare silver clusters Ag2+Hen and silver ion-snowball complexes produced via PI of doped HNDs. Shell closures at n = 10, 12, 32 and 44 are clear signs of icosahedral-shape solvation shells. The bottom excerpt mainly shows doubly charged silver Ag2+HeN displaying similar shell closures at n = 6, 10 and 12.
3.2.2.3 Meiwes-Broer and Tiggesbäumker Group (Rostock)
One of the first HND MS experiments utilizing a high-resolution time-of-flight mass spectrometer (HR-ToF-MS) was employed by the cluster and nanostructures group of K.-H. Meiwes-Broer and J. Tiggesbäumker at the University of Rostock, Germany. The capabilities of the apparatus originally constructed by the Toennies group were expanded greatly by adding different, versatile laser systems and a HR-ToF-MS (m/∆m ≈ 2000). While the group mainly focuses on spectroscopic studies of various clusters, HND MS was and is frequently utilized in combined studies of metal clusters (for a review of studies of metal clusters in HNDs see [76]). Since the early 2000s, the group has conducted a number of detailed studies on magnesium clusters. Diederich, Döppner and co-workers observed a highly structured Mgn+ abundance distribution with strong magic and antimagic cluster ions in both EI and PI (for a review of PI studies of HNDs see [77]) mass spectra (Fig. 2.5) [74, 78]. While some magic numbers agree well with shell closing predictions of the jellium model, others clearly do not. The authors attempt to explain the latter in terms of a level interchange model, where additional shell closings arise due to electron rearrangement caused by interchange of electronic levels. Furthermore, HenMg+ and HenMg2+ ions with up to n ≈ 150 were observed upon irradiation with femtosecond laser pulses [78]. The authors additionally studied ion snowball formation dynamics using the pump-probe technique (also see discussion on [75] below. In a subsequent study, Diederich and co-workers revisited their level rearrangement model and expanded their work on Mgn+ clusters towards larger clusters (n up to ~2500) as well as dications Mgn2+ (5 ≤ n ≤ 50) [79]. Note that the separation of overlapping isotopic patterns provided by the HR-ToF-MS is crucial for the identification of even-numbered Mgn2+. The authors find that the cluster abundance distributions of Mgn+ display the coexistence of electronic shell closures and geometrical packing schemes at n ≥ 92 between and clear evidence of icosahedral packing for n ≥ 147. Magic numbers of dicationic clusters such as n = 30 rather agree with the standard jellium electronic shell closures known e.g. from cationic sodium clusters [3], with only one level interchange at n = 41 (i.e. 80 electrons) observable within the limited range. Electronic shell effects are also found for smaller clusters of other divalent metals, namely cadmium and zinc. While some features agree with those of magnesium, the authors find the level interchange model inapplicable and clear signs of icosahedral packing such as an enhanced abundance of n = 147 missing for Cdn+ and Znn+. In a number of follow-up studies, the group further exploited the capabilities of their femtosecond laser setup for studying dynamics using the pump-probe technique. Döppner and co-workers extended their study of ion-induced snowballs [78] towards He-solvated Mg and Ag [75], observing He snowballs HenMgZ+ and HenAgZ+ (Z = 1, 2) with up to n ≈ 150 He atoms for the singly charged species upon femtosecond laser ionization with intensities of 1013–1014 W/cm2 (nanosecond PI as well as EI were found to produce similar results). The distributions of HenMgZ+ appear relatively feature-poor with steps at n = 4 (Z = 1) and n = 4, 8 (Z = 2) as well as kinks at n = 19–20 (Z = 1) and n = 10–11 (Z = 2), the latter of which are interpreted as closures of the first, liquid-like solvation shells [80]. On the other hand, the distributions of HenAgZ+ are much more structured and display indications of icosahedral packing schemes with pronounced steps at n = 10, 12, 32 and 44 (Z = 1) as well as n = 6, 10 and 12 (Z = 2). A subsequent study of HenPbZ+ finds magic numbers of n = 12 and 17 (Z = 1) as well as n = 12 (Z = 2), indicating a closure of the first shell at 17 and 12 He atoms, respectively [81]. In both cases, a higher charge state likely leads to a stronger interaction, resulting in smaller, more tightly bound structures. He-solvated ions are found to preferably form in HNDs predominantly doped with single metal atoms or small clusters [75]. Additionally, the authors study the dynamics of snowball formation and fragmentation of larger Mg and Ag clusters upon femtosecond laser ionization using the pump-probe technique. Increasing the pump-probe delay reveals a strong decrease in the yield of larger, multiply charged clusters with a minimum at ~30 ps, mirrored by a simultaneous increased production of HenAg+. Both signals are found to fully recover at pump-probe delays of ~100 ps. The first part of the signal progression up to 30 ps can be understood in terms of intense heating and fragmentation of the initial cluster, also leading to an increased number of single atoms/ions or small clusters being present in the HND and thus promoting snowball formation. The recovery of signals clearly illustrates the cage effect of the He environment, allowing fragments to dissipate excess energy and eventually recombine to form once again larger clusters and fewer snowballs. Similar trends are observed for Mg clusters, although on shorter timescales with local extrema located around 7 ps. Three studies by Döppner and co-workers further used the pump-probe technique to perform an extensive investigation of plasmon enhanced ionization of metal clusters (Ag, also Cd and Pb in [81]) embedded in HNDs, previously studied in free metal clusters [82]. The authors studied in detail how to manipulate the ion yield and maximum charge state of highly charged metal ions via laser pulse width and field strength [83], delay between two identical pulses [84] as well as a combination of all the above, additionally introducing asymmetry to dual delayed pulses [81]. Narrower pulses, stronger laser fields [83] and shorter delays in between pulses [84] are found to promote production of more highly charged ions, as is cluster irradiation by an initial weaker pulse, followed by a stronger one [81]. The highest achievable charge states were Z = 11 (Ag) [83, 84] and Z = 13 (Cd) [81]. Expanding on these experiments, Truong and co-workers utilized an ultrafast pulse shaper [85] and a feedback algorithm in order to further substantially increase the yield of highly charged AgZ+ ions, producing charge states up to Z = 20 [86]. The authors found the ideal pulse shape to have a double pulse structure, where a weaker (~ 15% intensity) pre-pulse is followed by a main pulse around 140 fs later, in good qualitative agreement with computations based on the nanoplasma model by Ditmire and co-workers [87]. A spectroscopic study by Przystawik and co-workers found that in a HND environment, Mg does not form classical clusters, but rather agglomerate in loosely bound, metastable complexes (‘foams’), each Mg atom separated by an estimated ~10 Å with a layer of He in between [88]. The proposed structure is in good agreement with a subsequent computational study by Hernando and co-workers [89] and found to collapse into hot, compact clusters upon laser excitation on a timescale of 20 ps [88]. This collapse was studied in detail by Göde and co-workers using femtosecond dual-pulse spectroscopy [90]. Mass spectra of Mg-doped HNDs subject to femtosecond multiphoton ionization (MPI) reveal Mgn+ (n ≤ 20) with enhanced signals of Mg5+ and Mg10+ as well as HenMg+ snowballs, in accordance with previous studies [74, 81]and [81], respectively. By studying the response of the various ion signals to changes in delay and intensity ratio of the dual femtosecond pulses, the authors were able to unravel the dynamics of cluster, snowball and even electronically excited, neutral complex (exciplex) formation following the light-induced collapse of Mg foams in HNDs.

Reproduced with permission from Ref. [91]. © copyright AIP Publishing. All rights reserved
Cluster size distribution of mixed potassium-cesium clusters from PI of co-doped HNDs as a function of total number of atoms. It appears that, to an extent, Cs atoms can replace K atoms in KN+ and still preserve the characteristic features of the cluster size distribution. However, the ion yield quickly drops off upon addition of further Cs atoms.
3.2.2.4 Stienkemeier and Mudrich Group (Freiburg)
Similar experiments to those of Meiwes-Broer and Tiggesbäumker were conducted since the early 2000s in Germany by F. Stienkemeier (University of Freiburg, previously University of Bielefeld), M. Mudrich (University of Freiburg, now University of Aarhus) and co-workers. While the group specializes in spectroscopic techniques, important mass spectrometric contributions involving alkali metals and PI of HNDs were also made. The basic setup consisted of a HND source (typically used to produce droplets with 5000–20,000 He atoms), femtosecond PI and QMS analysis. In an early investigation employing this setup, Schulz and co-workers studied complexes of up to 25 alkali atoms (Na and K), which were found to aggregate into highly spin-polarized, weakly bound van-der-Waals complexes instead of covalently bound or metallic clusters [92]. Upon PI of the alkali-doped HNDs, the authors observe the collapse and fragmentation of the system, evident in mass spectra featuring an exponential decrease of clusters sizes and displaying well-known characteristics such as pronounced odd-even oscillations and magic numbers (e.g. n = 5, 9, 21) corresponding to electronic shell closures. The authors observe a maximum cluster ion size of n = 3 and 5 for Rb and Cs, respectively, interpreting it as evidence for a lack of stable high-spin states. Droppelmann and co-workers expanded the investigation towards HNDs co-doped with two alkali metal species (lighter Na or K with heavier Rb or Cs) [91]. The authors observed pure clusters of both species as well as significantly less abundant mixed clusters upon PI of the doped HNDs, regardless of pickup order (Fig. 2.7). Species appear to be interchangeable to a degree, evident in the abundance/stability patterns of mixed clusters which appear to be governed by the total number of atoms (i.e. valence electrons), once more reproducing known odd-even oscillations and magic numbers such as n = 5 and 9. Incorporation of increasing numbers of heavier alkali metal atoms, however, tends to destabilize mixed clusters, resulting in quickly decreasing ion signals which is attributed to second-order spin orbit interaction. In two follow-up studies, Müller and co-workers shifted their focus towards helium snowballs around cationic alkali metal atoms and dimers (Na-Cs, Na2 and Cs2) [93] as well as reactions between alkali metal atoms (Na, Cs) and water [94]. The authors observed snowball formation with up to 3 and ~10 He atoms around light alkali cations (Na and K, respectively), with much further progressions of up to ~40 He atoms attached to the heavier Rb+ and Cs+ [93]. Additionally, weak signals of small snowballs around the dimers Na2+ and Cs2+ are detected. Local anomalies at n = 4 for K+Hen and n = 12 for Cs+Hen, hint at especially stable, possibly ring-like (cf. Mg2+ and Ag2+ [83]) and icosahedral structures, respectively. Shell closures are indicated at n = 14 for Rb+Hen as well as n = 16 for Cs+Hen. Generally, snowball formation is found to be favored around ion fragments produced from larger, multiply charged clusters rather than single atoms. In a separate study, reaction products of Na or Cs with H2O were observed upon PI of co-doped HNDs [94]. Whereas Na was found to form primarily weakly bound van-der-Waals complexes Nam(H2O)n, mass spectra of HNDs co-doped with Cs and H2O revealed a variety of compounds indicating efficient chemical reactions between Csm and (H2O)n prior to PI.

Another group utilizing HND MS in the early 2000s consisted of W. Lewis, R. Miller and co-workers who performed detailed studies of pickup and ionization processes in HNDs, developing clever techniques for manipulating dopant species in HND MS experiments along the way. In an early experiment, Lewis and co-workers employed a combination of a QMS and a Threshold PhotoElectron PhotoIon COincidence (TPEPICO) setup to study and control the fragmentation of triphenylmethanol and quantify the energetics of HND cooling [96]. In a subsequent study, Lewis and co-workers expanded the capabilities of the basic QMS setup by introducing an IR laser. The authors demonstrated the possibility of selecting specific isomers of dopant complexes in neutral HNDs using a technique called “optically selected mass spectrometry” (OSMS) [73]. By irradiating the HND with an IR laser tuned to an isomer-specific vibrational transition, the dopant complex is heated, resulting in the evaporation of He atoms from the droplet and a corresponding reduction of the EI cross section which manifests as a depletion of the ion signal in the mass spectrum. Using this technique to study the charge transfer processes in HNDs doped with HCN, HCCH and HCCCN (Fig. 2.8), the authors show that the charge transfer probability from He+ to a dopant molecule (or complex) is heavily dependent on the latter’s dipole and higher electrostatic moment(s). The authors explore possibilities of the developed techniques such as isomer selective mass spectrometry and controlling fragmentation patterns of complexes by charge-steering [73] and expand on the described findings in two follow-up studies of non-thermal ion cooling [97] as well as ionization and fragmentation processes in HNDs [95]. A different method of forming ionic complexes in HNDs, avoiding the large amounts of energy transferred to a dopant during ionization via He+ (or He*) and concomitant fragmentation, was developed by Falconer et al. The authors doped HNDs doped with Na+ ions and only then performed pick-up of neutral molecules, followed by desolvation to extract gas-phase analytes from the HNDs. The mass spectra revealed sodiated ion-molecule clusters [Na∙Mn]+, where M = H2O, HCN or N2, and could be explained by pickup statistics, suggesting no fragmentation occurs [98]. Lewis et al. also developed a calorimetry technique able to determine the binding energies of moderately to strongly bound clusters such as (H2O)n and Cn by observing the threshold HND size necessary to observe a certain cluster ion using a ToF-MS setup [99, 100].
3.2.2.5 Ellis and Yang Group (Leicester)
Most experiments produce HNDs via continuous (cw) expansion of pressurized, cold He into vacuum. In fact, all HND sources were continuous until 2002, when the first pulsed HND source was reported by Slipchenko, Vilesov and co-workers [101]. Pulsed sources achieve a much higher HND flux, have distinct advantages in combination with elements such as pulsed lasers and generally reduce pump load and consumption of high-purity He [101–103]. Besides technical issues like additional heat load on the nozzle region due to the valve operation, it quickly became apparent that pulsed HND sources generally behave differently from cw sources, so that well-established knowledge such as scaling laws could not easily be transferred. The pulsed HND source of Slipchenko et al. could only produce HNDs within a narrow average size range between 20000 and 70000 He atoms. The design was adapted by A. Ellis, S. Yang and co-workers in an attempt to improve the performance of pulsed HND sources. By experimenting with different nozzle shapes, the authors were successful in both widening the accessible size range of HNDs produced and achieving a more predictable behavior with varying stagnation pressure and nozzle temperature [104]. The authors utilized ToF-MS in combination with H2O and toluene doping to determine the average HND sizes and extract a scaling law, which suggests that the effect of stagnation pressure on the produced HND sizes is negligible, albeit in a limit temperature and pressure range (T = 10–16 K, p = 8–20 bar). Later, Yang and Ellis extended their studies to show that HNDs produced in a pulsed source exhibit velocity dispersion according to their size [105], in contrast to cw sources where the velocity spread of differently sized HNDs was found to be uniform for a given set of source conditions [36, 106]. The authors highlight the possibility of probing differently sized HNDs by simply probing the droplet beam at different times instead of changing the source conditions, as in a cw source. Yang, Ellis and co-workers were very productive in utilizing their pulsed HND source and ToF-MS setup to study a number of topics such as ionization and fragmentation/dissociation dynamics, atomic and molecular clusters and ion-molecule reactions. A summary of these extensive and rich experimental studies is attempted below.
The ionization of small dopant molecules via He+ charge transfer and subsequent fragmentation and dissociation products were investigated for several alcohols (C1–C6) and ethers [107], haloalkanes (C1–C3) [108] and diatomic molecules (O2, CO and N2) [109]. The authors found that for the small to medium-sized alcohols and ethers as well as the haloalkanes studied, the HND environment did alter the fragmentation patterns compared to the gas phase, but mostly quantitatively, enhancing certain channels like H-abstraction. Apart from the cyclic C5- and C6-alcohols, the parent ion remained a minor product, i.e. the HND was unable to prevent the excessive fragmentation. The authors concluded that while EI of doped HNDs could not be considered a soft ionization method suited for analytical mass spectrometry for the rather small molecules studied, it might still be worthwhile for larger species such as typical biomolecules [107, 108]. A similar, extensive study was carried out by Boatwright and co-workers on clusters of small molecules such as aliphatic alcohols (C1–C3), several halomethanes and inorganic triatomic molecules (H2O, SO2 and CO2) embedded in HNDs [110]. Again, the mass spectra suggest that the EI-initiated chemistry of both clusters and at least one of the single molecules differs significantly from gas-phase studies and proceed via direct bond fission processes instead of ion-molecule chemistry. The findings further support the authors’ conclusion that EI of HNDs doped with small molecules and their cluster cannot be considered a soft ionization route [110]. In contrast to the small molecules discussed so far, EI of HNDs doped with the diatomics O2, CO and N2 show a significant reduction of dissociation (fragmentation) compared to the gas phase reaction between the diatomic species and He+ [109]. The authors consider two possible explanations: suppression of the dissociation channel in the ion-molecule reaction by the HND or acting as a reservoir, allowing for recombination of the products after initial dissociation. In order to determine which is the case, the energetics of the ion-molecule reactions and the corresponding amount of He evaporation to dissipate the energy difference were calculated. The authors conclude that while the dissociation reaction itself must be suppressed in the case of O2 and CO, no conclusion can be drawn for N2 from the calculated energetics [109]. Shepperson and co-workers investigated the formation of small Hen+ cluster ions from HNDs with different sizes (average droplet size <N> between 4000 and 90000) and dopants (pristine, H2O and Ar) [111]. The authors find that the Hen+ / He2+ signal ratio increases with increasing HND size, reaching an asymptotic limit at around <N> = 50000. The authors conclude that larger HNDs favor the formation of Hen+ (n > 2) from He2+ due to the larger number of collisions of the latter and surrounding He atoms on its way of leaving the droplet. The introduction of Ar mainly leads to the asymptotic value being reached at a significantly smaller <N> = 10000, explained by a potential energy gradient, steering the charge hopping of the initially formed He+ (before formation of He2+) towards the impurity, which is most likely located close to the droplet center. The introduction of H2O also lowers <N> where the asymptotic limit is approached, although not as clearly. More interestingly though, the limit is approached from the opposite side, i.e., with H2O as a dopant, the Hen+ / He2+ signal ratio decreases with increasing <N>. The authors propose that the dipole moment creates as stronger gradient, which makes it likely that He2+ is formed in the vicinity of the impurity, additionally lowering the kinetic energy of the He2+ on its way to leaving the droplet, thus lowering the collision energy with He atoms, increasing the chance of additional He attaching to form Hen+ (n > 2) [111].
The first studies on clusters formed inside HNDs by Yang, Ellis and co-workers were performed on small molecules M such as aliphatic alcohols (C1–C5) [112] and H2O [113], using the pulsed HND source and ToF-MS setup. In both cases, mass spectra differed significantly from gas phase experiments, displaying higher abundances of unfragmented cluster ions Mn+, besides the dominant protonated [MnH]+ ions. Other important signals were due to dehydrogenated alcohol clusters [(ROH)n–H]+ and intact, He-tagged water clusters [(H2O)nHe]+. The emergence of the unfragmented, dehydrogenated and He-tagged species are attributed to the efficient cooling/quenching effects of the HND environment. In a follow-up study, Liu et al. formed and studied core-shell clusters of water and several different co-dopant species (Ar, O2, N2, CO, CO2, NO and C6D6), with similar results for both orders of doping [114]. Both binary and pure water clusters are observed in the mass spectra. The authors focused on pure water clusters and describe a “softening” effect on the charge transfer reaction for co-doping with non-polar molecules (O2, N2, CO2, and C6D6). The softer ionization process manifested in a higher ratio of intact water cluster ion signals (H2O)n+ compared to protonated (H2O)nH+ for all cluster sizes. The effect strength increased in the order N2, O2, CO2, C6D6 and is explained in terms of energy dissipation via evaporation of the co-dopant species. The different strength of the softening effect is caused by an interplay of the molecules’ dipole polarizabilities (i.e. binding energy to an ionic core), vibrational degrees of freedom and ionization energies. Whereas argon is fairly similar to the pure HND environment, showing no significant softening effect, the influence of the polar molecules CO and NO is more complicated. While a weaker softening effect is observed for some water cluster sizes, in most cases, fragmentation of intact water clusters to protonated (H2O)nH+ is actually enhanced by the presence of CO and NO. It is suggested (H2O)nH+ production is enhanced by CO and NO readily accepting OH radicals to form the stable HOCO and HONO in secondary ion-molecule reactions, which is supported by a subsequent ab initio theoretical study of Shepperson and co-workers [111].

Reproduced with permission from Ref. [115]. © copyright Royal Society of Chemistry. All rights reserved
Mass spectrum demonstrating the formation of binary gold/silver clusters grown in HNDs (<N> = 16000). The largest detected cluster ion consists of seven Au and four Ag atoms. Weak odd-even oscillations as well as distinct magic number clusters (e.g. AuAg2+, Au2Ag3+, Au3Ag2+, …) can be found, however, these are not further analyzed by the authors since the main scope of the study was the synthesis, deposition and ex situ of larger nanoparticles.
In addition to the previously discussed pulsed HND source and ToF-MS setup, the group of Yang and Ellis recently employed a different setup consisting of a classical cw HND source and a QMS, using it to conduct two studies on metal clusters, namely bi-metallic core-shell nanoparticles [115, 116]. In the former study, two temperature scan series illustrate the influence of HND source and oven temperatures on the yield of Nin+ cluster ions from EI of doped HNDs. While a certain size is required to ensure the pickup of multiple Ni atoms, charge transfer to the dopant cluster becomes less likely in larger droplets. Similarly, a certain dopant partial pressure is required for the capture of multiple Ni atoms, but HNDs are quickly evaporated by collisions if the pressure is too high. Additionally, a mass spectrum shows that mixed Au/Ag cluster cations with up to seven Au and six Ag atoms are formed (Fig. 2.9). Furthermore, various produced nanoparticles (Ag, Ni, Au, Ag/Au and Ni/Au) were also deposited on a substrate for ex situ analysis. Transmission electron microscope (TEM) images show that Ag-Au nanoparticles with a diameter of a few nm formed in HNDs adopt a crystalline structure, but do not allow conclusions about their structure. Evidence for a core-shell structure of Ni-Au nanoparticles is found via the absence of any Au 4f shift in X-Ray Photoelectron Spectroscopy (XPS) spectra characteristic for an Au/Ni alloy. A recent study by Spence and co-workers performed on the same setup demonstrates the formation of various aluminum cluster ions such as Aln+ (n ≤ 15), [(Al2O)Aln]+(n ≤ 12) as well as Al+Hen (n ≤ 17) and Al2+Hen (n ≤ 6) [116]. These observations contrast a previous study by Krasnokutski and Huisken, where no formation of Al clusters inside HNDs was observed [117]. Spence et al. suggest that inadequate pickup conditions prevented the formation of clusters in the experiment of Krasnokutski and Huisken [116]. The cw apparatus was further used to investigate complexes of and ion-molecule reactions between organic molecules and noble metal atoms in two recent studies. In the first study, complexes of tetrapyridyl porphyrin (5,10,15,20-tetra(4-pyridyl)porphyrin, H2TPyP) and gold were formed and analyzed by Feng and co-workers [118]. The mass spectrum of H2TPyP-doped HNDs shows excessive fragmentation of the molecules. Upon co-doping with Au, complexes of H2TPyP (fragments) and Au are detected and the H2TPyP fragmentation is generally reduced. The authors assume that Au atoms and small clusters attached to H2TPyP can dissipate excess energy from the charge transfer process and might additionally introduce a charge-steering/buffering effect where Au can act as a “buffer” in a sequential charge transfer (see earlier discussion about co-doping of amino acids and water by Ren et al. [70, 71]). The effect appears to be even stronger when Au is picked up after H2TPyP, attributed due to enhanced binding of Au (clusters) to multiple sites of H2TPyP instead of attachment of a previously formed Au cluster to a single site of H2TPyP. Additionally, the composition of fragments is altered, e.g. some protonated fragment channels and fragment “dimer” channels are clearly weakened. The second study by Sitorus and co-workers observed dissociative ion-molecule reactions in complexes of 1-pentanol and 1,9-decadiene, co-doped with gold or silver [119]. The mass spectra show a number of fragments as well as complexes of the organic molecule (fragments) and Au/Ag. However, in contrast to the previously discussed study, little to no softening effects on the fragmentation pattern of the organic molecules were observed upon co-doping with Au/Ag. An exception was the case of 1,9-decadiene and Au, where fragmentation was found to be efficiently reduced, especially for small fragments. The authors found that only for 1,9-decadiene and Au, the ionization potential of the metal atom was higher than for the organic molecules (Ag: 7.6 eV, Au: 9.2 eV [120], 1-pentanol: 10 eV [121] and 1,9-decadiene: 8.6 eV [119]), concluding that this was a prerequisite for a softening effect to occur.
3.2.2.6 Ernst Group (Graz)
The group of W. Ernst at TU Graz is dedicated to the study of optical and catalytic properties of metal clusters grown in HNDs. Whereas analysis is mostly performed using spectroscopy and microscopy methods, the group employs a ToF-MS as well (Fig. 2.10). While its primary use is the in-situ monitoring of cluster growth, some mass spectrometric studies involving alkali and earth-alkali as well as other metal species have also been conducted. Theisen and co-workers studied the submersion of and snowball formation around alkali metal ions (Rb and Cs) in HNDs by resonant two-photon ionization (R2PI) involving initial excitation to selected, non-desorbing states [122, 123]. The authors observed small Rb+Hen snowballs (up to n ~ 20) as well as intense ion signals at heavier masses, which are attributed to large Rb+Hen snowballs (n > 500) and well-described by a log-normal distribution [122]. While observations made for Cs are generally similar, Cs2+Hen snowballs are also detected. The authors tentatively assign possible shell closures of Cs+Hen around n = 17 and 50 while missing a noteworthily enhanced abundance of Cs+He12 [123]. A follow-up study by Theisen and co-workers covered in detail the ionization process for Rb and Cs at HNDs via R2PI and determined the (slight) lowering of ionization thresholds due to the HND environment [124]. Theisen and co-workers also expanded the studies of Schulz and co-workers on high-spin alkali clusters [92] towards larger oligomers Rbn+ and Csn+ with up to n = 30 and 21 atoms (in contrast to five and three in the previous study), respectively [125]. Whereas the authors observed these larger clusters using single-PI with photon energies of 3.3 and 4.1 eV, no ions larger than trimers were detected using multi-PI at 1.4 eV. Familiar magic numbers such as n = 5, 9 and 19 are found for both species, with additional anomalies at n = 13, 21 and 28 for Rbn+ [126] as well as n = 15 for Csn+. Barring explicit evidence, the authors consider several models of cluster formation to argue that neutral alkali clusters should be able to form in both high and low spin states on the larger HNDs (<N> ~20,000) used in the experiment. A cleverly constructed setup allowing for in-situ monitoring (EI/ToF-MS) of NPs while depositing them on a substrate for ex-situ analysis by methods such as electron microscopy was employed by the group in 2015. The setup was first used by Thaler and co-workers in an extensive study of metal (Ni, Cr or Au) nanoparticle (NP) formation of up to ~500 atoms (diameters up to 2 nm) in large HNDs (N = 105 to >108), with good agreement between in-situ and ex-situ analysis [127]. The presented mass spectra extending to m/z ~20,000 show well-known features of metal clusters such as odd-even oscillations and magic numbers corresponding to highly geometric structures such as a pentagonal bi-pyramid (Cr7+) or an icosahedron (Cr13+). Magic numbers of Au clusters are also evident throughout the mass spectrum (Fig. 2.11), but were not further analyzed by the authors. A more detailed analysis of small Aun+ (n ≤ 9) produced via R2PI can be found in [128] in the frame of a spectroscopic study. Messner and co-workers observe pronounced odd-even oscillations with even-numbered species practically absent, except for the dimer. Further mass spectrometric work can be found in studies that have a different general focus such as spectroscopy or characterization of an evaporation source. Mass spectra (EI/QMS) of neat and hydrated Crm(H2O)p+ (m ≤ 9, p = 0, 1), Cun(H2O)p+ (n ≤ 7, p = 0–2) as well as the detection of small, mixed CrCun+ (n = 1–3) were reported by Ratschek and co-workers [129] as well as Lindebner and co-workers [130]. Krois and co-workers present a mass spectrum demonstrating the efficient production of RbSr molecules via (R)2PI of co-doped HNDs in their spectroscopic study of the molecule [131]. Complexes of Li such as Li2+, LiHen+ (n ≤ 3) and LinOH+ (n = 1, 2) were observed by Lackner and co-workers in a laser spectroscopy study [132]. The detection of the latter species and the concurrent absence of Lin(H2O)p+ is interesting since Müller and co-workers observed the opposite in HNDs co-doped with Na and H2O [94].

source chamber (a), doping chamber equipped for sequential doping from various sources (b) and analysis chamber (c) containing a ToF-MS (10) with possibilities for both PI and EI as well as a substrate holder (11) and a quartz microbalance (12). Reproduced with permission from Ref. [127]. © copyright AIP Publishing. All rights reserved
Setup used by the Graz group for the production, in situ analysis and deposition of metal clusters. The main parts of the experiment are the

Reproduced with permission from Ref. [127]. © copyright AIP Publishing. All rights reserved
Mass spectrum of large gold clusters extending beyond 100 Au atoms demonstrating the capabilities of the setup shown in Fig. 2.10. The inserts show the prominent odd-even oscillations of small cluster ions and the resolution towards high masses exceeding m/z = 15000.

Reproduced with permission from Ref. [134]. © copyright American Astronomical Society. All rights reserved
Differential mass spectra from a study probing possible astrochemically relevant pathways of glycine formation in HNDs. The top figure shows the effect of adding atomic 13C to HNDs doped with two different gas mixtures (top, gas 1: 3:2:1 mixture of NH3, H2 and CO2, gas 2: ~2:1 mixture of H2 and CO2) The bottom figure shows the same mass spectrum for gas 1, overlaid with a gas-phase EI mass spectrum of glycine [133].
3.2.2.7 Krasnokutski and Huisken Group (Jena)
One of the most intriguing applications of HNDs as a “nano-cryo-reactor” is the possibility to study astrochemically relevant processes in the laboratory. F. Huisken, S. Krasnokutski and co-workers successfully employed a HND source and a QMS to conduct several clever mass spectrometric studies of such processes in the last decade. Additional techniques such as calorimetry (via shrinking of HNDs due to evaporation) and chemiluminescence (CL) measurements were used to complement mass spectrometry. The first studies investigated reactions of astronomically relevant atomic species (Mg, Al, Si and Fe) with O2, and in some cases H2O and C2H2 as well. Krasnokutski and Huisken performed an extensive study combining mass spectrometry and CL measurements, which found that single atoms of Mg react with O2 in the HND environment, but the most likely product MgO2 is expelled from the droplet, preventing detection by MS [135]. If on the other hand, Mg clusters were allowed to form prior to the reaction with O2, a number of MgxOy+ (x ≤ 4, y ≤ 2) compounds were detected in the mass spectra. The authors unexpectedly find that these reactions occur faster than 5 × 104 s–1 (with some ambiguity about the influence of the HND environment) suggesting a need to include this reaction in astrochemical modelling. Similar experiments were conducted on the reactions of Si with O2 (and their respective clusters) as well as H2O [136]. In a calorimetric approach, Krasnokutski and Huisken utilized pressure measurements to monitor the evaporation of HNDs due to the energy released during the reaction of Si and O2. The authors conclude that the entrance channel is barrierless and calculate the lower limit of the reaction rate to be 5 × 10–14 cm3 mol–1 s–1, indicating relevance in interstellar environments. Binary complexes SixOy+ (x ≤ 2, y ≤ 3) were detected in the mass spectra when Si clusters were allowed to form in larger HNDs (NHe ≥ 15,000). While SiOH+ are detected when droplets are doped with Si and (residual) H2O, the authors attribute this reaction to occur after EI of the doped HND. A subsequent study employs similar methods to study reactions of Al with O2 and H2O [117]. The authors found that Al atoms do not coagulate to form regular clusters in HNDs, a conclusion that has since been disputed (see [116] and previous discussion in this chapter). Despite this possible mishap, further findings are likely valid, such as the reaction of Al with O2 to form AlO2 occurring in HNDs, detection of AlxOy+ (x ≤ 2, y ≤ 3) in mass spectra of larger HNDs and the nonreactivity of single Al with H2O, whereas reactions occur if any of the species is allowed to form a cluster. Last in this series of studies is the exploration of iron reactivity with O2, H2O and C2H2 [137]. Combining mass spectrometry, R2PI spectroscopy [138] and quantum chemical simulations, the authors find that Fe atoms undergo reactions with the reactant species, but only form weakly bound complexes without significantly altering the molecular geometries. Since these complexes easily dissociate, weak bonding of Fe to interstellar ice and dust grains is also indicated.
In their most recent studies, Krasnokutski and co-workers employed a home-built atomic carbon source [139] to tackle two hot topics in astrochemistry—the chemical evolution of polycyclic aromatic hydrocarbons (PAHs) and the formation of glycine in astrophysical environments. PAHs are found to be a ubiquitous component of organic matter in space [140], suggested to be responsible for the IR emission features in the 3-to-15 µm range that dominate spectra of most galactic and extragalactic sources [140–143] and considered to be carriers of the diffuse interstellar bands (DIBs) [144, 145], a number of features in the UV-vis-IR-range first reported in 1922 by Heger [146], but remain largely unidentified to date except for a few bands recently attributed to C60+ [4, 147–150]. Motivated by the poorly understood chemical evolution of PAHs in astrophysical environments [143], Krasnokutski and co-workers investigated the reaction of C atoms with (deuterated) benzene [151] and PAHs (naphthalene, anthracene and coronene) [152] embedded in HNDs. Whereas the gas phase reaction C6D6 + C dominantly yields C7D5 [153, 154], mass spectra recorded by Krasnokutski and Huisken suggest that in the HND environment (NHe ≥ 4000), the reaction intermediate C7D6 is stabilized as the lone reaction product. Calorimetry measurements show that stabilization requires the dissipation of ~2.75 eV, considered feasible to occur on the surface of cold interstellar dust grains by the authors. Quantum chemical calculations reveal that the C atom is inserted into the aromatic ring, resulting in a seven-membered ring structure. Expanding their efforts to larger aromatic hydrocarbons, Krasnokutski and co-workers performed a similar study on naphthalene, anthracene and coronene [152]. Like the benzene study, mass spectra and calorimetry measurements show barrierless reactions resulting in the formation of C(PAH) in all three cases, however, the energy release detected via calorimetry is considerably less in the case of coronene. Quantum chemical calculations show that C atoms initially attach to CC bonds, but the binding energies for interior CC bonds (i.e. ones shared between two aromatic rings) are found to be < 1 eV, significantly lower than for peripheral CC bonds (i.e. belonging to form a single aromatic ring). This affects the subsequent reactions—whereas a C atom initially attaching to a peripheral bond leads to a ring opening and insertion into the carbon network to form a seven-membered ring structure in all examined PAHs, a ring opening is not possible for initial attachment to an interior CC bond. Consequently, the basic structure of the PAH is largely preserved, with a weakly bound out-of-plane carbon attached. The latter case is associated with a lower energy release, which, considering the higher ratio of interior versus peripheral CC bonds in larger PAHs such as coronene, explains the lower energy release evidenced by calorimetry measurements. The authors point out implications of their findings for the growth and destruction of PAHs in astrophysical environments. The described formation of seven-membered ring species from the reaction of “classical” PAHs) with atomic carbon could lead to a large number of “nonclassical” PAHs and furthermore facilitate formation and bottom-up growth of small PAHs by sequential reactions with carbon and hydrogen atoms. On the other hand, PAHs with a higher number ratio of interior to peripheral CC bindings, i.e. larger and more compact species are becoming increasingly chemically inert towards reactions with atomic carbon, limiting their growth.
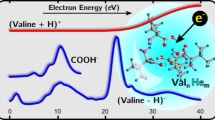
The latest study discussed here covers glycine (Gly), the simplest of amino acids, essential building blocks of proteins and imperative for the emergence of life. While Gly was detected in comets [155, 156] and meteorites [157], there is no convincing observational evidence for the existence of Gly in the interstellar medium yet. Lacking observational evidence, possible astrochemical formation pathways of Gly are being assessed in experimental and theoretical efforts. Chemical surface reactions occurring on ice surfaces of interstellar dust particles are thought to facilitate the formation of a large number of organic species, including amino acids [158–160]. Krasnokutski, Jäger and Henning studied two possible low-temperature routes of Gly formation considered feasible to occur on such interstellar dust particles by means of HND MS, calorimetry and theoretical calculations [134]. Both NH3 and H2 were calorimetrically shown to perform barrierless and highly exoergic reactions upon addition of C atoms, forming CH2NH and HCH, respectively, precursors for the formation of Gly. The pathway HCH + NH3 + CO2 \(\rightarrow\) Gly was investigated theoretically by calculating the potential energy surface of the three-body reaction, revealing a barrierless reaction pathway of Gly formation. Experimentally, the reaction was studied by adding C atoms to clusters of H2, NH3 and CO2 grown in HNDs and analyzing the reaction products via EI of HNDs and MS (Fig. 2.12). While some evidence for the formation of Gly was found, the results were not entirely conclusive due to low ion signals and numerous interference mass peaks on the positions of the parent and typical Gly fragment ions. The identification of mass peaks could be improved by using a high-resolution ToF-MS instead—e.g. for the mass spectrometric separation of NH213CH2COOH+ (m/z 76.035) and He19+ (m/z 76.049), a mass resolution of \(R=\frac{m}{\Delta m} \sim 5000\) is required.
3.2.3 Mass Spectrometry as a Complimentary Tool
Many groups conduct HND experiments that use MS as a complementary tool while relying on other techniques such as spectroscopy to achieve their scientific goals. Consequently, many authors have occasionally published MS studies or included valuable mass spectrometric work in studies with a different main focus. We attempt to summarize this body of work in the following section.

Reproduced with permission from Ref. [165]. © copyright AIP Publishing. All rights reserved
Average helium droplet sizes <N> as produced in a cw expansion (5µm nozzle diameter, p0 = 20 bar) and measured with different methods. The results of the titration method used by Gomez et al. is shown for two collision gases as filled squares (He) and circles (Ar). Early deflection measurements are shown as open triangles [106, 161] and stars [162–164]. The agreement between the methods is generally good, especially for the experimentally relevant intermediate temperatures between 6 and 10K.
3.2.3.1 Vilesov Group (Los Angeles)
Beginning in 2000, the group of A. Vilesov at the University of Southern California in Los Angeles has performed groundbreaking work around fundamental properties and applications of HNDs. Some notable examples are the development of pulsed HND sources (also see previous discussion about the work of Ellis, Yang and co-workers) [101, 103], spectroscopic studies of various dopant clusters [166–168] and studies of quantized vortices [169, 170]. The group has also developed and studied techniques for the determination of HND sizes that could be considered mass spectrometry in a broader sense. These serve as alternative or complementary methods to the common electrostatic deflection techniques which have difficulties with large HNDs due to their enormous mass and variety of charge state [171]. Gomez and co-workers measured the attenuation of a HND beam from a cw source upon introduction of He or Ar collision gas by monitoring the partial pressure in a downstream detection chamber and used their results for the determination of droplet sizes [165]. HND sizes determined by this titration method in the 7-to-10 K range (<N> ~ 105–107) are in good agreement with previous deflection measurements (Fig. 2.13) [161, 162]. The method finds that HNDs in a previously uncharacterized size range of 107–1010 He atoms are produced in the cw expansion at low nozzle temperatures (5.6–7 K). Furthermore, the ion yield of He4+ exhibits an anomalously large increase (relative to other Hen+) with HND size in the size range of <N> ~ 104–109, as previously observed [34, 172] and attributed to formation and ejection of He4+ at the HND surface via collision of two metastable He2* [34]. The authors propose that the yield of He4+ relative to He2+ can be used as a secondary method of determining HND sizes in the mentioned size range and apply it in the measurement of HND sizes produced in a pulsed source [165]. The process of He4+ formation in large HNDs was further investigated in a detailed study by Fine and co-workers by monitoring the ion yield of He4+ (relative to other Hen+) from EI of HNDs, produced in a pulsed source at temperatures between 9 and 19K [173]. The authors used two mass spectrometers, an in-line QMS and an orthogonal-extraction ToF-MS. Surprisingly, the observed He4+ intensity progression with HND size was in poor agreement between both mass spectrometers initially, with the one measured by ToF-MS considerably lower. Only upon increasing the time width of the electron pulse used for ToF-MS, the He4+ intensity approached that measured by the QMS. The authors concluded that two processes are involved in the formation of He4+. One is fast and produces He4+ as part of a “regular” Hen+ distribution. The other one, suspected to be responsible for the anomalous increase of He4+ intensity, is slower, occurring on timescales of the order of 10 µs, has an electron energy threshold of 40.5 ± 1.0 eV and selectively produces He4+ [172]. Fine and co-workers carefully elucidate possible formation mechanisms on the surface of large HNDs compatible with the observed energy threshold (40.5 ± 1.0 eV) and slow reaction times (~10 µs). The authors conclude that binary collisions of He* + He*, He* + He2* and He2* + He2* (provided He2* highly vibrationally excited) can all contribute to the selective formation of He4+, which consists of two He2+ cores in perpendicular orientation and a shared Rydberg electron [174].

Reproduced with permission from Ref. [175]. © copyright AIP Publishing. All rights reserved
Mass spectra of HNDs in pure condition (a) and doped with rare gases Ne-Xe (b–e) subject to PI at photon energies of 21.6 eV. The mass spectra reveal the formation of RgHen+ as well as Rg2Hen+ (Rg = Kr, Xe) complexes comparable to EI studies, but exhibit fewer features such as magic number or apparent shell closures.
3.2.3.2 Neumark Group (Berkeley)
The group of D. Neumark studies several different topics in chemical physics, one of which is spectroscopy and dynamics in HNDs. When first venturing into the field of HND research, three detailed studies on PI of pristine and doped droplets were conducted. Kim and co-workers utilized a synchrotron light source and a ToF-MS to study the PI characteristics of small HNDs (N ~ 8000) doped with rare gases (Rg = Ne–Xe) at photon energies 10–30 eV [175]. Mass spectra reveal small cluster ions Rgm+ (m ~ 1–3) and ion snowball complexes HenRgm+ (n, m ≥ 1) alongside the well-known distribution of Hen+ including features such as a magic He14+ ion (Fig. 2.14), in good agreement with a previous PI [35] as well as numerous EI studies. Ion yield curves recorded as a function of photon energy indicate that ionization of rare gas atoms and clusters embedded in HNDs is always mediated by He atoms, analogous to EI. The two possible methods are excitation transfer with the dominant resonance at 21.6 eV (atomic 1s–2p transition) below the He ionization threshold at 24.6 eV, or charge transfer following direct He ionization above it. Further notable features are the absence of a bare Ne+ or Ar+ signal from doped droplets as well as the magic number character of He12Kr2+ and He12Kr3+. In a subsequent investigation, Peterka and co-workers studied in detail the PI and photofragmentation of SF6 embedded in HNDs at photon energies of 21.8 and 25.5 eV [176]. Ionization was equally found to proceed via He as in the previous study of rare gas dopants. The recorded mass spectra were compatible with the EI study of Scheidemann and co-workers [43] in that SF5+ was the dominant fragment with SF3+ and SF4+ signals largely suppressed (less so in the 25.5 eV spectrum) compared to the gas phase. Additionally detected compounds were HenSF5+ (n ≤ 25), (SF6)mSF5+, H2O∙SF5+ and HenSF3+ (the latter only at 25.5 eV)—the suspected magic number ion He12SF5+ displayed no local abundance anomaly. Due to the similarity of photoelectron spectra of SF6 in HNDs and gas phase, the authors conclude that the dissociative states involved in the gas phase fragmentation are equally accessible in HNDs and consequently attribute the suppressed fragmentation to the rapid cooling of the hot nascent ion by the HND environment. Peterka and co-workers used the same setup to study the ionization dynamics of pristine HNDs at photon energies between 24.6 and 28 eV using photoelectron spectroscopy [177].
3.2.3.3 Von Helden Group (Berlin)
Many molecules of biological relevance, or biomolecules, are notoriously difficult to bring into the gas phase due to their large mass and/or low vapor pressure. A method capable of producing ions of such non-volatile compounds is electrospray ionization (ESI), which has developed into a tremendously successful and popular technique for analytical biochemists in the past decades [178–181]. The technique was employed by G. von Helden and his group to produce HNDs doped with large biomolecules such as amino acids, peptides and even proteins as large as ~12000 amu for spectroscopic investigations. In the experimental setup used by Bierau and co-workers, ions are produced in an ESI source, mass-selected in a QMS and finally transferred into either a ToF-MS for analysis or a hexapole ion trap for storage. After the trap is loaded, a beam of very large HNDs generated by a pulsed source is guided through the trap. Ions picked up by HNDs can leave the trap due to the HNDs’ high kinetic energy [182]. The authors demonstrated the doping of HNDs with singly charged phenylalanine as well as the much larger protein cytochrome C (~12,000 amu) in multiple charge states and determine the mean HND sizes as <N> ~ 1010–1012 by using a deflection method. In a subsequent study, Filsinger and co-workers exchange the deflection setup for two different ToF-MSs to monitor the HND size distribution (<N> ~105–107) as well as ions expelled from the HNDs upon laser irradiation in a photoexcitation/spectroscopy study of hemin, an iron-containing porphyrin in HNDs [183]. The setup was additionally used by Flórez and co-workers in an IR spectroscopy study of the peptide leu-enkephalin in its protonated form as well as its complex with a crown ether [184].
3.2.3.4 Drabbels Group (Lausanne)
The group of M. Drabbels studies the spectroscopic and dynamic properties of nanoscale systems using a variety of methods with his group at EPF Lausanne. For the purpose of studying atomic and molecular species in a HND environment and the interactions between the two, Drabbels and co-workers employ a versatile setup consisting of a HND source (typical droplet sizes of <N> ~103–104) followed by a pickup section and a multifunctional analysis region [185–187]. The cleverly designed analysis contains a QMS as well as a flight tube equipped with a set of electric lenses that can be used in a multitude of ways, e.g. as a ToF-MS, for photoelectron spectroscopy (PES) and velocity map imaging (VMI). Various lasers are used to excite and/or ionize dopant species. The setup has been used in several combined studies of mostly spectroscopic nature, but including mass spectrometric work as well. In a study of the excited state dynamics of silver atoms in HNDs upon photoexcitation, Loginov and Drabbels that AgHen exciplexes (n = 1, 2) are efficiently formed upon excitation of embedded Ag atoms to the 2P3/2 state [187]. These exciplexes generally form more efficiently in small droplets and are found to become solvated in the HND as AgHe2. Curiously, the natural isotope ratio of silver is not conserved in AgHe, which forms more efficiently with the 107Ag isotope. The authors suggest that tunneling is responsible for the effect which manifests in an abundance ratio of 107AgHe to 109AgHe that is ~20–50% above the natural abundance ratio, being higher in larger HNDs. Taking full advantage of the previously described setup’s capabilities, Braun and Drabbels conducted an extensive, three-part study about photodissociation (PD) of alkyl iodides in HNDs, investigating in detail the kinetic energy transfer [185], solvation dynamics [188] and fragment recombination [189]. Dopant alkyl iodides CH3I, CF3I and C2H5I are dissociated by 266nm laser irradiation, followed after 50 ns by non-resonant 780nm femtosecond PI of the products. Whereas the authors find that He-solvated fragment complexes HenI (from PD of CH3I and CF3I) and HenCH3+ (only from CH3I) were produced in the PD/PI of alkyl iodides with up to n = 15 or more attached He atoms, CF3 and C2H5 were only detected as bare fragments [188]. Based on the fragment velocity characteristics, Braun and Drabbels concluded that He-solvated fragment complexes are formed in the interior of the HND in a dynamic process balancing the formation of solvation shells around the escaping fragment and the encountered flow of He atoms due to the relative motion on their way through the HND. The He solvation and desolvation of barium (ions) as a probe for surface-generated ions was investigated by Zhang and Drabbels utilizing PI of Ba attached to small HNDs (<N> ~6000), analyzed via ToF-MS and excitation spectroscopy [186]. Judging from the recorded excitation spectra, which are practically identical to those of Ba+ in bulk liquid helium [190] as well as the mass spectra, the authors concluded that Ba+ becomes fully solvated in the HND upon PI before being ejected as bare Ba+ or Ba+Hen following photoexcitation of two different 6p  6s transitions (D1 and D2 lines). A follow-up study by Leal and co-workers in cooperation with the theoretical group around Barranco and Pi attempted to unravel in detail the desolvation dynamics of photoexcited Ba+ [191]. The authors find that half of the expelled ions are fully desolvated Ba+ while the rest are detected as Ba+Hen (n ≤ 25). Both the He attachment distribution of Ba+Hen exciplexes as well as the velocity of the desolvated Ba+ and Ba+Hen showed different characteristics for the different excited states, but was independent of droplet size or laser intensity. Whereas the performed time-dependent density functional calculations are able to reproduce the formation of Ba+Hen exciplexes and the measured excitation spectra very well, they fail to capture the experimentally observed desolvation of Ba+ and Ba+Hen.
3.2.3.5 Kong Group (Corvallis)
Electron diffraction is a widely used tool for structure analysis of crystals, surfaces and gas-phase molecules [192]. The group of W. Kong at Oregon State University is working towards the construction of a molecular goniometer, capable of collecting electron diffraction images of single, oriented macromolecules as well as complexes grown in HNDs. This would allow the structure determination of species that are difficult to access by conventional methods. The group relies on pulsed droplet sources and uses ToF-MS technology to monitor and adjust doping conditions, which is crucial for electron diffraction imaging of HNDs. He and co-workers conducted a study of methods for the characterization of a pulsed beam of pristine and halomethane-doped HNDs using two simple and cost-effective, home-built ToF-MSs utilizing EI and MPI [193]. In two subsequent studies, Alghamdi and co-workers investigated EI and MPI of large HNDs (N ~ 108) doped with aniline. The authors found that in both cases, the large HND environment suppressed the ionization mechanism via shielding of the neutral dopant against charge transfer from He2+ [194] and caging of photoelectrons allowing for recombination with photoionic (fragments) [195], respectively. A recent study demonstrated the feasibility of electron diffraction of smaller HNDs (N = 800–1.4×105) doped with CS2 [196].
3.2.3.6 Ichihashi Group (Tokyo)
The group of M. Ichihashi at Toyota Technological Institute conducts research on catalytic activity of charged metal nanoparticles. Odaka and Ichihashi used a recently constructed HND setup employing a pulsed HND source and a QMS in a study colliding size-selected cobalt clusters Com+ (m ≤ 5) with HNDs (N ~ 1600) to produce Com+Hen (m ≤ 5, n ≤ 21) [197]. The authors found several abundance anomalies, indicating increased stability of the corresponding configurations, such as Co2+Hen (n = 2, 4, 6 and 12), Co3+Hen (n = 3, 6), Co4+Hen (n = 4), and Co5+Hen (n = 3, 6, 8 and 10). By observing the total Com+Hen yield as a function of the relative velocity, different collision regimes are identified. The ion yield was highest at low relative velocity (~100 m/s) due to the attractive electrostatic van-der-Waals interactions between Com+ and HND, whereas a steep decline in yield towards higher relative velocities indicated a transition to hard-sphere collisions.
3.3 Review of Recent Work by the Scheier Group (Innsbruck)
We will now review the work performed by the group led by P. Scheier at the University of Innsbruck, which the authors of this chapter are currently part of. Much of the more recent work has already been discussed in three comprehensive review article about collisions, ionization and reactions in HNDs [22], the solvation of ions in helium [198] as well as the chemistry and physics of dopants embedded in HNDs [199]. We will thus focus on the most recent work performed in our group, including our own. The three following chapters are dedicated to the three experimental HND MS setups currently active in the group.
First, we will discuss the “ClusToF” experiment, a classical setup consisting of a HND source, followed by a pick-up region, an EI source and a high resolution ToF-MS. The main focus are mass spectrometric studies of chemical reactions and cluster growth inside HNDs, with the additional capability of performing action spectroscopy on He-tagged ions.
The “Snowball” experiment was designed to investigate the behavior of large, pristine HNDs. Utilizing a tandem MS setup consisting of two electrostatic SF-MSs, each equipped with an EI source, Laimer and co-workers recently demonstrated the existence of highly charged, stable HNDs [171]. The setup can be modified to allow the synthesis of nanoparticles in these large, highly charged and m/z-selected HNDs, followed by deposition on a substrate for ex situ analysis.
The third experiment, “Toffy” combines the newly developed technique of pickup into highly charged, m/z-selected HNDs [200] with a high-resolution ToF-MS. Tiefenthaler and co-workers constructed a highly versatile experimental setup which was able to produce considerable scientific output in a fairly short period of time. While both ClusToF and Toffy are similar in their scientific scope, the latter is additionally capable of performing collision-induced dissociation studies and controlling the helium attachment to analytes in an unprecedented manner. A more detailed description of each experiment is given in the respective chapter.
3.3.1 Classical HND MS Experiments
Early HND MS experiments by the Innsbruck group between 2006 and 2009 utilized either a QMS [201] or SF-MS [202–205] for mass analysis. It quickly became clear, however, that none of the instruments were ideal for HND MS. Whereas the QMS lacked in both resolution and accessible mass range, the SF-MSs’ low ion yields forced experimenters to slowly scan the mass range, which is problematic since experimental conditions changing over time may produce distorted mass spectra. These issues led to the construction of a new apparatus in 2010, the “ClusToF” experiment, now the longest running experiment in the Innsbruck group. It consists of an HND source (5µm nozzle diameter, typical stagnation pressure p0 ~ 20 bar, nozzle temperature T0 ~ 10K) followed by two differentially pumped pickup chambers, each equipped with an oven and gas inlet ports, and a third chamber housing a Nier-type (EI) ion source (Fig. 2.15). Ions created in this region are extracted via a weak electric field and focused by an electrostatic ion guide into an orthogonal extraction reflectron-ToF-MS. The ToF-MS can be mounted such that the extraction region is mounted in-line with the HND beam or perpendicular to it. A more detailed description of the experiment (in-line configuration) can be found in [172]. The employed ToF-MS is able to overcome the deficiencies of the previously used MSs by being able to record full-range mass spectra (m/z up to ~50 kDa) with a mass resolution of R ~6000 while maintaining a reasonable ion yield. An example mass spectrum of pure Hen+ fragments from larger droplets is shown in Fig. 2.16. The ion yield can be increased manifold if a high mass resolution is not required, but a higher ion yield is beneficial, e.g. for action spectroscopy measurements, which can be performed on the experiment as well by aligning a tunable OPO laser system with the ToF-MS extraction region [150, 206–208]. The ClusToF experiment has proven to be a reliable and versatile design, in large part responsible for the Innsbruck group’s successful application of HND MS in the last decade. In the following section, we will present the most recent studies performed with this experimental setup.

source chamber, follow by the ToF-MS. In the current version, the ToF-MS is rotated by 90°, mounted horizontally at the open flange of the ion source chamber facing the viewer, however, there were no significant differences found between the two alignments. Reproduced with permission from Ref. [22]. © copyright Elsevier. All rights reserved
Schematic setup of the “ClusToF” experiment. The HND beam traverses two differentially pumped pickup chambers before entering an ion

Reproduced with permission from Ref. [22]. © copyright Elsevier. All rights reserved
Mass spectrum of small Hen+ from EI of pristine HNDs (a) and extracted cluster size distribution (b) as recorded with the ClusToF setup. Magic numbers typically observed for Hen+ with this setup are as n = (7), 10, 14, 23 and 30 which can be seen in the cluster size distribution (b). Both insets demonstrate the capabilities of the high resolution ToF-MS by displaying the mass resolution around m/z = 3500 (a) and the deconvolution of an isobaric mass peak at m/z = 28 (b).
3.3.1.1 Cationic and Protonated Clusters of Rare and Inert Gases
The capabilities of the high resolution ToF-MS setup were recently demonstrated when Gatchell and co-workers were able to resolve an issue that had been a topic of debate for more than 30 years. Magic numbers found in early mass spectrometric studies of cationic rare gas, but especially Ar cluster ions, associated with shell closures of icosahedral structures resulting from sphere-packing models [11, 209], were not always reproducible [13, 210–214]. In a recent study, Gatchell et al. studied HNDs co-doped with H2 and Ar [215]. High-resolution mass spectra reveal both bare Arn+ and protonated ArnH+ cluster ion series which exhibit vastly different characteristics such as magic numbers. The bare Arn+ series exhibits relatively few features (magic n = 16, 19, 23, 27, 81, 87, antimagic n = 20) that agree well with previously reported measurements [13, 210–214], but miss key features of sphere-packing models such as the first and second icosahedral shell closures at n = 13 and 55. In contrast, magic numbers found in the protonated ArnH+ series are in essentially perfect agreement with previously mentioned early measurements [11] as well as the predictions of sphere-packing models (e.g. n = 13, 19, 26, 29, 32, 34, 49, 55, …). In addition, the Ar7H+ ion is found the be magic, a feature that is rarely found in the literature [215]. Ab initio structure calculations performed on Arn+ and ArnH+ systems up to n = 21 are able to reproduce very well the magic numbers found in the small cluster ion series. The calculated structures and magic numbers (where available) also show good agreement with previous theoretical work on pure cationic [216] and protonated Ar clusters [217–219]. The studies of protonated rare gas clusters were extended towards Ne and Kr by Gatchell and co-workers [220] (Kr was co-doped with D2) as well as towards He by Lundberg and co-workers [221]. In contrast to Ar, which is essentially monoisotopic, the richer isotopic patterns of Ne and especially Kr complicate the distinction between pure and protonated rare gas cluster series, preventing the authors from studying cluster sizes larger than n ~ 30. Similar to Ar, the mass spectra of Nen+ and NenH+ are found to be fairly different. Whereas the Nen+ and Arn+ series share a few magic numbers such as n = 14 and 21 (although both are much weaker in Arn+), other features are completely different, the magic n = 19 and 27 of Arn+ are both slightly antimagic in Nen+ and the prominent antimagic n = 20 of Arn+ does not present a noteworthy anomaly in Nen+ at all. In contrast, the agreement between the protonated NenH+ and ArnH+ series is much better. Some features become much more apparent upon calculating the second differences, defined as \(\mathrm{ln}\left[\frac{2 {I}_{n}}{({I}_{n-1}+{I}_{n-1})}\right]\) (where In denotes the yield of the nth ion in a series), as shown in Fig. 2.17b. It is now clear that all main features of ArnH+ up to n = 32, including n = 7, are equally found in NenH+. Calculations reveal that Ne7H+ consists of a linear Ne-H+-Ne core, surrounded by Ne atoms spanning a pentagon, which is oriented perpendicular to the axis of the ionic core and shares a common plane with the proton, a structure that is equally found for Ar7H+ [215, 217, 219].
Calculations performed by Gatchell and co-workers [215] also reveal the reason for the difference between shell closures at Ar13H+ and Ar14+ (instead of Ar13+ as would be expected from sphere packing schemes). The central ion present in smaller clusters is a linear Ar3+ molecule with reduced binding lengths between Ar atoms due to the presence of the charge. This compression of binding length leaves room in the icosahedral structure for a 14th Ar atom interacting relatively strongly with the ionic core, essentially forming an Ar4+ central ion, surrounded by two pentagonal rings of five Ar atoms. In the protonated Ar13H+ structure the proton, carrying most of the charge, can squeeze in between two of the central three Ar atoms without significantly distorting the icosahedral structure of the neutral cluster. Calculations of neutral, cationic and protonated Ne19 systems show analogous effects [220]. Again, Ne19 and Ne19H+ adopt similar, highly symmetric geometries with the proton squeezing in between two Ne atoms in the center of the cluster. The central ion in Ne19+ is a contracted, covalently bound Ne2+, which again distorts the highly symmetric structure. Assuming this allows for the addition of two strongly interacting Ne atoms along the axis of the central ion could explain the observed magic character of Ne21+.
In contrast to the lighter rare gas species, both the Krn+ and KrnH+ series share some key features of sphere packing, such as magic numbers at n = 13, 19 and 29. Other magic numbers found in all RgnH+, such as n = 17 and 26 appear to be shifted down to n = 16 (cf. Ar series) and 25. It appears as the structures of Krn+ are distorted less by the presence of the charge compared to the lighter rare gas species. This is supported by calculated structures of neutral, cationic and protonated Kr19+ [220], showing that indeed the charge-induced contraction of the Kr-Kr binding length is relatively smaller than in the lighter rare gas species.
The study of protonated He clusters by Lundberg et al. shows that the lightest rare gas presents a special case once again, with neither Hen+ nor HenH+ showing good agreement with icosahedral packing structures [221]. Besides the strong He2+ ion signal, the most prominent magic numbers of small Hen+ are n = 10, 14, 23 and 30, with the latter two being much less prominent, yet easy to identify upon careful analysis. Both n = 14 and 23 are found in other series as well, the former in bare ions such as Nen+ and Arn+, while the latter is seen in NenH+ and ArnH+. The mass spectrum of HenH+ is fairly unique among the studied rare gas species. While one magic number, n = 13, is shared between HenH+ and all other protonated cluster ion series as well as Krn+, none of the other magic numbers found, namely n = 6, 11, 22 (weak), 35 or 39 show enhanced abundance in any other series, except perhaps for n = 22, which has enhanced abundance in Krn+, but is not considered magic since Kr23+ has similar intensity. A detailed theoretical study of HenH+ (n ≤ 18) by Császár and co-workers shows significant drops in the calculated successive electronic evaporation energies (i.e. the energy difference of the reaction HenH+  Hen-1H+ + He) after n = 6 and 13 at the HF, MP2 and CCSD(T) (only up to n = 7) levels of theory levels of theory [222]. The calculated successive electronic evaporation energies are found to be comparable between MP2 and CCSD(T), but consistently lower at the HF level of theory. The values are lying between 25 (HF) and 44 meV (MP2/CCSD(T)) for n = 3–6 (this is incorrectly given as ~250 meV in [221]), dropping to between 5 and 17 meV for n = 8–13, with n = 7 slightly higher between 12 and 27 meV. Surprisingly, none of the theoretical methods indicate an enhanced binding energy of He11+, the strongest anomaly found by Lundberg et al., quite the contrary, as the MP2 calculations even display a slight local minimum. After n = 13, the successive electronic evaporation energies are dropping towards 0 for HF and 6 meV for MP2 and CCSD(T). The binding energy of additional He atoms can be expected to converge to the HND “bulk” value of 0.6 meV per atom [22], which is also recognized as a possible onset for microscopic superfluidity by Császár et al. [222].

Size distributions (a) and normalized second differences (see text for an explanation) (b) of pure and protonated rare gas clusters with common local anomalies indicated above. Many magic numbers corresponding to (partial) shell fillings are found to be shared between the different rare gas species. Reproduced from [220]. Licensed under CC BY 4.0
The findings of Gatchell, Lundberg and co-workers showed that the characteristics of cationic and protonated (as well as neutral) rare gas clusters, especially of the lighter species (He–Ar) can be fairly different, probably due to charge-induced distortion of the cluster geometries. Gatchell et al. [215] suggest that the discrepancies of magic numbers in early measurements of rare gas cluster ions, especially Ar, were due to erroneous identification of unresolved, protonated RgnH+ cluster ions where protonation was likely caused by water impurities. The studies further illustrate how sensitive cluster systems can be towards small changes such as the addition of a minor impurity.
While no further studies of the heaviest rare gases xenon and radon were conducted due to the increasingly difficult analysis of the very rich isotopic pattern of Xe and the radioactivity of Rn, respectively, Martini and co-workers studied a different van-der-Waals system, namely mixed cationic clusters of nitrogen and deuterium [223]. Again, the high resolution of the ToF-MS is crucial in the identification of the myriad ions such as (N2)n+, (N2)nDm+, HenDm+ and Dm+. The main ion series identified is due to (N2)nD+, which, in analogy to the discussed rare gas studies [215, 220, 221], shows very different characteristics from (N2)n+. Whereas the lone notable feature in the pure cationic series is the enhanced intensity of (N2)19+, an array of magic numbers, namely n = 2, 6, 7, 12, 17 is easily identified in the protonated (N2)nD+ series. Structures calculated for the magic number ions (N2)nD+ (n = 6, 7, 17) reveal a linear [N2–D–N2]+ central ion with a relatively even charge distribution between the three constituents. The structure of (N2)6H+ is found to be octahedral and slightly energetically favorable over the pentagonal bipyramid found for (N2)7H+. Similar to the rare gas studies, the magic numbers n = (2), 7, 12 and 17 can be explained in terms of packing molecules around the central [N2–D–N2]+ ion in icosahedral (sub-)shells. The calculated structure of the magic (N2)19+ is found to be similar, but the linear, central (N2)2+ ion is contracted in comparison to [N2–D–N2]+, allowing for two additional N2 molecules placed along the central axis. By increasing the D2 partial pressure, mixed (N2)nDm+ cluster ions were observed, predominantly with odd m. The data reveal a few indications that N2 molecules can be partly replaced by D2 in these mixed cluster ions. For one, the strong n = 6 anomaly in (N2)nD+ seemingly shifts down to n = 5 in (N2)nD3+, but remains at n = 5 for (N2)nD5+ instead of further shifting down. Additionally, the progression of the (N2)nDm+ (n = 0–12) series shows a strong drop in intensity after 2n + m = 35 (i.e., a total number of 35 atoms), except for n = 0 and 1. The interpretation of the authors is that any mixed cluster containing a total number of 17 molecules and at least two N2 molecules forms a magic cluster ion. The minimum of two N2 molecules suggests that a central [N2–D–N2]+ forms the ionic core, surrounded by a structure similar to the one found for (N2)17D+. These interpretations are supported by theoretical calculations. Structure optimizations of three possible (N2)16D2D+ isomers support this interpretation, since the structure of the (N2)17D+ cluster ion upon the exchange of N2 with D2 is found to be largely preserved in all cases. However, while N2 is found to prefer a linear orientation towards the charge center, D2 aligns perpendicular to it. While no further structure optimizations of mixed (N2)n(D2)mD+ cluster ions were attempted, potential energy surfaces of the interaction of a point charge (i.e., a proton) with N2 and D2 were calculated. The position with the lowest interaction energy is found to be along the molecular axis in the case of N2 and perpendicular to it for D2. Additionally, the interaction energy is found to be higher for N2 compared to D2, which explains why a [N2–D–N2]+ ionic core is formed where possible. Furthermore, by considering the atomic van-der-Waals radii and the D–D bond length, the authors conclude that the spacing of molecules in the structural lattice remains very similar upon exchange between the two species, explaining the observed efficient mixing of N2 and D2 in mixed (N2)n(D2)mD+ cluster ions.
3.3.1.2 Anionic Complexes of Hydrogen
It is no surprise that hydrogen is one of the most widely investigated chemical elements and perhaps the one that is best understood. Its simplicity, being comprised of only protons and electrons (as well as neutrons in the case of its isotopes deuterium and tritium), makes hydrogen appealing for theoretical studies. It is ubiquitous, both in the wider universe as well as a constituent of countless chemical compounds vital to life on earth. In the past decades, there has been growing interest in and rapid development of the possibilities of using hydrogen for energy storage due to the rising demand for renewable and sustainable energy sources in the face of global warming [224]. Hydrogen complexes are also of astrophysical relevance and have been considered [225, 226] as potential carriers of the diffuse interstellar bands (DIBs) [146, 227]. Here, we take a closer look at anionic complexes of hydrogen. Since H2– is unstable except for highly rotationally excited metastable states [228], the simplest stable polyatomic anion is H3–. First theoretical investigations were published in 1937 [229] and suggested the existence of a stable H–(H2) anionic complex. It took close to 40 years until the first reports of its detection were made [230, 231]. Since the first observations were disputed due to poor signal quality [232], it was only relatively recent that H3– was [32, 33] shown to be stable using high-quality theoretical calculations [233, 234] and experimental detection in a dielectric discharge plasma experiment [235]. Motivated by the possible formation of anionic hydrogen clusters in space, the structure and stability of Hn– anions with odd n between 3 and 13 was first investigated around 1980 at the Hartree-Fock level of theory [226, 236] and more recently in 2011 by DFT [237]. Despite the efforts, there was no consensus on the equilibrium geometries of Hn– anions or the stability of such systems.

Reproduced with permission from Ref. [238]. © copyright American Physical Society. All rights reserved
Size distributions of anionic hydrogen (deuterium) clusters from doped HNDs as measured with the ClusToF setup. Both species produce essentially identical features such as apparent shell closures at n = 25, 65 and 89, corresponding to 12, 32 and 44 H2 molecules surrounding an H– ionic core.

Reproduced with permission from Ref. [239]. © copyright AIP Publishing. All rights reserved
Classical (top) and quantum structures (bottom) of anionic hydrogen clusters H–(H2)n as calculated using PIMD. The strong symmetry and heavy localization of solvating H2 molecules in the magic number clusters (n = 12, 32 and 44) is clearly visible in comparison with the non-magic n = 18 cluster.
In 2016, Renzler et al. reported the first direct observation of anionic hydrogen and deuterium clusters Hn– (Dn–) with n ≥ 5 formed by doping large HNDs (~106 He atoms) with molecular hydrogen and deuterium using the ClusToF setup [238]. The authors observed exclusively Hn–clusters with odd n (Fig. 2.18) and concluded that these clusters consist of an anionic H–core, formed by dissociative electron attachment (DEA) to H2, with two to well over 100 H2 molecules attached, bound by ion-induced dipole interaction. No significant differences in the mass spectra were found between Hn– and clusters. The authors also obtained equilibrium geometries from DFT calculations for Hn– (n = 3, 5 and 7), finding that H5– adopts a linear structure, in agreement with [226, 236] but in contrast to the bent structure found by [237]. Meanwhile, H7– is predicted to have trigonal pyramid shape, agreeing with [237], but contradicting [226, 236]. Furthermore, clusters with n = 25, 65 and 89 (i.e. 12, 32 and 44 H2 (D2) molecules attached to the Hn– core) were found to be anomalously abundant, suggesting increased stability of these clusters, or magic character. It was found that these clusters consist of the anionic core, surrounded by up to three solid-like solvation shells of 12, 20 and 12 molecules in an icosahedral-like geometry, a structure that was previously found for cations solvated in helium, such as theoretically in Na+Hen [240] and experimentally in Ar+Hen [241]. Calvo and Yurtsever modeled the structure of H–(H2)n and D–(D2)n clusters for a wide range of n = 1-54 using path integral molecular dynamics (PIMD) with a potential optimized for yielding accurate geometries [239]. Their findings clearly show the icosahedral-like shape of the magic cluster anions while also being able to qualitatively reproduce the geometries found by DFT for n = 1–6 (Fig. 2.19) [237]. In a separate follow-up study, Joshi et al. used dispersion-corrected DFT to determine the geometries of the experimentally found magic anionic hydrogen clusters, again confirming the icosahedral-like structure and increased stability [242]. The authors further calculated the binding energies and HOMO-LUMO gaps for systems of the form X(H2)n, for n = 12, 32 or 44 and a variety of metal and non-metal anionic and neutral atomic impurities X. The findings suggest that many of the investigated X(H2)n systems exhibit similar (icosahedral) geometries and binding energies as the corresponding anionic hydrogen clusters, predicting similar stability and feasibility of experimental detection. Another recent study by Mohammadi et al. attempted to more accurately predict structures and energies of small H–(H2)n clusters (n = 1–5) reported by previous publications. While the structures for n = 1–4 agree well with those found by [237, 239], the structures for n = 5 differ from those previously reported. The authors also report structural isomers for n = 3–5 and different binding energies and bond lengths compared to the previous DFT and PIMD studies. Huang et al. recently suggested that small anionic hydrogen clusters H–(H2)n clusters (n = 1–7) could be carriers of DIBs [225]. The authors identified 25 absorption bands between 440 and 620 nm which coincide with DIBs within the uncertainty (which is rather large at ~±5nm) of the calculated absorption wavelength. Since the energies involved in these transitions are much larger than the binding energies of such clusters, an excited cluster would be metastable and dissociate shortly after excitation. Thus, an effective production mechanism must exist for the clusters to contribute to any DIBs, which the authors propose to occur on interstellar dust grains.
In a follow-up study, Renzler et al. studied mixed oxygen and hydrogen (as well as deuterium) cluster anions produced by attachment of low-energy electrons (0–30 eV) to large helium droplets (N ~ 5x105) co-doped with oxygen and hydrogen [243]. The experiment produced HnOm– anions with a wide variety of combinations of n and m. The authors find that anions, even pure Om–, are predominantly produced at electron energies associated with resonances of electron attachment (EA) to H2. In contrast to the pure hydrogen experiment, however, anions with even n are not only readily observed, but in many cases more abundant than neighboring clusters with odd n. It appears that electron-induced chemistry between the two dopants leads to the production of different central anions in mixed HnOm– anion clusters, most notably HO2– and H2O–. While the former can certainly be expected due to DEA to H2 and subsequent reaction of H– with O2, H2O– is unstable with respect to autodetachment and thus its detection is surprising. The authors concluded that a rapid electron transfer mechanism from H2– to O2 must exist to initiate DEA to O2, producing O– that subsequently reacts with H2 to produce the H2O– anion, which is stabilized in the presence of at least one other molecule. Surprisingly, HnOm– anions with both even n and m are also observed, leading the authors to conclude that the HND environment is able to efficiently quench both DEA processes and stabilize the resulting cluster anions. All the phenomena and processes described above are likely unique to mixed clusters formed in helium droplets.
3.3.1.3 Anionic Complexes of Nitrogen
Motivated by their possible application as non-polluting high-energy-density materials (HEDMs), polynitrogen compounds (i.e., compounds comprised primarily or exclusively of nitrogen) have received growing interesting in past decades [244–248]. While theoretical investigations led the way predicting structures of possible polynitrogen compounds, experimental verification generally proved difficult. Pure cationic nitrogen clusters have been widely produced and studied using various methods such as supersonic molecular jets [249–253], sputtering of solid N2 [254, 255] and ionization of doped helium droplets [49]. Studies of pure anionic nitrogen clusters, however, are rare. Reports of nitrogen cluster anions Nn– with n > 3 were made by Tonuma et al., who observed Nn– (n = 1-9, 11, 13) upon sputtering of frozen N2 with highly charged, energetic Ar ions from a linear particle accelerator [254], Vij et al., who isolated cyclic N5– in an ESI-MS-MS experiment [256], Vostrikov and Dubov, who measured the cross sections for attachment of low energy electrons to large (N2)n clusters [257] and Pangavhane et al. in 2011, who detected Nn– (n = 6, 10–15) upon laser desorption ionization of phosphorus nitride (P3N5) [258].
Weinberger et al. recently published a detailed study on the formation of nitrogen cluster anions Nn– with n ≥ 3 upon EA to large helium droplets (N ~ 105) doped with N2 [259]. The most prominent anion series observed was Nm– with 3 ≤ m odd < 140. Further anion series arising due to impurities are odd-numbered nitrogen clusters complexed with water and C2N2– complexed with intact nitrogen molecules. Evidence for the existence of even-numbered Nm– with m > 2 can be found, but the corresponding anion signals are roughly two orders of magnitude weaker than those of the nearest odd-numbered clusters, barely exceeding background noise levels. No evidence is found for N– or N2–, in accordance with the short lifetimes of N2– (on the order of 10–15s [260]) and negative electron affinity of N (–0.07 eV [261]). The authors concluded that odd-numbered Nm– are comprised of a central N3– azide anion solvated by \(n=\frac{m-3}{2}\) N2 molecules. The N3–(N2)n series exhibits distinct anomalies in the ion intensity after n = 4 and 11, which indicates increased stability of these compounds. A follow-up study by Calvo and Yurtsever employed a variety of quantum chemical methods to find the stable structures of odd-numbered nitrogen anion clusters [262]. The calculations confirm the assignment of an N3– anionic core solvated by N2 molecules and yield optimized geometries for N3–(N2)n for n = 1–9. The first five N2 molecules are found to arrange in parallel around the linear N3– and form the first solvation shell. Calculations of binding and dissociation energies confirm the higher stability of N3– (N2)5 expected from the calculated equilibrium geometries. While the general agreement between the two studies is good, this finding is at odds with the experimental observations of Weinberger et al., who find N3–(N2)4 to be particularly stable. Unfortunately, the pronounced enhanced stability of N3–(N2)11 could not yet be investigated theoretically due to the size of the system.
3.3.1.4 Solvation of Alkali Clusters in HNDs
Alkali atoms and clusters have been a popular study target in connection with HNDs, owing largely to their simple electronic structure and exceptional doping characteristics. Whereas an interior location is favorable for most other dopants, alkali atoms and small clusters are found to reside in dimples at the HND surface due to short range Pauli repulsion between their and the surrounding He atoms’ s valence electrons [62, 63, 263–266]. Cationic alkali atoms and clusters, however, are solvated due to the strong, charge-induced van der Waals interaction with the surrounding He atoms. This causes the closest He atoms to form a snowball structure with a high degree of localization around the ion. The solvation of alkali ion impurities and the structure of snowballs around them was studied in detail theoretically by Reatto et al. [80, 267, 268], Gianturco et al. [269–271] and other authors [272, 273] with a wide variety of methods. Experimental work can be found in a photoionization study of HNDs doped with Na–Cs [93] and an EI study of Na- and K-doped HNDs [274]. Lithium is often omitted from HND MS studies due to complications in the evaluation caused by numerous mass interferences. Recently, Rastogi et al. conducted a combined study of Li+ solvation combining high-resolution MS and different theoretical methods [275]. The mass spectra revealed Lim+ (m ≤ 3) and HenLi+ (extending to n > 50), with prominent local anomalies in the ion abundance of HenLi+ at n = 2, 6, 8, 14 and 26, a weaker anomaly at n = 4 as well as pronounced minima at n = 21, 24, 27 and 28. Theoretical calculations suggest particularly strongly bound structures of HenLi+, n = 4, 6 and 8, in decent agreement with the experimental observations and are in good agreement among each other, regardless of whether quantum or classical approaches are used. However, the calculations failed to reproduce the strong n = 2, 14 anomalies and mostly indicate a structure of slightly increased stability at n = 10 which is not reflected in the mass spectra.
Interestingly, creating a positively charged alkali cluster is not the only way leading to its solvation. Using a classical model, Stark and Kresin predicted that, upon growing larger, the relative strength of attractive and repulsive forces between dopant alkali clusters and HNDs shifts, eventually tipping the scale in favor of the submersion of alkali clusters into the HND beyond a certain, element-specific, critical size nc [276]. The authors calculated nc = 23, 21, 78 and >100, for Li, Na, K and Rb, respectively, while for cesium a failure of submersion is indicated by the exceedingly large nc value of 625. Shortly after, An der Lan et al. conducted studies utilizing EI MS of HNDs doped with sodium [277] and potassium [278] in an attempt to experimentally determine nc. The authors followed the suggestion given by Stark and Kresin [276] to exploit the fact that dopants can be selectively ionized via Penning ionization if they are located at or near the HND surface. By monitoring the cluster ion yield as a function of both cluster size and incident electron energy, An der Lan et al. were able to measure nc, exp = 21 for Na and nc, exp ≈ 80 for K, in excellent agreement with the theoretical predictions. While the transition from an exterior to an interior location was observed sharply for Na clusters, it is more diffuse for K and was expected to be even more diffuse for rubidium [278].

Cluster size distributions extracted of cationic Rb clusters grown in large HNDs at different electron energies (a) and ratio of ion yields recorded at 21.6 and 40 eV for Rb (b) [126]. The gradual submersion of Rb clusters into the HND is evident from the decrease in ion yield between 60 and 100 atoms in the 21.6 eV mass spectrum in (a). A gradual decline of ion yield ratio corresponding to a submersion into the HND is noticeable starting around 50 Rb atoms is evident in (b). Rubidium clusters larger than ~100 atoms are assumed to be fully submerged. Also shown is a similar transition for K clusters suggesting submersion into the HND around 80 K atoms [278]. Reproduced from [126]. Licensed under CC BY 4.0
Recently, Schiller et al. expanded the experimental evidence of alkali cluster submersion with a study of rubidium [126]. The authors recorded EI mass spectra of clusters with up to 200 Rb atoms grown on or in HNDs at electron energies between 8 and 160 eV (Fig. 2.20a). For determination of nc, they followed the same principle proposed by Stark and Kresin and implemented by An der Lan et al. and indeed found a diffuse transition suggesting full submersion of Rb clusters larger than ~100 atoms (Fig. 2.20b). While this observation is still compatible with nc > 100 predicted by Stark and Kresin (the exact calculation yielded nc = 131 which was interpreted carefully by the authors), the agreement is worse than for the lighter alkali species Na and K. It will most likely be difficult to further extend the series of studies towards the heaviest (nonradioactive) and lightest alkali species in cesium and lithium, respectively. Since Stark and Kresin’s model slightly overestimates the critical submersion size of Rb clusters, it is possible that this is also true and perhaps exaggerated for cesium. This could, in principle, warrant revised theoretical work and an attempt of experimental observation of Cs cluster submersion. Lithium, on the other hand, does not form large enough clusters on HNDs to allow for the experimental observation with the previously used method. A possible solution would be the production of larger, neutral Li clusters, e.g. in a gas aggregation source followed by HND pickup and analysis via the discussed or other methods.
3.3.1.5 Studies of Gold Complexes
Gold has always been precious to humans ever since its discovery. This is in part due to the chemical inertness of bulk gold under normal conditions, enabling pieces of art crafted from the noble metal to retain their beauty over long periods of time [279]. Gold has been a source of fascination, if not obsession for alchemists who attempted to transmute lead into gold. Gold nanoparticles (AuNPs) have been used for millennia, e.g. for their optical [280] and medical properties [281], long before the nature of the phenomena could be explained. The first publication starting to recognize AuNPs was Faraday’s 1857 report, mentioning a “finely divided metallic state” [282]. Faraday’s work laid the foundation for expanding the knowledge about nanoparticles via colloid science, especially during the first half of the 20th century [283]. It was the 1980s ground-breaking inventions of the scanning tunneling (STM) and atomic force microscopes (AFM), which allowed for the routine analysis and manipulation of nanoscale materials down to atomic precision, that helped colloid science to develop into today’s ever-present field of nanotechnology. Gold became the metal of choice in many nanotechnology experiments and applications since it is not, like most other metals, readily covered by a passivating oxide layer [283]. Its widespread use in nanotechnology ultimately lead to the discovery that while bulk gold is inert, nanoscale gold actually exhibits significant catalytic activity, be it as ions [284] or neutral nanoparticles [285]. Ironically, while gold’s inertness long prevented chemists from developing a real interest, it also promoted its widespread use in nanotechnology and, in turn, helped open up the field of gold catalysis. It has since become clear that in many cases gold is not only a good, but the best known catalyst [279, 283, 286–289]. Additionally, metal nanoparticles emerged as good candidates for hydrogen storage as part of a green energy revolution [279, 290291292293]. These discoveries have sparked great interest in the fundamental research of nanoscale gold and gold chemistry with the goal to better understand and further develop gold catalysis.
Gold clusters were mainly produced using sputtering [294–296] and laser evaporation methods [297, 298]. In a recent study, Martini et al. presented mass spectra of cationic and anionic gold clusters produced via EI/EA of gold-doped helium droplets [299]. The ion abundance distribution extracted from the cationic mass spectrum is roughly shaped like a log-normal distribution, caused by the statistics of picking up dopant atoms in helium droplets, superimposed with a pronounced odd-even oscillation up to n = 21, with odd-numbered Aun+ being significantly more abundant (Fig. 2.21a). Becker et al. performed CID on Aun+ (2 ≤ n ≤ 23) and found that clusters with n ≤ 15 dissociate by dimer evaporation if n is odd, while larger clusters dissociate by monomer evaporation [300], which was later supported by ion mobility measurements and theoretical calculations [301]. The observed odd-even oscillations were also found by Katakuse et al. in mass spectra of cationic gold clusters produced by sputtering a gold foil with 10 keV Xe+ ions [294]. Although the general shape of the ion abundance distribution presented by Katakuse et al. is an exponential decay versus a log-normal distribution in the helium droplet experiment, the most prominent features are in excellent agreement—apart from the discussed odd-even oscillations, both studies reveal local anomalies at n = 21, 35 and 58 as well as a local minimum at n = 30. Katakuse et al. applied a one-electron shell model used in modeling Nan+ systems [3], explaining the enhanced stability of odd-numbered (even-electron) Aun+ and the magic numbers, which mark electronic shell closures [294]. Apart from monocationic gold clusters, dicationic Aun2+ (only odd-numbered are detected due to interference from Aun+) are also detected with roughly two orders of magnitude lower abundance and an appearance size of n = 9. The appearance size agrees with previously reported ones [302], however, there are also reports of appearance sizes as low as n = 2–5 [303–305].

Reproduced with permission from Ref. [299]. © copyright Elsevier. All rights reserved
Cluster size distributions of Aun± produced from EI of gold-doped HNDs (solid symbols) and by bombardment of a gold sheet with 10 keV Xe+ (open symbols) [294]. Both methods produce similar results in terms of observed odd-even oscillations and shell closures. The fact that features are less apparent in the HND mass spectra can be attributed to the suppressed fragmentation via cooling of nascent ions by the HND environment.
Besides the previously discussed cationic clusters, Martini et al. also report mass spectra of anionic gold clusters revealing Aun– with up to n = 35 atoms (Fig. 2.21b). No dianionic Aun2– could be detected. The ion abundance distribution also features a log-normal shape with similar, but much less pronounced, odd-even oscillations between 5 and 21 gold atoms [299]. Anionic mass spectra by Katakuse et al. extending to n = 67 reveal more pronounced odd-even oscillations between n ~ 3–25 as well as abrupt drops at n = 35, 58 [295], neither of which could be confirmed from the mass spectra recorded by Martini et al. since, unfortunately, the recorded mass spectrum ends at n = 35. Interesting trends are noted in the helium droplet experiment upon scanning the ion yield of Aun± as a function of electron energy. For both Aun± species, the maximum ion yield shifts to a different electron energy value with increasing cluster size, however, the shifts are opposite in cationic and anionic clusters. For Aun+, the maximum shifts to lower electron energies for larger gold clusters and vice versa for Aun–. Both can be explained in terms of the dopant cluster ionization processes. In helium droplets, dopant clusters are usually not directly ionized, but rather via an intermediate He+, He*– or He* species. While the energy transferred by these species to the dopant cluster is independent of the incident electron energy, the energy available to dopant clusters from inelastic scattering of secondary electrons is increased. This energy is assumed to enhance fragmentation of larger clusters, resulting in a higher yield of small cluster (fragment) ions. In the case of anionic Aun–, the trend is reversed—higher electron energy leads to an increased yield of larger cluster anions. The authors propose that larger helium droplets produce larger clusters, but also require larger amounts of helium to be evaporated to produce gas-phase ions—the energy for this being provided by the incident or secondary electrons [299].
A key step to understanding gold catalysis on small clusters is determining their structure. It was shown that in the regime of small (n = 1–20) gold clusters, the addition or removal of a single atom can drastically alter the cluster’s catalytic activity [306]. Methods employed for structure determination of small Aun+ are ion mobility spectroscopy of bare Aun+ [301, 307] or analysis of Aun+ using different probing techniques (e.g. spectroscopy or mass spectrometry) and tagging species, such as CH3OH [308], Ar [309, 310], H2 [311] and CO [312, 313]. A key disadvantage of the tagging methods is that the interaction of the tagging species might alter the underlying gold cation structure (the “gold skeleton”) that is being probed, making a weakly interacting tagging species such as He desirable. Two recent studies combined aspects of both structure determination of Aun+ and Au-Rg (Rg = He–Xe) chemistry in a series of helium droplet experiments. The first study examined the adsorption of He to Aun+ (n ≤ ~15) [21], while the second one focused on the adsorption of rare gases He through Xe on Au+ [314]. In the latter study, large helium droplets (n ~ 106) were doped, first with the rare gas (except in the case of He), then with Au, followed by EI and analysis of the ejected ions via ToF-MS. The experimental findings are shown to be reproducible in three separate measurements with different experimental conditions and are complemented by MP2 calculations for n = 1–6, aiming to resolve the structure of the complexes detected by mass spectrometry. By analyzing the ion abundance of AunHem+ as a function of m for a given n, magic numbers can be identified, indicating structures of enhanced stability. The abundance distribution of AuHem+ reveals a pronounced maximum at m = 12, indicative of the formation of an icosahedral solvation shell, as observed for other atomic ions such as the previously discussed Rg+ (Rg = Ar–Xe) [241], anionic hydrogen and deuterium clusters [238] or Cu+ [315] and. The distribution of AuHem+ bears close resemblance to that of KrHem+, which, like AuHem+, additionally displays a local anomaly at m = 14 that is not found for other RgHem+ [241]. In principle, further solvation structures of dodecahedral and icosahedral geometry, so-called anti-Mackay layers [16, 17] can be formed. These are easily assigned via distinct magic numbers at m = 32, 44 in the mass spectra of ArHem+ [241] as well as anionic hydrogen and deuterium clusters [238], but are not clearly seen in other RgHem+ or AuHem+. Analysis of the magic numbers obtained from the abundance distributions of AunHem+ for n ≥ 2 in connection with the MP2 calculations can be used to investigate the structure of the underlying cationic gold clusters. The gold skeleton structures for n = 2–6 obtained by MP2 calculations agree well with those presented by Schooss et al. [307]. The first He atoms attached to these structures are found to locate in the molecular plane, binding directly to gold atoms, thus maximizing the charge-induced dipole interaction. The binding energies are strongest for Au2Hem+ (m = 1, 2) and Au3Hem+ (m = 1–3) with 13 and 12 meV, respectively. Binding energies for the first He atoms in AunHem+ (n = 3–6 , m ≤ n) range between 0.9 and 6 meV. For gold skeletons where the charge is distributed unevenly between the gold atoms (e.g. n = 4, 6), binding energies vary by a factor of up to ~5 depending on the binding site, which is reflected by corresponding drops in the ion abundance distributions after the more strongly bound sites are occupied. No MP2 calculations were attempted for n ≥ 7, but assuming the gold skeleton is identical to the predicted hexagonal Au7+ structure [301, 307], an observed anomaly in Au7Hem+ at m = 8 could be explained by assuming that six He atoms bind to the corner Au atoms with two additional He are bound to the central Au atom above and below the molecular plane. An enhanced ion abundance of m = 1, 2 may indicate that the latter are even more strongly bound than the He atoms located in the molecular plane. It is interesting to note that for Au5Hem+, the authors only show four He atoms bound to the corner atoms of the X-shaped Au5+, but the abundance distribution of Au5Hem+ indicates increased stability of the m = 5–8 ions. The additional four He atoms could be speculated to locate around the central Au atom which would be unoccupied in the presented Au5He4+ structure [21]. The structure of Aun+ changes from planar or near-planar to a 3-dimensional geometry around n = 8. While most of the literature agrees that Au7+ is planar and Au9+ is 3-D, there is no conclusive evidence for Au8+. Most theoretical predictions conclude that the global minimum structures are 3-D conformers, however, planar isomers are found to be rather close in energy [301, 307, 316, 317]. Whereas ion mobility spectroscopy measurements show the best agreement with a 3-D Au8+ [301, 307], the abundance distributions of Au8Hem+ show an anomaly at 3-4 attached He atoms that cannot be reconciled with any of the predicted 3-D structures, but could be explained by a planar structure with He atoms bound to four equivalent corner Au atoms [316, 317]. It should be noted that this feature is not seen as clearly in one of the measurements, which indicates it might be dependent on experimental conditions. One should also keep in mind that in a helium droplet environment, nascent clusters and cluster ions can be trapped in states other than the global minimum configuration. It is also conceivable that a mix of structures is produced. For n = 9–14, the abundance distributions of AunHem+ do not show any anomalies at small values of m, consistent with a predominance of 3-D structures.
The interest in gold chemistry led to the discovery of, amongst many other, complexes that curiously feature bonds between a noble metal and rare gas atoms. The first report of such a complex was made in 1977 by Kapur and Müller [318] who detected NeAu+ using a magnetic sector instrument with a field evaporation/ionization source, however, not reproducibly so. In 1995, Pyykkö predicted the cationic gold-rare gas complexes AuRg+ (Rg = He-Xe) and RgAuRg+ (Rg = Ar-Xe) to be stable and calculated the bond dissociation energies (BDE) of AuRg+ ranging from 0.02 eV for up to 0.9 eV and atomization energies of RgAuRg+ (Rg = Ar-Xe) ranging from 0.9 to 2.25 eV, with the heavier rare gases more strongly bound [319]. The author states that “one can expect a degree of covalent bonding between Au+ and the heavier [noble] gases”, referring to Ar–Xe. Three years later, the existence of AuXe+ (and AuXe2+) was experimentally confirmed by Schröder et al. via reaction of Au+ produced in a laser-desorption/laser-ionization source with C6F6 and Xe in an FT-ICR mass spectrometer [320]. The authors estimate 1.3 eV as the BDE of AuXe+. Two years later, Seidel and Seppelt attempted to produce AuF, but the reaction unexpectedly produced AuXe42+ as AuXe42+-(Sb2F11−)2 crystals instead [321]. Relatively strong interactions between coinage metal atom clusters and rare gas atoms have also been found in neutral Aun and mixed AunAgm clusters complexed with a few Ar and Kr atoms [322–327] and cationic CuNem+ and CuArm+ complexes [315].
Martini et al. recently published a mass spectrometric study of cationic atomic gold ions complexed with all non-radioactive rare gases by analyzing the abundance distributions of AuRgm+ (Rg = He–Xe), supported by CCSD(T)/def2-TZVPP ab initio calculations [314]. Large helium droplets were doped, first with the ligand rare gas species (except in the case of He), then with gold, and subsequently submitted to EI and analysis via ToF-MS. The mass spectra reveal a local anomaly of AuRg2+ for all rare gas ligands except He. While this is consistent with theoretical studies by Li, Zhao and co-workers, predicting a drop in BDE after m = 2 for Rg = Ar–Xe, it is surprising for Ne, where AuNe3+ is expected to actually have a slightly higher BDE than AuNe2+ [328–332]. The bond nature of AuRgm+ (m = 1, 2) is predicted to exhibit significant covalent character for Rg = Kr and Xe, slight covalent character for Ar [319, 329, 333–335], but to be entirely physical for Ne [328, 333, 335]. Alkali metal ions embedded in rare gas clusters A+Rgm, can be used as an entirely physically bound model system, but no anomalies at m = 2 can are predicted for A = Li–K and Rg = Ar, Xe [336–339]. It is conceivable that the theoretical treatment of AuNem+ is inaccurate, failing to reproduce the observed anomaly at m = 2, which might also infer that the character of binding in AuNem+ is not entirely physical in the end.
The mass spectra reveal further magic numbers of AuRgm+, namely m = 12 for He and Ne as well as 6 and 9 for Ar. No further magic numbers after m = 2 are detected for either Kr or Xe. The magic numbers of He and Ne suggest the well-known icosahedral cage structure to be formed around the central Au+ ion. Magic numbers at m = 6, 9 for Ar suggest that these complexes adopt a different packing scheme. A hard sphere model assuming pair-wise Au+–Rg and Rg–Rg interaction is adapted from previous work [339, 340] and employed to predict the energetically most favorable packing scheme for the rare gas solvation shells around the central ion, with the ratio between Au+–Rg and Rg–Rg bond lengths [335, 341, 342] as the only parameter. The model predicts close-packed (fcc or hcp crystal structure), icosahedral and octahedral structures for He, Ne and Ar, respectively, which leaves the magic numbers of these ligands in good agreement with shell closure predictions (m = 12 for He/Ne and 6 for Ar). The higher degree of covalent bonding in the Kr and Xe complexes likely prevents the simple model from making accurate predictions for the corresponding AuRgm+ [314]. Additional support for these findings is provided by molecular dynamics simulations performed by Martini et al. for AuHem+ and AuArm+ [314]. Three distinct solvation shell closures after m = 12, 32 and 44 are clearly observed in radial density function plots extracted from the simulation of AuHe110+, indicating formation of the previously mentioned anti-Mackay cage structure. Although the general shape of the distribution indicates a more liquid-like behavior, weak corresponding drops in the ion abundance of AuHem+ at m = 32, 44 can be made out [21]. Simulations of AuAr108+ at 0 K reveal further distinct shell closures at m = 10, 22 and 34. These do not have corresponding local anomalies in the mass spectra, although 10 is close to the anomaly observed at m = 9. While the outer solvation structures are no longer distinct in simulations of AuAr108+ at a finite temperature of 10 K, the first and second shells remain distinct, with a total of 9 atoms in them, matching the observed magic number. The same m = 9 magic number is also found in dissociation energy calculations performed on much smaller systems, which additionally predict a drop after m = 14 that is not reflected in the mass spectra of AuArm+. Furthermore, the simulation fails to reproduce the m = 2 anomaly since it does not correctly account for covalent bonding.
We will now take a look closer look at the chemistry of gold (clusters) with a reactant more common than rare gases, namely hydrogen. Early reports of bond formation between gold and hydrogen were made more than a century ago [343, 344], followed by numerous further studies [345, 346]. Lundberg et al. recently extended the study of gold chemistry towards the chemical bonding of hydrogen to gold clusters and their binding capacities for molecular hydrogen by investigating mixed AunHm+ (n ≤ 8, m ≤ 20) complexes via HND MS and MP2 calculations [347]. The presented mass spectra are in good agreement with previously discussed studies, showing the known odd-even oscillations of pure gold clusters as well as hydrogen attachment to clusters with 8 and fewer gold atoms. The ion abundance distributions as a function of m for a given n (Fig. 2.22a) display a rich structure including odd-even oscillations and intense magic numbers for n ≤ 5. Several local anomalies are found in complexes with n = 6–8 gold atoms. For n ≤ 5, the most abundant ions by far are AunHn+3+. The MP2 calculations reveal that, for n = 2–5, the structures of the odd-numbered magic number ions are essentially identical to previously discussed AunHem+, with each He replaced by one H2 molecule (Fig. 2.22b) [21]. In even-numbered (i.e. n = 2, 4) magic ions, one H atom is chemically bound between two gold atoms to form a structure reminiscent of the next-highest odd-numbered gold skeleton—these structures can also be considered as protonated gold clusters. Again, up to n = 4, the adsorption of H2 is equivalent to He adsorption (in the even-numbered gold skeletons, H replacing Au does not offer a binding site for H2). For n = 6, 7 there are “unoccupied” gold atom sites where no H2 is adsorbed, whereas for magic ions in the AunHem+ experiment, gold sites bind one or even two (in Au7He8+) He atoms [21]. The calculations also yield binding energies for the first few (up to 5) attached H2 molecules, which support the notion of enhanced stability of the magic number ions. For n = 1,2, the binding energies of the first two H2 molecules range between 0.8 and 1.1 eV, with a steep drop to 0 eV or even slightly negative energy for the third H2. In complexes with larger gold skeletons (i.e., n = 3-7), the binding energies gradually decrease to around 0.5 eV and the stepwise drops after a magic number ion become less pronounced.
In a follow-up study, Lundberg et al. showed that water molecules will efficiently replace H2 in AunHm(H2O)p+ complexes, evident from a clear, systematic shift up to n = 6 in the magic numbers of the ion abundance series [348]. Binding energies obtained from MP2 calculations for attached H2O are roughly twice as large than for H2 on average. While these binding energies are substantial, no significant changes in the underlying gold skeletons are found in the calculated geometries. The findings of the study indicate that water contaminations could be detrimental for the hydrogen storage capabilities of gold nanoparticles but could also be deliberately used to trigger the release of stored hydrogen by adding water to the system.

a Cluster size distributions of AunHx+ (n = 1–8) as a function of x, measured via EI of co-doped HNDs and b structures of particularly abundant cluster ions as calculated using MP2. The cluster size distributions reveal intense magic numbers and odd-even oscillations. Structures calculated for even numbers of gold atoms are reminiscent of the next highest odd-numbered gold skeleton where one gold atom is replaced by a hydrogen atom. Reproduced from [347]. Licensed under CC BY 4.0
3.3.1.6 Studies of Imidazole
Imidazole (C3H4N2, Im), is an aromatic, nitrogen-containing compound and ubiquitously found in nature as an important building block of various biomolecules [349]. Kuhn et al. recently studied the EI/EA-induced intra-cluster chemistry of Im clusters grown in HNDs by monitoring cationic and anionic fragments utilizing HND MS [350]. Anions were found to predominantly form via collision of the dopant cluster with He*–, evident from electron energy scans exhibiting a strong, well-known 22 eV resonance typical for anion formation in the HND environment [351–354]. The dominant anion series consists of dehydrogenated [Imn–H]– (n up to ~25), with several weaker series also observed consisting of intact Imn–, either pure or complexed with different combinations of CN and C2H4 fragments. The observed anions likely consist of an ionic [Im–H]– (if no CN is fragment is present) or CN– core based on the respective electron affinities of 2.67 (Im–H) and 3.8 eV (CN), solvated by n–1 neutral Im molecules. The existence of the more complex species with two fragment hints at complex reactions taking place in the Im cluster upon EA. Cationic species are formed via He+ as evident from the onset of formation at the He ionization threshold (24.6 eV) and predominantly exist as protonated ImnH+. Other cationic series observed are intact ImnH+ and [ImnCH3]+. The latter could consist of either CH3+ solvated by n Im molecules or methylated [ImCH3]+ solvated by n–1 Im molecules. While both protonated and dehydrogenated species [Imn±H]± were always more abundant than the corresponding pure Imn±, the ion abundance distributions of the latter decay less quickly towards larger cluster sizes in comparison, indicating that larger clusters are more stable against intra-cluster protonation and dehydrogenation reactions. Interestingly, local abundance anomalies hinting at magic number clusters are few and inconsistent. The only examples given are notable abundance drops in [(Imn–H)C2H4]– after n = 6 and 11. While no further magic numbers are discussed by the authors, these can also be found, albeit less pronounced, in Imn–, perhaps along with n = 8. The cationic ion abundance distributions are notably smoother, with the only possible magic numbers at n = 6 and perhaps 10 in Imn+.
A different recent study investigated organometallic compounds formed by co-doping HNDs with gold and imidazole and analyzing the mixed AumImn+ (m ≤ 6, n ≤ 15) complexes ejected upon EI with high-resolution ToF-MS (Fig. 2.23) [355]. The recorded mass spectra reveal that for every number of gold atoms, there is a number of Im molecules corresponding to a particularly abundant compound (for m = 4, both n = 3 and 4 are particularly abundant). It is tempting to assign these as magic number ions corresponding to particularly stable structures, however, DFT calculations performed for up to m = 4 gold atoms reveal that this is not universally accurate. Whereas the optimized structures of AuIm2+ and Au3Im3+ are indeed found to be particularly stable compared to their n ± 1 counterparts, the structures calculated for the anomalously abundant compounds Au2Im3+, Au4Im3+ and Au4Im4+ were not (the binding energy of Au4Im4+ is incorrectly given as 1.7 eV in Fig. 2.7 of [355]. The correct value of 1.26 eV is displayed in Fig. 2.8 of the same publication). The authors find a different explanation in that compounds with more Im molecules than the experimentally observed magic numbers generally fragment by loss of neutral Im, thus enriching these sizes. Compounds with a lower number of Im molecules tend to fragment by loss of neutral Au atoms, perhaps along with additional Im molecules, possibly further contributing to the strong ion signals of the particularly stable AuIm2+ and Au3Im3+.

a Mass spectrum of organometallic AumImn+ with different ion series marked in color and b extracted cluster size distribution (m = 1–6) as a function of n, with vertical lines indicating the most abundant ions. While the pure ImnH+ series (gray) exhibits a rather smooth distribution, distinct maxima are found in the AumImn+ series with m = 1–6. Reproduced from [355]. Licensed under CC BY 4.0
3.3.1.7 Studies of Buckminsterfullerene C60
Fullerenes, the cage-shaped molecular allotropes of carbon have created tremendous interest in their unique physical and chemical properties since their discovery by Kroto et al. in 1985 [4], recognized in 1996 with the Nobel Prize in Chemistry. A short comprehensive review of fullerenes was published by Klupp et al. [356]. The most common fullerene, Buckminsterfullerene (C60), has been widely investigated by a variety of techniques, including mass spectrometry of C60-doped helium droplets. Notable findings include the first dianions detected in a helium droplet environment [357] and the independent confirmation [150, 206] of the assignment of C60+ as the first (and so far only) diffuse interstellar band (DIB) carrier, first assigned by Campbell, Walker and co-workers in their ion trap experiment [147, 148, 358]. Mauracher and co-workers give a detailed summary of C60 experiments performed in helium droplets by the Innsbruck group in their comprehensive review from 2018 [22]. Besides the interaction of fullerenes with the helium environment, the experiments investigated in detail the adsorption of atoms and small molecules such as He, H2, N2, O2, H2O, NH3, CO2, CH4 and C2H4, to positively or negatively charged C60 and C70 as well as fullerene aggregates (see [22, 359] and references therein). Additionally, complexes of C60 with atomic carbon [360] as well as sodium and cesium [361, 362] were investigated. While former experiments aimed to understand the growth processes involved in the formation of fullerenes themselves and their role in extraterrestrial chemistry, research of metal-doped (especially with alkali and earth alkali species) fullerides is driven by hot topics such as potential applications as superconductors [363–366] and high capacity hydrogen storage materials [367–370]. A peculiar structure formed by reaction of C60 with carbon atoms is the particularly stable C60=C=C60 “dumbbell” structure, which was first synthesized in the late 1990s [371, 372] and recently formed in helium droplets and detected as [C60=C=C60]± [360]. Goulart and co-workers recently observed small ionic complexes between gold and C60 formed in helium droplets and found a similar, highly stable [C60AuC60]± dumbbell structure, both in anionic and cationic form (Fig. 2.24) [373]. The structure may be reminiscent of the linear [XeAuXe]+, which has also been found to be surprisingly stable, considering it consists of a noble metal and two rare gas atoms [319, 374]. A follow-up study by Martini and co-workers examined larger anionic and cationic clusters of up to ~10 C60 molecules and up to ~20 metal atoms (either gold or copper) formed in helium droplets and subsequently ionized by electron impact or EA [375]. The observed ion intensities show several other local anomalies indicating ions of increased stability. As expected from the previously described experiment, (C60)2Au± exhibit strong ion signals, which are interpreted as the previously discussed [C60AuC60]± dumbbell structure. Similarly, strong ion signals observed for (C60)2Cu± may lead to the assumption of a similar structure, however, there is no theoretical evidence to support this conclusion and the observed ion abundances are not as dominant as in the Au series. Most (C60)mMn± (M = Cu or Au) ion series with m ≤ 3 exhibit some form of odd-even oscillations, where ions with an odd number of metal atoms n are generally found to be more stable (n = 3 is often prominent). A few exceptions where compounds show increased stability for even n are C60Aun– (n = 2, 4), (C60)3Aun+ (n = 4, 6, 8), (C60)3Aun– (n = 4, but also 1) and (C60)2Cun+ (n = 4, 6, 10, but also 1). Two more noteworthy observations can be made for Cu compounds. First, the ion intensity series of (C60)mCun± exhibits a sudden change from heavily structured distributions for m ≤ 2 to a smooth ones for m ≥ 3, with only (C60)3Cu4– showing slight magic character. A similar change is observed in the Au series as well, but for significantly bigger ensembles between m ≤ 4 and m ≥ 5). The reason for this sudden change in small (C60)mCun± compounds remains unclear. Second, di-anionic compounds (C60)mCun2– with m = 2, 4, 6 and odd n can be observed in the mass spectra (compounds with even n might also be present, but would be buried in the ion signals of singly charged species), significantly smaller than the smallest bare C60 dianion observed in helium droplets, (C60)52– [357, 376]. Interestingly, no (C60)mCun2– with odd m are observed, even though such ions should be discernible if the ion signal intensities were comparable with dianions of neighboring, even m. This circumstance hints at a pronounced odd-even effect in the formation or stability of (C60)mCun2–, which unfortunately cannot be explained at this point. While the measured ion abundances for small (C60)mAun± (m ≤ 2, n ≤ 2 and m = 1, n = 3) agree well with theoretical predictions [373], there are only few other theoretical investigations of small (C60)mMn compounds that would help to explain the observations made.

a Cationic and b anionic mass spectra of HNDs co-doped with gold atoms and C60 molecules. The most striking feature is the dramatically increased abundance of both cationic and anionic (C60)2Au. These ions were found to adopt a highly stable dumbbell-shaped structure similar to [C60=C=C60]+ [371372360]. Reproduced from [373]. Licensed under CC BY 4.0
3.3.1.8 Complexes of Formic Acid
Formic acid (HCOOH, abbreviated FA) is the simplest carboxylic acid and can be used as a model system for the larger, more complex RCOOH compounds (where R is one of many possible organic substituents). FA is ubiquitous in earth’s atmosphere and plays an important role in atmospheric chemistry [377], it was the first organic acid detected in interstellar space [378], has further been detected in galactic hot molecular cores, regions of increased density and temperature within molecular clouds that feature rich chemistry [379] and was considered to be involved in the formation of glycine precursors, albeit with a negative result [380]. In terms of technical applications, FA has been demonstrated to be an excellent fuel for use in direct fuel cells, achieving superior performance compared to direct methanol fuel cells at ambient conditions [381, 382]. The extensive research devoted to finding suitable catalysts for direct formic acid fuel cells, a key problem in their commercial application, has found palladium-based catalysts as the best-performing candidate so far [382, 383]. Recent investigations have considered nanostructures such as hollow Pd-nanospheres [384], various core-shell nanoparticles [385] as well as Pd and La nanoparticles supported on modified C60 [386–388], which show promise for the use in direct FA fuel cells. Interestingly, little research has been devoted to the study of pure complexes of FA and C60.
Two recent studies by Mahmoodi-Darian et al. investigated the interaction of HNDs doped with FA [389] as well as FA and C60 with low-energy electrons (Fig. 2.25) [390]. The most abundant anion cluster series in the purely FA-doped droplet experiment was dehydrogenated [FAn–H]–, whereas for cations, protonated [FAn+H]+ dominates the mass spectrum. Less abundant, but also observed are undissociated [FAn]±, deprotonated [FAn–H]+, and the more abundant ions complexed with one or more water molecules [389].

Reproduced with permission from Ref. [390]. © copyright Elsevier. All rights reserved
Size distributions of various mixed cluster ions composed of FA, water (W) and C60. By far the most intense ion series belongs to [FAnH]+, which does not exhibit any noteworthy features, however. Series with added W and C60 molecules reveal a richer structure. Interestingly, the magic character of WFA5 and W2FA6 indicated in (b) appears to be preserved when adding one C60 molecule, as evident in (d), (e).
Whereas residual H2O is a very common impurity in mass spectra recorded in helium droplet experiments, the amount of water in the present mass spectra is much larger than in previous experiments with other hydrocarbon, C60 or metal clusters on the same apparatus, but comparable to a study of methanol and ethanol clusters, where the large amounts of water observed were attributed to be produced an intra-cluster reaction [391]. While no such attempt has been made in the present study, Bernstein and co-workers added small amounts of water to the FA sample without observing a significant difference in soft x-ray ionization mass spectra of pure FA clusters, concluding that the presence of (H2O)[FAn+H]+ can be explained by loss of a CO molecule from [FAn+1+H]+ [392]. While a contamination of either the vacuum chamber and/or the sample cannot be ruled out, it is likely that the large amount of water in the mass spectra arises from intra-cluster ion-molecule reactions [389]. The findings are in good general agreement with a study utilizing sputtering of frozen FA films by energetic 252Cf fission fragments—the anion mass spectra are dominated by [FAn–H]– with minor contributions of (H2O)m[FAn–H]– (m ≤ 2), similarly the cationic spectra feature mostly [FAn+H]+, with (H2O)m[FAn+H]+ (m ≤ 2) also being detected [393, 394]. In this study, undissociated compounds or [FAn–H]+ could not be detected due to poor mass resolution. A study of low energy (1 eV) EA to FA clusters produced by supersonic expansion agrees well in observing primarily [FAn–H]– and [FAn]– in the mass spectra ranging up to m/z 200 [395]. A notable difference compared to the helium droplet and sputtering experiments in the mass spectra is the absence of (H2O)[FAn–H]– for n = 2 and 3, accompanied by the appearance of peaks 13 and 21 u above [FAn]– (n = 2, 3), whose composition remains unclear. In the helium droplet experiment, the anion series of [FAn]– and (H2O)[FAn–H]– show abrupt onsets following n = 2 and n = 5, respectively [389]. The observed onset of [FAn]– is in good agreement with the pure FA cluster experiment [395]. In addition, theoretical studies predict a substantial negative adiabatic electron affinity (AEA) of around -1 eV for both the formic acid monomer [396, 397] and dimer [396]. The negative electron affinities likely render the corresponding anions short-lived, whereas the trimer is found to have an AEA closer to 0 eV [396] and can thus be assumed to possess a longer lifetime, explaining the observed onset in the abundance distribution. While calculations are available to explain the sudden onset in (H2O)[FAn–H]–, the sputtering experiments reveal a similar onset [393, 394]. Whereas most anionic and cationic cluster series in the helium droplet experiment show a rather smooth abundance distribution, some noteworthy magic numbers such as n = 5 for [FAn–H]– and (H2O)[FAn+H]+ as well as n = 4, 6 for (H2O)2[FAn+H]+ can be found. The magic number character of [FA5–H]– is rather weak, but can be found in the sputtering experiments (potentially together with n = 6) [393, 394]. The magic number character of (H2O)[FA5+H]+ is much stronger and universally found in other experiments utilizing fission fragment sputtering of frozen FA [393, 394], soft x-ray photoionization of pure FA clusters [392], a variable temperature and pressure electron ion source (though it should be noted that the distributions depend strongly on the source conditions) [398], and an electron impact study of mixed FA-water clusters [399]. Theoretical studies found that the composition of (H2O)[FAn+H]+ probably changes from a central FA·H+ ion to a central H3O+ between n = 3 and 4 with (H2O)[FA5+H]+ adopting a structure where the central H3O+ is surrounded by a cyclic arrangement of 5 FA molecules [399, 400]. While the magic character of (H2O)2[FAn+H]+ (n = 4, 6) is also found in the soft x-ray photoionization [392] and variable temperature and pressure electron ion source [398] experiments, no theoretical work has been published on this ion. Magic numbers in the series of [FAn+H]+ were found in the soft x-ray experiment (n = 5) if a sufficient delay between laser pulse and ion extraction is introduced, allowing for slow decays of [FAn+H]+ into [FAn-1+H]+ and [FAn-2+H]+ to occur [392], as well as in the variable temperature and pressure ion source experiment, where the magic number varied between n = 4–6 depending on source conditions [398]. None of these additional magic numbers were found in the helium droplet and sputtering experiments or theoretical investigations [389, 399–402]. An interesting feature, unsurprisingly exclusive to the helium droplet experiment, is the detection of FA anions complexed with up to 15 helium atoms, namely Hem[FA–H]– and Hem[FA2–H]– [389]. The abundance distribution of Hem[FA–H]– shows several local anomalies at m = 5, 7 and 11 helium atoms attached. A possible anomaly at m = 9, which would complete a weak odd-even oscillation, is unfortunately obscured by the dominating interference of (H2O)2[FA–H]–. While the authors do not draw further conclusions from these observations, and no theoretical work has been devoted to FA anions complexed with helium, a comparison can be made with of acetic acid (AA) anions complexed with heliums observed in a previous study by da Silva et al. Attachment of up to 14 He atoms to AAn– and [AAn–H]– (n ≥ 2) was observed, but no helium attachment to the (dehydrogenated or undissociated) AA monomer anions was detected [403]. The authors assumed that the attachment of He atoms is likely hindered by the excess charge in AA–, but favored by the neutral AA moieties present in larger AAn– clusters. This is contrasted by the FA study, where Hem[FA–H]– actually presents the strongest signal among the He attachment series. Since FA and AA are chemically similar and it can be very tricky to find the right conditions for helium attachment to any ions, it seems more plausible that experimental conditions are responsible for the lack of helium attachment to AA monomer anions.
In a follow up study, Mahmoodi-Darian et al. utilized EA and EI of large helium droplets co-doped with FA and C60 to record mass spectra of mixed complexes of FA and C60 (as well as H2O) [390]. Again, the presence of large amounts of water in complexes involving FA can be noticed, however, the relative intensity of C60 complexed with H2O is much lower, which strengthens the interpretation of H2O in FA complexes arising mainly from intra-cluster reactions. Some interesting features can be found in the abundance distributions of [(C60)p(H2O)mFAn]± complexes (along with their hydrogenated and dehydrogenated counterparts) when compared to complexes without C60. Whereas [FAn+H]+ and [FAn–H]– are the dominant series of bare FA clusters in the positive and negative mass spectra of pure FA clusters, the hydrogenation and dehydrogenation reactions are found to be strongly suppressed in the presence of C60 for low numbers (≲ 6) of FA molecules. A comparison of the ionization energies (7.6 vs. 11.3 eV [133]) and electron affinities of C60 and FA (2.68 [133] vs. -1.27 eV [396]) suggests that in both cases it is more favorable for the additional charge to reside on C60 rather than FA, thus suppressing the autoprotonation and dehydrogenation reactions. The distributions of [C60FAn]+ and [C60FAn+H]+ show an enhanced abundance at n = 4 and n = 4–6, respectively, in contrast to the smooth, featureless distributions of the compounds without C60. While [C60FA6H]+ exhibits the strongest enhancement in these series, its magic character is lost upon further addition of C60 molecules, whereas the anomaly at n = 4 persists for up to at least four C60 molecules. The strong magic character found in [(H2O)FA5H]+ is preserved, if not strengthened, upon addition of at least up to four C60 molecules. It is not obvious, however, whether the presumed structure of a central H3O+ surrounded by a cyclic arrangement of five FA molecules [399, 400] changes when C60 is added, since the proton affinity of C60 (8.75 eV [404]) is significantly higher than that of FA and water (7.69 and 7.16 eV, respectively [133]). In the [C60(H2O)2FAnH]+ series, the magic character of [(H2O)2FA6H]+ is not only clearly preserved, but further enhanced upon addition of C60. The magic character of [(H2O)2FA4H]+ is probably preserved since after n = 4, a strong drop in the ion abundance can be observed, however, the local anomaly is more clearly seen in the bare [(H2O)2FAnH]+ series, perhaps due to different experimental conditions shaping the size distribution.
In the anionic mass spectra, the distributions of [C60FAn]– exhibits a local anomaly at n = 4, which is not reflected in the bare cluster [FAn]– distribution. The local anomaly is not found in [(C60)2FAn]–, but reappears in [(C60)3FAn]–. The shape of the [FAn–H]– distribution changes from a plateau-shaped maximum at n = 3–5 in the bare FA study to a maximum at n = 3 in the C60 study, which likely coincides with the maximum of the envelope of the distribution and is thus not considered a local anomaly. Additionally, [FA5–H]– no longer presents a local anomaly, illustrating the weakness of its magic character described earlier. The changes in the distribution shape are presumably caused by different experimental conditions. Interestingly, n = 1 represents the strongest ion signal in the [C60FAn]+ series, but becomes strongly suppressed in [(C60)pFAn]+ for p = 2–4 where the strongest signal is found at n = 2. Similar can be observed in the [(C60)pFAn]– series (p = 1–3) although [(C60)2FA]– is not suppressed as strongly, and, to a lesser extent, in the [(C60)pFAnH]+ and [(C60)pFA–Hn]– series. In complexes of C60 and ammonia, the signal of [(C60)p(NH3)]+ is also found to be strongly suppressed for n = 1 and p = 2, 3 [405]. Certain small ionic complexes are also found to be absent in complexes of C60 and either Cu or Au), although no consistent, simple pattern is observed for these compounds [375] (also see previous discussion on C60-metal complexes).
3.3.1.9 Complexes of Adamantane and Water
Adamantane (C10H16, Ad) is basically a hydrogenated unit cell of the diamond crystal structure and the smallest of the diamondoids, a group of hydrocarbons sometimes referred to as nanodiamonds. They have intriguing physical and chemical properties as well as a wide array of applications in chemistry, material science and medicine [406–409]. Recently, Goulart et al. conducted the first study of Ad clusters synthesized in HNDs [410]. The authors observed Adnz+ clusters with n ≤ 140, z ≤ 3 and the series for z = 2 and 3 starting at n = 19 and 52 molecules, respectively (Fig. 2.26). Several magic numbers are identified which correspond to particularly stable, geometric packing schemes and that mostly persist regardless of the charge state. Whereas the first magic numbers n = 13 and 19, only observed in Adn+, suggest icosahedral packing schemes, further magic numbers associated with icosahedral packing like n = 55 are notably absent in the mass spectra. Instead, magic numbers of n = 38, 52, 61, 68, 75 and 79 are observed for all charge states (beyond their appearance size in the case of trications), which line up perfectly with predictions for face-centered cubic (fcc) packing structures [411]. Apparently, the preferred packing scheme of Ad clusters changes from icosahedral to fcc between n = 19 and 38. The study of Ad was extended with an investigation of HNDs co-doped with Ad and water by Kranabetter et al. [412]. The authors recorded mass spectra of both pure Adn+ and protonated (H2O)mH+ as well as mixed (H2O)mAdn+ clusters. Notable magic numbers observed in these experiments were again n = 13 and 19 for Adn+ as well as m = 4, 11, 21, 28 and 30 for (H2O)mH+, with m = 21 clearly showing the strongest magic character. These features were observed regardless of the doping order. The mixed cluster series displayed an interesting interplay between the magic numbers of both pure series. For one, the exceptional magic character of (H2O)21+ is preserved when complexed with any number of Ad molecules between 6 and 19, growing relatively stronger with larger numbers of Ad molecules. Additionally, (H2O)mAd12+ and (H2O)mAd18+ are particularly abundant for 5 < m < 21, suggesting that intermediate size water clusters are able to effectively replace a single Ad molecule in the icosahedral magic number structures of Ad13+ and Ad19+. The strongest magic character is observed when both these features are combined in (H2O)21Ad12+ and (H2O)21Ad18+. The authors also attempted, but were unable to find, evidence for the formation of clathrate hydrates [413–415] in which small numbers of Ad are trapped within a structured water cluster.

Reproduced with permission from Ref. [411]. © copyright American Chemical Society. All rights reserved
Ion abundance distributions of adamantane cluster ions Adnz+ (z = 1–3) [410]. Interestingly, all magic numbers appear independent of the charge state. While small magic numbers (n = 13 and 19, only observable for z = 1) indicate cluster ions adopting icosahedral shape, the higher magic numbers of n = 38, 52, … are in perfect agreement with predictions for fcc packing structures.
3.3.2 Multiply Charged Droplets
The consequences of electron impact ionization in pristine helium droplets have recently been studied using a tandem mass spectrometer setup that is able to resolve the charge state of ionized droplets [171, 416]. In the referenced setup, neutral helium droplets are produced through expansion of helium gas (Messer 99.9999% purity) into vacuum at 20 bar stagnation pressure through a 5 µm nozzle, cooled down to 4-10 K. After leaving the cluster source through a skimmer, the neutral droplets are ionized by electron impact before entering a 90° spherical electrostatic analyzer, where charged droplets can be selected by their mass-per-charge ratio. In the second stage of the tandem mass spectrometer, previously selected charged droplets can again be ionized and analyzed for their final mass-per-charge ratio in an identical electrostatic analyzer. By scanning the second electrostatic analyzer and detecting the charged droplets that pass the analyzer with a channel electron multiplier, mass-per-charge spectra for positively and negatively charged droplets can be obtained.
Positive charge carriers in helium droplets can be formed if an electron above the ionization threshold of helium [IE(He) = 24.6 eV] collides with a He atom and forms a \({\text{He}}^{+}\) ion [22]. Resonant charge hopping processes and subsequent formation of \({\text{He}}_{2}^{+}\) upon charge localization [57] will lead to the formation of an Atkins Snowball [25], an ionic core of \({\text{He}}_{3}^{+}\) [417] surrounded by a rigid shell of helium atoms. An electron that has a kinetic energy higher than 48 eV can also produce multiple positive charges in a droplet. Negative charge carriers in helium droplets can be introduced upon impact of electrons with energies below the ionization threshold of helium. At a kinetic electron energy of 22 eV excited \({\text{He}}^{*}\) can be formed which, contrary to ground state helium, can capture a slow electron and form highly mobile, heliophobic \({\text{He}}^{*-}\) [352]. The resonant formation of molecular \({\text{He}}_{2}^{\text{*}-}\) in helium droplets has also been observed [22], however, the production of the molecular anion seems to be far less efficient. At even lower electron energies of a few eV, which resembles the energy necessary for an electron to penetrate the surface of a helium droplet [418], slow electrons can form voids in the droplet and reside as electron bubbles [419].
Subjecting a precursor droplet to subsequent ionization leads to the appearance of product droplets detected at rational fractions of the precursor peak as shown in Fig. 2.27. The individual peaks originate from multiple additional charges added to the precursor droplets and therefore reducing their mass-per-charge ratio in the second ionizer. For instance, the peak at 1/2 in the anionic spectrum is produced by adding one additional charge to the singly charged precursor droplet, respectively adding two additional charges for the peak at 1/3. For all peaks in the shown cationic spectrum to appear, the initially negatively charged precursor must be positively charged multiple times while passing the second ionizer to bear a net positive charge. Peaks above the ratio of 1/2 can only be seen when the precursor droplet already carries multiple charges before being subjected to repeated ionization in the second ionizer. In Fig. 2.28a a charge state spectrum of such a positively multiply charged droplet is shown. To resolve the individual charge states in the second analyzer, the kinetic energy of the electrons in the second ionization stage is set as to introduce negative charges into the droplets that recombine with already present positive charge centers and therefore decrease the net charge state of the droplet. As the charge state of a droplet decreases, the mass-per-charge ratio increases and therefore the droplet is detected at a higher mass-per-charge ratio than the precursor droplet.

Mass-per-charge spectra for positively (red) and negatively (blue) charged droplets obtained by subjecting anionic precursor droplets (green) with a droplet size of 25×106He/z to subsequent electron impact ionization. Precursor droplets were produced at 6K by ionizing neutral droplets with electrons of 25.9 eV kinetic energy and 615 µA electron current in the first ionizer. Product droplets at rational fractions relative to the precursor result from ionizing selected precursor droplet with 167 µA electron current at 23.0 eV (anions) and 38.0 eV (cations)
As described above, negative charges can either be introduced into a droplet by capturing slow electrons that form electron bubbles or through the formation of \({\text{He}}^{*-}\). Electrons that have a kinetic energy of 22 eV or 44 eV can inelastically scatter with helium atoms, leading to excitation of a helium atom and capture of the free electron by the excited He*. The corresponding resonances in the kinetic energy spectrum of incident electrons in the second ionizer is shown in Fig. 2.28b. While both curves of the charge reduction process show a resonance around 3 eV electron energy, which corresponds to the formation of an electron bubble, only the reduction to 3/2 has two distinguishable resonances at and above 22 eV, the latter resembling the formation of \({\mathrm{He}}_{2}^{*-}\). In the region between 23 and 34 eV the formation of negative and positive charge carriers is possible, leading to a competition between both processes in the droplets. Charge reduction of multiply charged anions follows the same mechanisms as in cations. However, as observed autodetachment of negative charges in precursor droplets, shown in Fig. 2.29a, suggests, negative charges in helium droplets are not as stable as positive charges during the approximately 2 ms of flight time from the first ionizer to the detector.

a Charge state analysis of multiply charged cationic droplets with a droplet size of 1.89x106He/z, produced at 7 K with an electron current of 294 µA at 39.6 eV in the first ionizer. Using an electron current of 195 µA at 23 eV in the second ionizer to introduce negative charges into the droplets and decrease net positive charge shows droplets that had their charge state reduced from up to +12 to +1 (integer positions) and +17 to +2 (half-integer positions) respectively. b Energy spectra of a charge reduction process in positively charged precursor droplets resulting in product droplets with a relative mass-per-charge ratio of 2/1 and 3/2. The electron energy range where resonances of negative charge formation, i.e. electron bubbles and He*-,He2*- are observed are highlighted in grey

a Autodetachment of negative charge carriers in anionic precursor droplets with a droplet size of 3.78×106He/z, produced at 7 K with an electron current of 685 µA at 28.5 eV in the first ionizer and second ionizer off. b Charge state of droplets versus the minimum droplet size at which the charge state can be observed for anionic (blue triangles) and cationic (red diamonds) droplets
When changing the charge state of precursor droplets, all observed patterns in resulting product droplets are strongly dependent on nozzle temperature, precursor size and ionization settings. Increasing electron current or the kinetic energy of electrons generally leads to a relative increase in intensity of lower mass-per-charge ratios in product droplets. An increase in precursor droplet size results in a higher number of achievable charge states upon secondary ionization. The possible share of already multiply charged droplets when selecting a droplet mass-per-charge ratio as precursor droplet is governed by the neutral droplet size distribution at a given temperature. For lower temperatures, more droplets double the size of the selected precursor droplet will be available, which, when being doubly charged upon ionization will appear at the same mass-per-charge ratio as the precursor droplet. The same principle holds for triply as well as more highly charged droplets.
The highest number of charges a droplet can carry for the ms timescale of the experiment is limited by the size of the droplet. In Fig. 2.29b the minimum droplet size for the observed charge states is shown both for positive and negative charges. While the threshold size for doubly charged cations is (1.00 ± 0.05)×105 helium atoms, doubly charged anions can only be observed above a threshold size of (3.95 ± 0.20)×106 atoms, at which a positively charged droplet can already hold more than 30 charges. This trend continues for further thresholds and illustrates a remarkable difference in charge stabilization mechanisms between negatively and positively charged droplets. As the threshold sizes for observable positively charged droplets scale in a linear fashion with the square of the droplet radius, it is likely that the positive charge carriers arrange near the surface of the droplets.
The experimental findings presented above show that helium droplets are capable of stabilizing multiple positive [171] and negative [416] charge carriers due to sufficiently large cohesive forces in helium droplets. In previous experiments, only doubly positively charged helium droplets [420] could be observed. The liquid droplet model [10] that takes possible multiply charging of helium droplets into account [421] underestimates the stability threshold sizes measured up to a factor of four [422]. Previous works with multiply charged Ne droplets also show a distinct discrepancy [423] in predicted threshold sizes by the liquid droplet model, which is explained by quantum effects.
3.3.2.1 Size Distributions
The size distributions of helium nano droplets produced by expansion of precooled helium through a pinhole into vacuum have been investigated under various conditions by means of titration [165] and deflection of charged droplets [161] during the last 30 years. Here we present recent results [424] that have been obtained by producing helium droplets in continuous supersonic expansion with 20 bar stagnation pressure, ionizing via electron impact and deflecting the charged droplets in an electrostatic sector analyzer (90° spherical, 0.07 m radius, 0.02 m electrode distance; 3.67° cylindrical, 5m radius, 0.01 m electrode distance, used for temperatures below 8 K).
As presented in the previous section “Multiply Charged Droplets”, especially large droplets are able to stabilize multiple charge centers upon electron impact ionization. To minimize the influence of multiply charging on the droplet size distributions, the electron current is reduced to 50 nA, the lowest current that can be achieved while still guaranteeing stable electron emission from the ionizer filament. Even at this low electron current, due to the large geometrical cross sections of helium droplets produced at temperatures below 9 K, multiple electron hits per droplet are expected. Therefore, the electron energy of the ionizer was set to 22 eV to ensure the formation of negative droplet ions that have substentially larger critical droplet sizes for stabilizing multiple charges than cationic droplets.
To calibrate the results of electrostatic deflection measurements with regards to droplet size, the characterization of the beam velocity for each expansion condition is obligatory. Prior investigations by Buchenau [36] and Henne [425] showed that the velocity of individual droplets at a given temperature is dependent on the mass-per-charge ratio of the droplet. Therefore, droplet velocities were recorded for different selected mass-per-charge ratios. The velocities were obtained by time-of-flight measurements over the distance from the ionizer to the detector. The charged droplet beam was chopped by switching off the acceleration voltage of the electrons in the ionizer with a high-speed voltage switch. The switching pulse was used as trigger to start the time measurement. Figure 2.30a shows a set of droplet velocities for 5 K nozzle temperature. While the individual time-of-flight measurements with a peak width of only 5% justify the use of our electrostatic sector as a mass-per-charge selector, the overall velocity span in the range of 20% at a given expansion temperature underlines the importance of mass-per-charge selected velocity measurements for the evaluation of droplet size distributions in deflection measurements. The beam velocities at the mean size of the recorded log-normal shaped droplet size distributions are shown in Fig. 2.30b in comparison to experimental results reported in the literature. Possible sources for the discrepancies between our measurements and the literature could be a high fraction of fast small droplets that are suppressed by our sector geometry or a larger fraction of droplets in a high angular momentum state due to the used laser drilled nozzle that would lower the overall forward momentum of the droplet beam.
The resulting droplet size distributions shown in Fig. 2.31 follow a clear log-normal shape, as has been observed for small droplets in previous experiments [161]. Contrary to previous studies, we report a log-normal shaped distribution in the whole temperature range from 4 to 9 K and corresponding mean droplet sizes. The average sizes of these log-normal shaped distributions are compared with previous measurements by the group of Vilesov on neutral droplets [165] and deflection measurements by Henne and Toennies [161] in Fig. 2.32. In the temperature range of 6.5–9 K, our measurements are in reasonable agreement with previously reported average sizes. Below this range, we observe a plateauing of the mean droplet size, contrary to He titration measurements by the group of Vilesov [165]. The He titration method, in which the average He droplet size is deduced from the measured attenuation of a droplet beam colliding with collisional helium atoms, shows an exponential trend of increasing average size of neutral droplets, suggesting the existence of droplets having average diameters in the micrometer range. Such micrometer sized droplets have recently been identified optically [426] by the same group. A possible source for the deviation from titration measurements lies in the charged nature of the helium droplets used in our experiment. As discussed before, the possibility of a droplet carrying multiple charge centers can lead to a lower apparent droplet size than if the droplet would be singly charged. Assuming the geometrical cross section of a droplet resembling the ionization cross section, an exponential rise in droplet size for temperatures below 7 K would promote multiply charging if the ionization current, as in our case, is kept constant.

a Droplet velocity distribution for different mass-per-charge values selected from a negatively charged droplet beam produced at 5 K. b Measured temperature dependent helium beam velocities determined by various experimental means. Time of flight velocity measurements by pulsing the helium beam via heating the nozzle with a pulsed laser by the group of Vilesov [165] are shown as green circles, random chopper disk measurements by Henne and Toennies [161] as red diamonds. Data obtained via pulsed ionization in our setup, as described in the text, are shown as blue triangles

Distribution of helium droplet size recorded at 5 K with an electron current of 50 nA at 22 eV. A log-normal fit to the data gives a mean droplet size of 1.316(3) × 108 He atoms

Mean helium droplet sizes for expansion temperatures that have been measured by different experimental methods and groups. Measurements produced with the presented experimental setup are shown as blue triangles, neutral He titration measurements by the group of Vilesov [165] as red circles and plate deflection measurements by Henne and Toennies [161] as green diamonds
Another plausible cause for the deviation from the literature could be the pickup of dopant molecules, such as residual water [113] in the vacuum vessel. A charge transfer from He*- to dopant species could create charge centers that are not able to trigger secondary electron emission due to a lack of sufficient ionization energy for overcoming the work function of the surface of the channel electron multiplier detector.
3.3.3 Pickup with Charged HNDs
Experiments using helium droplets for pickup have been performed for many years now [427]. Helium droplets have proven to be very suitable for experiments at ultracold conditions as well as for spectroscopy studies [206, 427]. Furthermore, HNDs have been found to be a very sophisticated way of growing nanoparticles of a desired composition and size [428]. Measured size distributions of HNDs, produced via free jet expansion, exhibit a strong correlation between ionization conditions and mean droplet size. As discussed previously, Laimer et al. developed a new experimental setup to investigate the underlying mechanisms. The tandem mass spectrometry experiment was designed to study possible fragmentation channels of highly charged HNDs. To the surprise of the experimentalists, the HNDs were found to be stable up to very high charge states. If HNDs are charged beyond a certain maximum charge density, low mass singly charged ions are ejected, leading to a negligible reduction in the amount of helium of the residual HNDs not visible within the resolution of the experiment [171]. The knowledge about the stability of highly charged HNDs was pivotal in the process of the development of further experiments performing pickup into (highly-) charged HNDs. Pickup of dopants into multiply charged HNDs promises a higher yield of dopant cluster ions since every charge center located close to the surface of a HND acts as a seed for dopant cluster growth. Thus, multiple dopant clusters are formed in every HND as opposed to only one, as is the case for pickup into neutral HNDs.
In this chapter, an experimental setup is described where pickup of neutral dopants into stable, highly charged HNDs is realized. A rendered picture of the source used to form such HNDs is shown in Fig. 2.33, while a schematic of the apparatus is shown in Fig. 2.34. The focus of this work lies on the technical realization as well as on the characterization of the experiment. A more detailed description of this setup and its performance in comparison to conventional techniques was published recently [200].

Reproduced with permission from Ref. [200]. © copyright AIP Publishing. All rights reserved
A cutaway rendering of the experimental setup using charged pickup. Including the helium droplet source, ion source, quadrupole bender, and high temperature oven.

source used for charged pickup. The green lines represent simulated trajectories of helium droplets. Reproduced with permission from Ref. [200]. © copyright AIP Publishing. All rights reserved
Schematic of the HND
3.3.3.1 Overview of the Experimental Setup
Neutral HNDs are produced via a continuous free jet expansion of pressurized and precooled helium through a pinhole nozzle (5 µm diameter) as described previously [172, 429]. After passing a skimmer, the droplets travel a distance of about 10 cm before being ionized in an electron impact ion source. A DC quadrupole is used to select HNDs of a specific mass-per-charge ratio and deflect these by 90° in either of two directions. On one side (left), a channel electron multiplier (CEM) is installed for monitoring the helium droplet beam. On the opposite side (right), the HNDs enter a differentially pumped pickup region. In addition to the oven shown in Fig. 2.34, a second pickup chamber can be installed between the first oven and the gas barrier, housing a second oven as well as a gas inlet. This source for the production of highly charged, mass-selected and doped HNDs is connected to a modified QMS/ToF-MS tandem mass spectrometer (Q-Tof Ultima, Waters/Micromass). Figure 2.35 depicts a schematic of the entire experimental setup.

Schematic of the experimental setup including the QMS/ToF-MS tandem mass spectrometer. The blue line indicates the beam of neutral HNDs; the purple line represents the trajectory of the charged mass-selected HNDs and the red line indicates the path of the extracted dopant cluster ions
Following the pickup region, dopant ions are extracted by evaporating excess helium from the HNDs in collisions with a gas of variable pressure (typically He) in an evaporation cell equipped with a RF hexapole ion guide (a detailed description of this method can be found in 3.3.4). The dopant ions now enter a region housing a RF quadrupole mass filter followed by another RF hexapole, designed to perform collision induced dissociation (CID) measurements. The RF quadrupole mass filter is used to select precursor ions of a specific mass-to-charge ratio which are guided into the collision cell. Here, the ions perform collisions of controlled energy with a monoatomic gas (typically Ar). When no CID measurements are performed, the collision gas is removed and the quadrupole mass filter simply acts as an ion guide. The final analysis is performed by an orthogonal extraction reflectron ToF-MS with a resolving power m/Δm of up to 10,000.
3.3.3.2 Pickup and Growth of Dopant Clusters
Pickup of single neon atoms in HNDs was first reported by Scheidemann et al. [32]. Since then, HNDs have been used to study numerous different species, all of which have been picked up by neutral HNDs [22]. The growth of a dopant cluster within or at the surface of a neutral helium droplet depends almost exclusively on the pickup probability. Dopants are attracted to each other via van-der-Waals interaction which leads to the formation of dopant clusters within or at the surface of a HND [22].
Samples with high vapor pressures can be introduced into our machine via a stainless-steel gas line. For samples with lower vapor pressures, two different ovens were constructed. For organic samples, a CNC-machined molybdenum holder is constructed around a glass cup that holds the sample, which is ideal for fragile samples such as biomolecules since it is highly unreactive. Molybdenum was chosen for its thermal stability and to prevent surface charges due to its conductivity. The holder is heated via a tantalum filament able to generate temperatures of up to 800K. For samples with even lower vapor pressures, e.g. metals with high melting points such as gold, a CNC-machined aluminium nitride ceramic oven is used. It is heated via a tantalum-rhenium filament and can be used at temperatures of up to 1800 K. The disadvantages, compared to the molybdenum/glass oven, are that it is harder to clean, it is more delicate and one must face the issue of surface charges on the ceramic.
3.3.3.3 Ionization and Stability of the Dopant Clusters
In order to analyze dopants picked up by neutral HNDs via mass spectrometry, the doped HNDs have to be ionized. Based on the knowledge obtained from the formation and stability of highly-charged HNDs, we have to conclude that dopant cluster ions can only be released from large HNDs upon multiply-charging of the doped HNDs and subsequent asymmetric Coulomb explosion [171]. Although superfluid helium is one of the best matrices to dissipate excess energy, the ionization process via charge transfer from He+ or He2+ is so exothermic (typically more than 10 eV) that the dopant clusters created are often fragmented.
This effect is illustrated in Fig. 2.36. A comparison between pickup into charged HNDs (Fig. 2.36a) and conventional pickup into neutral HNDs (Fig. 2.36b) demonstrates the effect of fragmentation of neutral gold clusters inside large HNDs upon charge transfer from He+ or He2+. Both methods produce Aun+ distributions featuring similar log-normal envelopes, governed by Poissonian pickup statistics. However, whereas the distribution of Aun+ produced by pickup into charged HNDs is smooth, indicating little or no fragmentation (Fig. 2.36a), intense fragmentation of Aun+ upon EI of neutral gold-doped HNDs is indicated by intense odd-even oscillations and pronounced drops in the ion yield after n = 9 and 21, suggesting magic number character of these ions (Fig. 2.36b). The very same intensity anomalies have been observed for alkali [3, 14] or noble metal cluster ions [294] when formed via conventional techniques. High ion yields relative to neighboring cluster sizes are the result of an increased stability of a specific cluster ion, either due to electronic [3] or geometric [430] reasons. The stability of Aun+ and many small metal cluster ions is predominantly determined by their electronic structure. Spin pairing of the single 6s electrons of gold explains the odd-even oscillation while closure of the 1p and 1d orbitals for 8 and 20 electrons is responsible for the intensity drops at n=9 and 21 (one extra gold atom is required to obtain a positively charged cluster). Therefore, what we observe in the experiments upon pickup into neutral HNDs, is not the size distribution of neutral dopant clusters but rather their charged fragments.

Reproduced with permission from Ref. [200]. © copyright AIP Publishing. All rights reserved
A comparison of gold cluster series produced utilizing pickup into a charged and b neutral HNDs.
In the experiment using pickup into charged HNDs, no such oscillations are observed. This means that species created after pickup into charged HNDs grow by exclusively following the statistics of the pickup process, and do not fragment in the ionization process. This effect is especially beneficial when the species of interest is rather fragile. Performing pickup into charged HNDs enables the production of high yields of fragile ions such as even-numbered gold clusters or biomolecules which are prone to fragmentation when using conventional pickup into neutral HNDs followed by an ionization process.
3.3.3.4 Extracting Dopant Clusters from Charged HNDs: An Efficient Method to Obtain Helium-Tagged Ions
In conventional HND experiments, the pickup section of the experiment is usually followed by an ionization device, the most common being an EI or PI source. When a HND becomes highly charged, some low mass singly charged ions are ejected from the HND. These ejected ions are then accessible for mass spectrometric analysis.
In the case of pickup into charged HNDs, dopant cluster ions are ejected from the HND only if the droplet shrinks below the critical size for its charge state due to the evaporation of helium atoms. The evaporation of helium atoms is caused by pickup events and the release of binding energy during dopant cluster formation [171]. In the case of strongly bound gold clusters, every pickup and addition of a gold atom to a cluster will evaporate between 5000 and 8300 helium atoms [171, 431].
In order to extract the dopant cluster ions from the HNDs in the setup shown in Fig. 2.34, the following approach is chosen. The HNDs are continuously evaporated in a collision cell where the HNDs are heated by collisions with room temperature stagnant helium gas. In the process, the HNDs shrink until they are no longer stable with respect to their charge state. As a result, charged dopant cluster ions are ejected from the HNDs which often remain solvated by a small number of helium atoms, referred to as helium-decorated or helium-tagged ions [198]. From the helium decoration, one can obtain valuable information about the dopant (cluster) ions. The temperature of these ions can be estimated very conveniently since helium evaporates at temperatures above the binding energy of the weakest bound helium atom. Apart from that, observed shell closures contain information about the geometric structure of the dopant cluster [299]. Most importantly, He-decorated species are perfectly suitable for messenger-type action spectroscopy. When light at a certain wavelength is absorbed by a helium tagged ion, energy transfer to the adsorbed helium will lead to its evaporation. Measuring either the depletion of a helium tagged ion or the formation of its photoproduct while scanning the photon energy of a tunable laser provides an absorption spectrum of ions at very low temperatures [206, 432–434]. Hence, being able to produce large amounts of helium-decorated sample species is highly desirable.
The process of helium decoration in neutral pickup regimes where dopant ions are ejected from the HND is not easily controlled. The internal energy of the dopant cluster is often high at the time it is ejected and therefore most of the dopants are ejected with very little to no helium attached. As mentioned above, in a setup using pickup into charged HNDs, the ejection of dopant molecules is triggered by relatively slow evaporation of the HNDs in a collision cell. In the present setup, this evaporation cell is a 25 cm long guiding RF hexapole filled with stagnant helium gas at room temperature. When the HNDs collide with gaseous helium atoms, the collision energy leads to evaporation of helium from the droplet. The decreasing size of the HNDs lead to the ejection of cationic dopants, often solvated by up to a few hundred He atoms [25, 171, 435] whenever the droplet size shrinks below the critical size of the current charge state. The hexapole serves to collect and guide the small ejected ions since the ejection occurs in all directions. In further collisions of the ejected dopants with gaseous helium, the helium attached to the ions can be gradually removed. Thus, the amount of helium that is still attached to the dopants can be controlled by varying the pressure in the evaporation cell. Figure 2.37 shows a comparison of different helium decoration measurements made in a setup using pickup into neutral HNDs and the described apparatus.
The upper panel of Fig. 2.37 represents a measurement where helium-decorated fullerene C60 cations were produced via pickup into charged HNDs. For the measurement, C60 was evaporated in the high temperature ceramic oven. The mass spectrum shown in the lower panel represents a measurement of helium-decorated C60+ using electron ionization of neutral HNDs doped with C60 in an action spectroscopy measurement of C60+ [206]. The comparison shows that the production efficiency of C60+ with one helium attached is increased by more than a factor of 1000 using pickup into charged HNDs. Please note that the intense peaks at m/z=724 and m/z=725 are predominantly due to isotopes of the bare fullerene cation containing four and five 13C. The high production efficiency of helium-tagged ions using charged pickup facilitates action spectroscopy of cold ions with a minimal matrix shift. Furthermore, the knowledge of the matrix shift as a function of the attached helium atoms provides information on the structure and size of the solvation layer. For a sufficiently large number of attached helium atoms, a constant matrix shift is observed.

Reproduced with permission from Ref. [200]. © copyright AIP Publishing. All rights reserved
A comparison of helium-decorated fullerenes produced in the described experiment using pickup into charged HNDs (top panel) and an experiment using pickup into neutral HNDs (bottom panel).
3.3.3.5 Tailoring the Size Distribution of Dopant Clusters Produced
By preselecting a mass-per-charge ratio of HNDs for the pickup into charged HNDs, the size distribution of dopant clusters produced can be manipulated in a very convenient way. A demonstration of this technique is shown in Fig. 2.38. Using this method to optimize the experimental parameters to produce the desired dopant cluster size distribution is especially interesting when sublimation sources are used where the partial pressure of the sample cannot be controlled easily. An example is the evaporation of sensitive amino acids which are destroyed by too high temperatures during sublimation of the sample.

Demonstration of the altering of size distributions of gold clusters created by tuning the mass-per-charge ratio and the size of the HNDs used for pickup
Not only can the mean size of the dopant clusters be altered by the selection of different mass-per-charge ratios of HNDs (see Fig. 2.36), but also the width of the dopant cluster distribution, as can be seen in Fig. 2.38. As discussed earlier, a small mass-per-charge ratio of HNDs produces smaller dopant cluster ions. When the partial pressure of the sample in the pickup cell is increased, the dopant cluster sizes produced are increased as well. The width of the dopant cluster size distribution, however, does not increase proportionally.
Figure 2.38 demonstrates this effect using gold clusters as an example. The different partial pressures of gold in the pickup cell are realized by adjusting the oven temperature. The measurement shown in the left panel was taken at an oven heating power of 60 W, whereas the measurement shown in the right panel was taken at an oven power of 75 W.
For the measurements shown in Fig. 2.38, the nozzle temperature was held at 9.2 K for the left panel and at 9.6 K for the right panel. As shown in [171], a HND can only hold more than one charge when its size exceeds 105 helium atoms. The plot in the right panel in Fig. 2.38 shows a measurement where pickup is performed predominantly with singly charged HNDs. The left panel shows a measurement with HNDs that are well above the threshold for multiply charging. In this panel, a Poissonian distribution with an expected value of λ = 16 is plotted with a red line together with the mass spectrum (black line) to clarify that the distribution follows the pickup statistics. In the right panel a Poissonian distribution is shown (red line) that has an expected value of λ=65 and was divided by a factor of 5. The size distribution of singly charged gold clusters produced is five times narrower than expected for a “normal” pickup process. Our tentative explanation is that the gold cluster ions produced in this measurement are very close to the maximum size of gold clusters that can be produced with HNDs of this size. With every gold atom picked up by the HND, a substantial part of the droplet is evaporated, hence, the HND gradually shrinks. Since the partial pressure of gold in the pickup region is rather high, every HND that passes the cell is expected to have picked up the largest number of gold atoms possible for its size. The measured distribution is therefore not determined by the pickup probability but rather the maximum number of gold atoms that a HND can pick up before it evaporates almost entirely. The tail to the right side can be explained by pickup with HNDs bigger than 50,000 helium atoms per charge. A few of these bigger droplets also exit the DC quadrupole since it has limited resolving power. Droplets exceeding 50000 helium atoms per charge can be doubly charged, according to [171]. As soon as these HNDs pick up a few gold atoms, they drop below the stable size for holding two charges and eject a small, singly charged unit. These fragment ions can be detected as monomer ions Au+. The remaining, singly charged HND now has a mass-to-charge ratio that is far bigger than the initially selected one of 40,000 helium atoms per charge, thus creating gold clusters that do not fit the main distribution, explaining the origin of the secondary, low-intensity size distribution of gold cluster ions observed between n=17 and n=30.
3.3.3.6 Proton Transfer Ionization in HNDs Pre-doped with H2
Since our experiments are typically performed with HNDs containing more than 105 helium atoms per cluster, ionization of the dopant occurs almost exclusively via charge transfer from helium. This is also the case for experiments utilizing pickup into neutral HNDs. For EI, the ionization probability is dependent on the geometrical cross section of the target helium droplet. The following equations represent the most common way for cation formation within a HND
Helium has the highest ionization energy of all elements at 24.6 eV [436]. Charge transfer from He+ or He2+ to all other elements and molecules is therefore energetically possible. Ionization energies of molecules are typically around or below 10 eV, which results in a charge transfer from He+ being exothermic by more than 14 eV in most cases. This excess energy is initially absorbed by the dopant in the form of electronic and ro-vibrational excitation. If this excitation energy is not sufficiently dissipated by the HND, it may lead to fragmentation of the dopant ion. Rovibrational excitation of dopants can be cooled very efficiently via evaporation of helium atoms due to the exceptionally high thermal conductivity of superfluid helium. However, electronic excitation into an antibonding state of a molecular dopant cannot be dissipated by the sourrounding helium. As a result, such excited dopant molecules are likely to be observed as fragments, even when picked up into a charged HND. To this end, a soft ionization process for doped HNDs was recently developed in our group utilizing the instrument shown in Figs. 2.33 and 2.34. During the time the multiply charged droplets require to travel from the ionization region to the pickup cell, all charge centers will localize as He2+ or He3+ ionic cores [417] surrounded by a dense, solid-like layer of He atoms [25]. As in the case of a conducting sphere, the charge centers will reside close to the surface of the HNDs and mutual repulsion will arrange them in some form of a two-dimensional Wigner crystal on the surface of the droplet. The ionization energy of He2+ [22] or He3+ [437] is still higher than the ionization energies of any dopant. Whereas the first dopant molecule colliding with an ionic core might fragment due to the excess energy, remaining excitation energy will be dissipated rapidly by the HND environment. Thus, the next dopant molecule which attaches to the fragmented molecular ion will not undergo fragmentation. This way, mixed clusters consisting of a fragment ion that is solvated by several intact molecules are formed. In order to produce intact dopant cluster ions, the fragmentation process needs to be suppressed.
Charge transfer from He2+ or He3+ transfers one electron from the first dopant molecule M to the He2+ or He3+ and leads to the formation of an excited cationic molecule M+*. If the cation M+ has an open electronic shell it can be highly reactive and unstable towards fragmentation. Adding a proton to M leads to a protonated cation MH+ which has a closed electronic shell and thus produces a more stable cation. Proton transfer reaction is one of the softest ways of forming cations from neutral bio molecules [438]. Proton transfer to a molecule M from a hydrogen containing cation that has a lower proton affinity than M is a very efficient process. In gas phase, exothermic proton transfer often is associated with unwanted fragmentation of the proton acceptor molecule which requires proton donors with only slightly lower proton affinity than the reaction partner to minimize the transfer of excess energy. The enormous cooling power of HNDs, however, should be capable of dissipating excess energy originating from proton transfer processes, which is relatively slow process compared to an electron transfer reaction. If molecular hydrogen H2 is picked up by charged HNDs, the following reactions will lead to the formation of (H2)nH+ ions [171].
The small proton affinity of these ions makes them perfect proton donors for almost every molecular dopant. With these pre-doped, charged HNDs, pickup of bio molecules is performed. Thus, the charge transfer process that forms sample ions within the HNDs is no longer
but rather a proton transfer process
where typically ∆E’ < ∆E, reducing ionization-induced fragmentation. Figure 2.39 shows a comparison of two mass spectra of HNDs doped with the amino acid valine. Both mass spectra were obtained via pickup into charged HNDs utilizing the setup shown in Figs. 2.33 and 2.34. Proton transfer ionization by hydrogen pre-doping (lower diagrams) can be compared with charge transfer ionization from He2+ (upper diagrams).

A comparison of two measurements with identical settings apart from the pre-doping with \({\mathrm{H}}_{2}\) in the measurement shown in the lower panels. The residual pressure of helium in the hydrogen pickup region was 0.08 mPa. The pressures are corrected for the gas type, according to the manual
For this measurement, D-valine was vaporized in a low temperature oven heated to 110 °C. In the upper left-hand panel, strong fragmentation of the biomolecule with a mass of 117 amu to its main fragment with 72 amu can be observed, corresponding to the loss of the carboxylic group COOH. As a comparison, in the lower left-hand panel, a measurement with hydrogen pre-doping at a pressure of 0.08 mPa and otherwise identical conditions as above is shown. The ionization process clearly appears to be softer indicated by the strongly suppressed COOH loss. A satellite peak that is 18 amu higher than the main ion series can be assigned to protonated valine clusters complexed with a water molecule. Since the ion signal is not distributed among the different fragmentation channels as is the case with charge transfer ionization from He2+, the signal intensity of the pure valine clusters has more than quadrupled for proton transfer ionization. It should be noted that from the mass spectra alone it is not clear whether the peaks corresponding to valine or protonated valine clusters are still intact or fragmented with the COOH neutral product still attached.
A closeup look at the monomer region is provided in the right-hand panels. In the top right panel, one can see that charge transfer from helium cations to valine, also produces protonated valine with similar intensity as the parent molecular cation. In this case, the likely proton donor is another a valine molecule. There are also peaks located one and two mass units below the parent cation, indicating hydrogen loss as a fragmentation channel, which can be explained by the large amount of excess energy available after charge transfer from He2+. In the case of H2 pre-doping, protonated valine is by far the most abundant ion and no hydrogen loss is observed. Additionally, hydrogen tagging with up to ten H2 molecules bound to protonated valine can be observed. The low binding energy of such taggants is a measure of the internal energy of the valine cluster ions. They also provide suitable messengers for action spectroscopy of cold ions [432, 433, 439–441].
3.4 Conclusion/Outlook
Many important discoveries and developments in HND research have been propelled by mass spectrometry since the emergence of the field. We attempted to review the work performed in the last decades by groups all over the world as well as our own group’s most recent research in this chapter. However, while a lot of experimental techniques and components have been refined in various ways over the last decades, the vast majority of HND MS experiments never diverted from the tried-and-true basic principle of doping neutral helium droplets with an analyte, followed by ionization and analysis. Building on our recently gained knowledge about highly charged HNDs, further experiments showed that a simple re-ordering of these steps and ionizing HNDs before pickup opens up a plethora of exciting possibilities and applications, extending far beyond the field of HND MS. The ability to control the produced dopant cluster size distribution and efficiently produce helium-tagged ions are only two examples of what could be in store for the future.
References
P. Jena, A.W. Castleman, in Science and Technology of Atomic, Molecular, Condensed Matter & Biological Systems, ed. by P. Jena, A.W. Castleman (Elsevier, 2010), pp. 1–36
O. Echt, K. Sattler, E. Recknagel, Phys. Rev. Lett. 47, 1121 (1981)
W.D. Knight et al., Phys. Rev. Lett. 52, 2141 (1984)
H.W. Kroto et al., Nature 318, 162 (1985)
M.G. Mayer, Phys. Rev. 74, 235 (1948)
O. Haxel, J.H.D. Jensen, H.E. Suess, Phys. Rev. 75, 1766 (1949)
G. Audi, Int. J. Mass Spectrom. 251, 85 (2006)
P.G. Lethbridge, A.J. Stace, J. Chem. Phys. 89, 4062 (1988)
T.P. Martin et al., J. Phys. Chem. 95, 6421 (1991)
M. Pellarin et al., Chem. Phys. Lett. 217, 349 (1994)
I.A. Harris, R.S. Kidwell, J.A. Northby, Phys. Rev. Lett. 53, 2390 (1984)
J.A. Northby, J. Chem. Phys. 87, 6166 (1987)
W. Miehle et al., J. Chem. Phys. 91, 5940 (1989)
W.A. de Heer, Rev. Mod. Phys. 65, 611 (1993)
M. Brack, Rev. Mod. Phys. 65, 677 (1993)
M. R. Hoare, in Advances in Chemical Physics (Wiley, 2007), pp. 49–135
J.P.K. Doye, D.J. Wales, R.S. Berry, J. Chem. Phys. 103, 4234 (1995)
M.B. Knickelbein, W.J.C. Menezes, J. Phys. Chem. 96, 6611 (1992)
E. Janssens et al., Phys. Rev. Lett. 99, 063401 (2007).
W. Huang, L.-S. Wang, Phys. Rev. Lett. 102, 153401 (2009)
M. Goulart et al., Phys. Chem. Chem. Phys. 20, 9554 (2018)
A. Mauracher et al., Phys. Rep. 751, 1 (2018)
L. Meyer, F. Reif, Phys. Rev. 123, 727 (1961)
L. Meyer, F. Reif, Phys. Rev. 110, 279 (1958)
K.R. Atkins, Phys. Rev. 116, 1339 (1959)
M.W. Cole, R.A. Bachman, Phys. Rev. B 15, 1388 (1977)
A.P.J. van Deursen, J. Reuss, J. Chem. Phys. 63, 4559 (1975)
J. Gspann, H. Vollmar, J. Chem. Phys. 73, 1657 (1980)
J. Gspann, Surf. Sci. 106, 219 (1981)
J. Gspann, Physica B 169, 519 (1991)
P.W. Stephens, J.G. King, Phys. Rev. Lett. 51, 1538 (1983)
A. Scheidemann, J.P. Toennies, J.A. Northby, Phys. Rev. Lett. 64, 1899 (1990)
A. Scheidemann et al., Physica B 165–166, 135 (1990)
H. Buchenau, J.P. Toennies, J.A. Northby, J. Chem. Phys. 95, 8134 (1991)
R. Fröchtenicht et al., J. Chem. Phys. 104, 2548 (1996)
H. Buchenau et al., J. Chem. Phys. 92, 6875 (1990)
M. Lewerenz, B. Schilling, J.P. Toennies, J. Chem. Phys. 102, 8191 (1995)
A. Bartelt et al., Phys. Rev. Lett. 77, 3525 (1996)
S. Yang, A.M. Ellis, Chem. Soc. Rev. 42, 472 (2012)
C. Jackschath, I. Rabin, W. Schulze, Z. Phys. D Atoms Mol. Clust. 22, 517 (1992)
D. Rayane et al., J. Chem. Phys. 90, 3295 (1989)
J.P. Toennies, A.F. Vilesov, K.B. Whaley, Phys. Today 54, 31 (2001)
A. Scheidemann, B. Schilling, J.P. Toennies, J. Phys. Chem. 97, 2128 (1993)
V.H. Dibeler, F.L. Mohler, J. Res. Natl. Bur. Stan. 40, 25 (1948)
B.P. Pullen, J.A.D. Stockdale, Int. J. Mass Spectrom. Ion Phys. 21, 35 (1976)
T. Stanski, B. Adamczyk, Int. J. Mass Spectrom. Ion Phys. 46, 31 (1983)
D. Margreiter et al., Int. J. Mass Spectrom. Ion Process. 100, 143 (1990)
B.E. Callicoatt et al., J. Chem. Phys. 108, 9371 (1998)
M. Fárnı́k and J. P. Toennies, J. Chem. Phys. 122, 014307 (2004)
N.G. Adams, D. Smith, J. Phys. B: At. Mol. Phys. 9, 1439 (1976)
B.R. Rowe et al., Chem. Phys. Lett. 113, 403 (1985)
D. Smith, N.G. Adams, Int. J. Mass Spectrom. Ion Phys. 23, 123 (1977)
B.E. Callicoatt et al., J. Chem. Phys. 109, 10195 (1998)
T. Ruchti et al., J. Chem. Phys. 109, 10679 (1998)
T. Ruchti, B.E. Callicoatt, K.C. Janda, Phys. Chem. Chem. Phys. 2, 4075 (2000)
B.E. Callicoatt et al., J. Chem. Phys. 105, 7872 (1996)
A. M. Ellis and S. Yang, Phys. Rev. A 76, 032714 (2007).
A.A. Scheidemann, V.V. Kresin, H. Hess, J. Chem. Phys. 107, 2839 (1997)
Y. Ren and V. V. Kresin, Phys. Rev. A 76, 043204 (2007).
S. Vongehr et al., Chem. Phys. Lett. 353, 89 (2002)
F. Dalfovo, Z. Phys. D Atoms Mol. Clust. 29, 61 (1994)
F. Ancilotto, P.B. Lerner, M.W. Cole, J. Low Temp. Phys. 101, 1123 (1995)
F. Ancilotto et al., Z. Physik B Condens. Matter 98, 323 (1995)
P.B. Lerner, M.W. Cole, E. Cheng, J. Low Temp. Phys. 100, 501 (1995)
F. Stienkemeier, F. Meier, H.O. Lutz, J. Chem. Phys. 107, 10816 (1997)
J. Reho et al., J. Chem. Phys. 112, 8409 (2000)
F. Stienkemeier, F. Meier, H.O. Lutz, Eur. Phys. J. D 9, 313 (1999)
M. Mella, G. Calderoni, F. Cargnoni, J. Chem. Phys. 123, 054328 (2005).
A. Hernando et al., J. Phys. Chem. A 111, 7303 (2007)
Y. Ren, R. Moro, V.V. Kresin, Eur. Phys. J. D 43, 109 (2007)
Y. Ren, V.V. Kresin, J. Chem. Phys. 128, 074303 (2008).
B. Bellina, D.J. Merthe, V.V. Kresin, J. Chem. Phys. 142, 114306 (2015)
W.K. Lewis et al., J. Am. Chem. Soc. 127, 7235 (2005)
T. Diederich et al., Phys. Rev. Lett. 86, 4807 (2001)
T. Döppner et al., J. Chem. Phys. 126, 244513 (2007)
J. Tiggesbäumker, F. Stienkemeier, Phys. Chem. Chem. Phys. 9, 4748 (2007)
M. Mudrich, F. Stienkemeier, Int. Rev. Phys. Chem. 33, 301 (2014)
T. Döppner et al., Eur. Phys. J. D 16, 13 (2001)
Th. Diederich et al., Phys. Rev. A 72, 023203 (2005)
M. Rossi et al., Phys. Rev. B 69, 212510 (2004)
T. Döppner et al., Phys. Chem. Chem. Phys. 9, 4639 (2007)
L. Köller et al., Phys. Rev. Lett. 82, 3783 (1999)
T. Döppner et al., Eur. Phys. J. D 43, 261 (2007)
T. Döppner et al., Phys. Rev. Lett. 94, 013401 (2005)
F. Verluise et al., Opt. Lett., OL 25, 575 (2000)
N.X. Truong et al., Phys. Rev. A 81, 013201 (2010)
T. Ditmire et al., Phys. Rev. A 53, 3379 (1996)
A. Przystawik et al., Phys. Rev. A 78, 021202 (2008)
A. Hernando et al., Phys. Rev. B 78, 184515 (2008)
S. Göde et al., New J. Phys. 15, 015026 (2013)
G. Droppelmann et al., Eur. Phys. J. D 52, 67 (2009)
C. P. Schulz et al., Phys. Rev. Lett. 92, 013401 (2004)
S. Müller, M. Mudrich, F. Stienkemeier, J. Chem. Phys. 131, 044319 (2009)
S. Müller et al., Phys. Rev. Lett. 102, 183401 (2009)
W.K. Lewis, C.M. Lindsay, R.E. Miller, J. Chem. Phys. 129, 201101 (2008)
W.K. Lewis et al., J. Am. Chem. Soc. 126, 11283 (2004)
W. K. Lewis, R. J. Bemish, and R. E. Miller, J. Chem. Phys. 123, 141103 (2005).
T. M. Falconer et al., Rev. Sci. Instrum. 81, 054101 (2010)
W. K. Lewis et al., Rev. Sci. Instrum. 83, 073109 (2012)
W. K. Lewis et al., Rev. Sci. Instrum. 85, 094102 (2014)
M.N. Slipchenko et al., Rev. Sci. Instrum. 73, 3600 (2002)
R. Katzy et al., Rev. Sci. Instrum. 87, 013105 (2016)
D. Verma, A.F. Vilesov, Chem. Phys. Lett. 694, 129 (2018)
S. Yang, S.M. Brereton, A.M. Ellis, Rev. Sci. Instrum. 76, 104102 (2005)
S. Yang, A.M. Ellis, Rev. Sci. Instrum. 79, 016106 (2008)
U. Henne, Untersuchung großer durch Elektronenstoß erzeugter negativer und positiver Helium-Clusterionen, PhD dissertation, University of Göttingen/Max-Planck-Institut für Strömungsforschung, 1996
S. Yang et al., Phys. Chem. Chem. Phys. 7, 4082 (2005)
S. Yang et al., J. Phys. Chem. A 110, 1791 (2006)
A.M. Ellis, S. Yang, Chin. J. Chem. Phys. 28, 489 (2015)
A. Boatwright, J. Jeffs, A.J. Stace, J. Phys. Chem. A 111, 7481 (2007)
B. Shepperson et al., J. Chem. Phys. 135, 041101 (2011)
S. Yang, S.M. Brereton, A.M. Ellis, Int. J. Mass Spectrom. 253, 79 (2006)
S. Yang et al., J. Chem. Phys. 127, 134303 (2007)
J. Liu et al., Phys. Chem. Chem. Phys. 13, 13920 (2011)
A. Boatwright et al., Faraday Discuss. 162, 113 (2013)
D. Spence et al., Int. J. Mass Spectrom. 365–366, 86 (2014)
S.A. Krasnokutski, F. Huisken, J. Phys. Chem. A 115, 7120 (2011)
C. Feng et al., Phys. Chem. Chem. Phys. 17, 16699 (2015)
B. Sitorus et al., AIP Conf. Proc. 2049, 020066 (2018)
H.-P. Loock, L.M. Beaty, B. Simard, Phys. Rev. A 59, 873 (1999)
F.S. Ashmore, A.R. Burgess, J. Chem. Soc. Faraday Trans. 2 73, 1247 (1977)
M. Theisen, F. Lackner, W.E. Ernst, Phys. Chem. Chem. Phys. 12, 14861 (2010)
M. Theisen, F. Lackner, W.E. Ernst, J. Chem. Phys. 135, 074306 (2011)
M. Theisen et al., J. Phys. Chem. Lett. 2, 2778 (2011)
M. Theisen, F. Lackner, W.E. Ernst, J. Phys. Chem. A 115, 7005 (2011)
A. Schiller et al., Eur. Phys. J. D 75, 1 (2021)
P. Thaler et al., J. Chem. Phys. 143, 134201 (2015)
R. Messner et al., J. Chem. Phys. 149, 024305 (2018)
M. Ratschek, M. Koch, and W. E. Ernst, J. Chem. Phys. 136, 104201 (2012).
F. Lindebner et al., Int. J. Mass Spectrom. 365–366, 255 (2014)
G. Krois et al., Phys. Chem. Chem. Phys. 16, 22373 (2014)
F. Lackner et al., J. Phys. Chem. A 117, 11866 (2013)
P.J. Linstrom, W.G. Mallard, NIST Chemistry WebBook, NIST Standard Reference Database Number 69 (National Institute of Standards and Technology, 2018)
S.A. Krasnokutski, C. Jäger, T. Henning, ApJ 889, 67 (2020)
S.A. Krasnokutski, F. Huisken, J. Phys. Chem. A 114, 7292 (2010)
S.A. Krasnokutski, F. Huisken, J. Phys. Chem. A 114, 13045 (2010)
S.A. Krasnokutski, F. Huisken, J. Phys. Chem. A 118, 2612 (2014)
S.A. Krasnokutski, F. Huisken, J. Chem. Phys. 142, 084311 (2015)
S.A. Krasnokutski, F. Huisken, Appl. Phys. Lett. 105, 113506 (2014)
F. Salama, Proc. Int. Astron. Union 4, 357 (2008)
A. Leger, J.L. Puget, Astron. Astrophys. 137, L5 (1984)
L.J. Allamandola, A.G.G.M. Tielens, J.R. Barker, Astrophys. J. Lett. 290, L25 (1985)
A.G.G.M. Tielens, Annu. Rev. Astron. Astrophys. 46, 289 (2008)
M.K. Crawford, A.G.G.M. Tielens, L.J. Allamandola, Astrophys. J. Lett. 293, L45 (1985)
A. Leger, L. D’Hendecourt, Astron. Astrophys. 146, 81 (1985)
M.L. Heger, Lick Observatory Bull. 10, 146 (1922)
E.K. Campbell et al., Nature 523, 322 (2015)
G.A.H. Walker et al., ApJL 812, L8 (2015)
H. Linnartz et al., J. Mol. Spectrosc. 367, 111243 (2020)
S. Spieler et al., ApJ 846, 168 (2017)
S.A. Krasnokutski, F. Huisken, J. Chem. Phys. 141, 214306 (2014)
S.A. Krasnokutski et al., ApJ 836, 32 (2017)
R.I. Kaiser et al., J. Chem. Phys. 110, 6091 (1999)
I. Hahndorf et al., J. Chem. Phys. 116, 3248 (2002)
J.E. Elsila, D.P. Glavin, J.P. Dworkin, Meteorit. Planet. Sci. 44, 1323 (2009)
K. Altwegg et al., Sci. Adv. 2, e1600285 (2016)
A.K. Cobb, R.E. Pudritz, ApJ 783, 140 (2014)
H. Linnartz, S. Ioppolo, G. Fedoseev, Int. Rev. Phys. Chem. 34, 205 (2015)
G.M. Muñoz Caro et al., Nature 416, 403 (2002)
E. Congiu et al., J. Chem. Phys. 137, 054713 (2012)
U. Henne, J.P. Toennies, J. Chem. Phys. 108, 9327 (1998)
M. Lewerenz, B. Schilling, J.P. Toennies, Chem. Phys. Lett. 206, 381 (1993)
B. Schilling, Bericht 14/1993. Ph.D. dissertation, University of Göttingen/Max-Planck-Institut für Strömungsforschung, 1993
B. Samelin, Bericht 16/1998. Ph.D. dissertation, University of Göttingen/Max-Planck-Institut für Strömungsforschung, 1998
L.F. Gomez et al., J. Chem. Phys. 135, 154201 (2011)
M.N. Slipchenko et al., J. Chem. Phys. 124, 241101 (2006)
K. Kuyanov-Prozument, M.Y. Choi, A.F. Vilesov, J. Chem. Phys. 132, 014304 (2010)
E. Loginov et al., Phys. Rev. Lett. 106, 233401 (2011)
L.F. Gomez, E. Loginov, A.F. Vilesov, Phys. Rev. Lett. 108, 155302 (2012)
L.F. Gomez et al., Science 345, 906 (2014)
F. Laimer et al., Phys. Rev. Lett. 123, 165301 (2019).
H. Schöbel et al., Eur. Phys. J. D 63, 209 (2011)
J. Fine et al., J. Chem. Phys. 148, 044302 (2018)
P.J. Knowles, J.N. Murrell, Mol. Phys. 87, 827 (1996)
J.H. Kim et al., J. Chem. Phys. 124, 214301 (2006)
D.S. Peterka et al., J. Phys. Chem. B 110, 19945 (2006)
D.S. Peterka et al., J. Phys. Chem. A 111, 7449 (2007)
C.G. Edmonds, R.D. Smith, in Methods in Enzymology (Academic Press, 1990), pp. 412–431
J.B. Fenn et al., Mass Spectrom. Rev. 9, 37 (1990)
C. Ho et al., Clin Biochem Rev 24, 3 (2003)
J.J. Pitt, Clin Biochem Rev 30, 19 (2009)
F. Bierau et al., Phys. Rev. Lett. 105, 133402 (2010)
F. Filsinger et al., Phys. Chem. Chem. Phys. 14, 13370 (2012)
A.I.G. Flórez et al., Phys. Chem. Chem. Phys. 17, 21902 (2015)
A. Braun, M. Drabbels, J. Chem. Phys. 127, 114303 (2007)
X. Zhang, M. Drabbels, J. Chem. Phys. 137, 051102 (2012)
E. Loginov, M. Drabbels, J. Phys. Chem. A 111, 7504 (2007)
A. Braun, M. Drabbels, J. Chem. Phys. 127, 114304 (2007)
A. Braun, M. Drabbels, J. Chem. Phys. 127, 114305 (2007)
H.J. Reyher et al., Phys. Lett. A 115, 238 (1986)
A. Leal et al., J. Chem. Phys. 144, 094302 (2016)
B.K. Vainshtein, Structure Analysis by Electron Diffraction (Elsevier, 2013)
Y. He et al., Review of Scientific Instruments 86, 084102 (2015)
M. Alghamdi, J. Zhang, W. Kong, J. Chem. Phys. 151, 134307 (2019)
M. Alghamdi et al., Chemical Physics Letters 735, 136752 (2019)
J. Zhang et al., J. Chem. Phys. 152, 224306 (2020)
H. Odaka, M. Ichihashi, Eur. Phys. J. D 71, 99 (2017)
T. González-Lezana et al., Int. Rev. Phys. Chem. 39, 465 (2020)
S. Albertini, E. Gruber, F. Zappa, S. Krasnokutski, F. Laimer, P. Scheier, Chemistry and physics of dopants embedded in helium droplets. Mass Spectrom. Rev. (2021)
L. Tiefenthaler et al., Rev. Sci. Instrum. 91, 033315 (2020)
S. Denifl et al., J. Chem. Phys. 124, 054320 (2006)
K. Głuch et al., J. Chem. Phys. 120, 2686 (2004)
S. Feil et al., Int. J. Mass Spectrom. 252, 166 (2006)
F. Zappa et al., Eur. Phys. J. D 43, 117 (2007)
S. Denifl et al., Phys. Rev. Lett. 97, 043201 (2006).
M. Kuhn et al., Nat. Commun. 7, 13550 (2016)
M. Gatchell et al., Faraday Discuss. 217, 276 (2019)
L. Kranabetter et al., Phys. Chem. Chem. Phys. 21, 25362 (2019)
I.A. Harris et al., Chem. Phys. Lett. 130, 316 (1986)
T.A. Milne, F.T. Greene, J. Chem. Phys. 47, 4095 (1967)
A. Ding, J. Hesslich, Chem. Phys. Lett. 94, 54 (1983)
P. Scheier, T.D. Märk, Int. J. Mass Spectrom. Ion Process. 76, R11 (1987)
N.E. Levinger et al., J. Chem. Phys. 89, 5654 (1988)
F.F. da Silva et al., Phys. Chem. Chem. Phys. 11, 9791 (2009)
M. Gatchell et al., Phys. Rev. A 98, 022519 (2018)
T. Ikegami, T. Kondow, S. Iwata, J. Chem. Phys. 98, 3038 (1993)
K.T. Giju, S. Roszak, J. Leszczynski, J. Chem. Phys. 117, 4803 (2002)
T. Ritschel, P.J. Kuntz, L. Zülicke, Eur. Phys. J. D 33, 421 (2005)
D.C. McDonald et al., J. Chem. Phys. 145, 231101 (2016)
M. Gatchell et al., J. Am. Soc. Mass Spectrom. 30, 2632 (2019)
L. Lundberg et al., Molecules 25, 1066 (2020)
A.G. Császár et al., Mol. Phys. 117, 1559 (2019)
P. Martini et al., J. Chem. Phys. 152, 014303 (2020)
F. Zhang et al., Int. J. Hydrogen Energy 41, 14535 (2016)
L. Huang et al. arXiv:1912.11605 [astro-ph, physics:physics] (2019)
A.M. Sapse et al., Nature 278, 332 (1979)
G.H. Herbig, Ann. Rev. Astron. Astrophys. 33, 19 (1995)
B. Jordon-Thaden et al., Phys. Rev. Lett. 107, 193003 (2011)
D. Stevenson, J. Hirschfelder, J. Chem. Phys. 5, 933 (1937)
R.E. Hurley, Nucl. Inst. Methods 118, 307 (1974)
W. Aberth, R. Schnitzer, M. Anbar, Phys. Rev. Lett. 34, 1600 (1975)
Y.K. Bae, M.J. Coggiola, J.R. Peterson, Phys. Rev. A 29, 2888 (1984)
J. Stärck, W. Meyer, Chem. Phys. 176, 83 (1993)
M. Ayouz et al., J. Chem. Phys. 132, 194309 (2010)
W. Wang et al., Chem. Phys. Lett. 377, 512 (2003)
K. Hirao, S. Yamabe, Chem. Phys. 80, 237 (1983)
L. Huang, C.F. Matta, L. Massa, J. Phys. Chem. A 115, 12445 (2011)
M. Renzler et al., Phys. Rev. Lett. 117, 273001 (2016)
F. Calvo, E. Yurtsever, J. Chem. Phys. 148, 102305 (2017)
D.E. Galli, D.M. Ceperley, L. Reatto, J. Phys. Chem. A 115, 7300 (2011)
P. Bartl et al., J. Phys. Chem. A 118, 8050 (2014)
M. Joshi, A. Ghosh, T.K. Ghanty, J. Phys. Chem. C 121, 15036 (2017)
M. Renzler et al., J. Chem. Phys. 147, 194301 (2017)
P.C. Samartzis, A.M. Wodtke, Int. Rev. Phys. Chem. 25, 527 (2006)
V.E. Zarko, Combust. Explos. Shock Waves 46, 121 (2010)
B. Hirshberg, R.B. Gerber, A.I. Krylov, Nat. Chem. 6, 52 (2014)
M.J. Greschner et al., J. Phys. Chem. A 120, 2920 (2016)
S. Liu et al., Adv. Sci. 7, 1902320 (2020)
H.T. Jonkman, J. Michl, J. Am. Chem. Soc. 103, 733 (1981)
L. Friedman, R.J. Beuhler, J. Chem. Phys. 78, 4669 (1983)
P. Scheier, A. Stamatovic, T.D. Märk, J. Chem. Phys. 88, 4289 (1988)
T. Leisner et al., Z. Phys. D Atoms Mol. Clust. 12, 283 (1989)
Y.K. Bae, P.C. Cosby, D.C. Lorents, Chem. Phys. Lett. 159, 214 (1989)
T. Tonuma et al., Int. J. Mass Spectrom. Ion Process. 135, 129 (1994)
F.A. Fernández-Lima et al., Chem. Phys. 340, 127 (2007)
A. Vij et al., Angew. Chem. Int. Ed. 41, 3051 (2002)
A.A. Vostrikov, DYu. Dubov, Tech. Phys. 51, 1537 (2006)
S.D. Pangavhane et al., Rapid Commun. Mass Spectrom. 25, 917 (2011)
N. Weinberger et al., J. Phys. Chem. C 121, 10632 (2017)
J.S.-Y. Chao, M.F. Falcetta, K.D. Jordan, J. Chem. Phys. 93, 1125 (1990)
J. Mazeau et al., J. Phys. B: At. Mol. Phys. 11, L557 (1978)
E. Yurtsever, F. Calvo, J. Phys. Chem. A 123, 202 (2019)
F.R. Brühl, R.A. Trasca, W.E. Ernst, J. Chem. Phys. 115, 10220 (2001)
F. Stienkemeier et al., Z. Phys. D Atoms Mol. Clust. 38, 253 (1996)
F. Ancilotto, G. DeToffol, F. Toigo, Phys. Rev. B 52, 16125 (1995)
A. Leal et al., Phys. Rev. B 90, 224518 (2014)
M. Buzzacchi, D.E. Galli, L. Reatto, Phys. Rev. B 64, 094512 (2001)
D.E. Galli, M. Buzzacchi, L. Reatto, J. Chem. Phys. 115, 10239 (2001)
E. Coccia et al., J. Chem. Phys. 126, 124319 (2007).
C. Di Paola et al., J. Chem. Theory Comput. 1, 1045 (2005)
F. Sebastianelli et al., Comput. Mater. Sci. 35, 261 (2006)
N. Issaoui et al., J. Chem. Phys. 141, 174316 (2014)
S. Paolini, F. Ancilotto, F. Toigo, J. Chem. Phys. 126, 124317 (2007)
L. An der Lan et al., Chem. A Eur. J. 18, 4411 (2012)
M. Rastogi et al., Phys. Chem. Chem. Phys. 20, 25569 (2018)
C. Stark, V.V. Kresin, Phys. Rev. B 81, 085401 (2010)
L. An der Lan et al., J. Chem. Phys. 135, 044309 (2011)
L. An der Lan et al., Phys. Rev. B 85, 115414 (2012)
G.J. Hutchings, M. Haruta, Appl. Catal. A 291, 2 (2005)
P. Mulvaney, MRS Bull. 26, 1009 (2001)
C.L. Brown et al., Gold Bull. 40, 245 (2007)
M. Faraday, Philos. Trans. R. Soc. Lond. 147, 145 (1857)
G.J. Hutchings, M. Brust, H. Schmidbaur, Chem. Soc. Rev. 37, 1759 (2008)
G.J. Hutchings, J. Catal. 96, 292 (1985)
M. Haruta et al., Chem. Lett. 16, 405 (1987)
M.-C. Daniel, D. Astruc, Chem. Rev. 104, 293 (2004)
M. Haruta, Nature 437, 1098 (2005)
A. Corma, H. Garcia, Chem. Soc. Rev. 37, 2096 (2008)
G.C. Bond, D.T. Thompson, Catal. Rev. 41, 319 (1999)
Y. Li, R.T. Yang, J. Phys. Chem. C 111, 11086 (2007)
M. Yamauchi, H. Kobayashi, H. Kitagawa, ChemPhysChem 10, 2566 (2009)
T. Hussain et al., Appl. Phys. Lett. 100, 183902 (2012)
C.M. Ramos-Castillo et al., J. Phys. Chem. C 119, 8402 (2015)
I. Katakuse et al., Int. J. Mass Spectrom. Ion Process. 67, 229 (1985)
I. Katakuse et al., Int. J. Mass Spectrom. Ion Process. 74, 33 (1986)
I. Rabin, C. Jackschath, W. Schulze, Z. Phys. D Atoms Mol. Clust. 19, 153 (1991)
A. Herlert et al., J. Electron Spectrosc. Relat. Phenom. 106, 179 (2000)
H. Sik Kim et al., Chemical Physics Letters 224, 589 (1994)
P. Martini et al., Int. J. Mass Spectrom. 434, 136 (2018)
St. Becker et al., Computational Materials Science 2, 633 (1994)
S. Gilb et al., J. Chem. Phys. 116, 4094 (2002)
W.A. Saunders, Phys. Rev. Lett. 64, 3046 (1990)
W.A. Saunders, Phys. Rev. Lett. 62, 1037 (1989)
W.A. Saunders, S. Fedrigo, Chem. Phys. Lett. 156, 14 (1989)
J. Zieglera et al., Int. J. Mass Spectrom. 202, 47 (2000)
A. Sanchez et al., J. Phys. Chem. A 103, 9573 (1999)
D. Schooss et al., Philosophical transactions of the royal society a: mathematical. Phys. Eng. Sci. 368, 1211 (2010)
R. Rousseau et al., Chem. Phys. Lett. 295, 41 (1998)
A. Schweizer et al., J. Chem. Phys. 119, 3699 (2003)
A. N. Gloess et al., J. Chem. Phys. 128, 114312 (2008)
K. Sugawara, F. Sobott, A.B. Vakhtin, J. Chem. Phys. 118, 7808 (2003)
M. Neumaier et al., J. Chem. Phys. 122, 104702 (2005)
A. Fielicke et al., J. Am. Chem. Soc. 127, 8416 (2005)
P. Martini et al., J. Phys. Chem. A 123, 9505 (2019)
G.E. Froudakis et al., Chem. Phys. 280, 43 (2002)
F. Remacle E.S. Kryachko, J. Chem. Phys. 122, 044304 (2005)
E. M. Fernández et al., Phys. Rev. B 70, 165403 (2004)
S. Kapur, E.W. Müller, Surf. Sci. 62, 610 (1977)
P. Pyykkoe, J. Am. Chem. Soc. 117, 2067 (1995)
D. Schröder et al., Inorg. Chem. 37, 624 (1998)
S. Seidel, K. Seppelt, Science 290, 117 (2000)
L. M. Ghiringhelli et al., New J. Phys. 15, 083003 (2013)
A. Shayeghi et al., Angew. Chem. Int. Ed. 54, 10675 (2015)
Z. Jamshidi, M.F. Far, A. Maghari, J. Phys. Chem. A 116, 12510 (2012)
L.A. Mancera, D.M. Benoit, J. Phys. Chem. A 119, 3075 (2015)
A. Ghosh, T.K. Ghanty, J. Phy. Chem. A 120, 9998 (2016)
L.M. Ghiringhelli, S.V. Levchenko, Inorg. Chem. Commun. 55, 153 (2015)
X. Li et al., Mol. Phys. 107, 2531 (2009)
X.-Y. Li, X. Cao, Y. Zhao, J. Phys. B At. Mol. Opt. Phys. 42, 065102 (2009)
L. Xinying, C. Xue, Z. Yongfang, Theor. Chem. Acc. 123, 469 (2009)
L. Xinying, C. Xue, Z. Yongfang, Aust. J. Chem. 62, 121 (2009)
P. Zhang et al., J. Mol. Struct. (Thoechem) 899, 111 (2009)
S.J. Grabowski et al., Chem. A Eur. J. 22, 11317 (2016)
L. Belpassi et al., J. Am. Chem. Soc. 130, 1048 (2008)
W.H. Breckenridge, V.L. Ayles, T.G. Wright, J. Phys. Chem. A 112, 4209 (2008)
M. Al-Ahmari et al., J Clust Sci 26, 913 (2015)
M. Slama et al., Eur. Phys. J. D 70, 242 (2016)
M. Slama et al., Mol. Phys. 115, 757 (2017)
J. Hernández-Rojas, D.J. Wales, J. Chem. Phys. 119, 7800 (2003)
D. Prekas, C. Lüder, M. Velegrakis, J. Chem. Phys. 108, 4450 (1998)
A. Yousef et al., J. Chem. Phys. 127, 154309 (2007).
C. Kittel, Introduction to Solid State Physics, 8th edn. (Wiley, 2004)
J. Chem. Soc., Abstr. 112, ii199 (1917)
E. Hulthèn, R.V. Zumstein, Phys. Rev. 28, 13 (1926)
G.N. Khairallah, R.A.J. O’Hair, M.I. Bruce, Dalton Trans. 3699 (2006)
H. Schmidbaur, H.G. Raubenheimer, L. Dobrzańska, Chem. Soc. Rev. 43, 345 (2013)
L. Lundberg et al., J. Am. Soc. Mass Spectrom. 30, 1906 (2019)
L. Lundberg et al., Eur. Phys. J. D 74, 102 (2020)
H. Rosemeyer, Chem. Biodivers. 1, 361 (2004)
M. Kuhn et al., Eur. Phys. J. D 72, 38 (2018)
D. Huber et al., J. Chem. Phys. 125, 084304 (2006)
A. Mauracher et al., J. Phys. Chem. Lett. 5, 2444 (2014)
N. Weinberger et al., Eur. Phys. J. D 70, 91 (2016)
E. Jabbour Al Maalouf et al., J. Phys. Chem. Lett. 8, 2220 (2017)
M. Gatchell et al., Phys. Chem. Chem. Phys. 20, 7739 (2018)
G. Klupp, S. Margadonna, K. Prassides, in Reference Module in Materials Science and Materials Engineering (Elsevier, 2016)
A. Mauracher et al., Angew. Chem. Int. Ed. 53, 13794 (2014)
E.K. Campbell et al., ApJ 822, 17 (2016)
O. Echt et al., ChemPlusChem 78, 910 (2013)
S.A. Krasnokutski et al., J. Phys. Chem. Lett. 7, 1440 (2016)
M. Harnisch et al., Eur. Phys. J. D 70, 192 (2016)
M. Renzler et al., J. Phys. Chem. C 121, 10817 (2017)
A.F. Hebard et al., Nature 350, 600 (1991)
T.T.M. Palstra et al., Solid State Commun. 93, 327 (1995)
A.Y. Ganin et al., Nat. Mater. 7, 367 (2008)
A.Y. Ganin et al., Nature 466, 221 (2010)
M. Yoon et al., Phys. Rev. Lett. 100, 206806 (2008)
Q. Wang, P. Jena, J. Phys. Chem. Lett. 3, 1084 (2012)
M. Robledo et al., RSC Adv. 6, 27447 (2016)
A. Kaiser et al., Int. J. Hydrog. Energy 42, 3078 (2017)
J. Osterodt, F. Vögtle, Chem. Commun. 547 (1996)
N. Dragoe et al., Chem. Commun. 85 (1999)
M. Goulart et al., J. Phys. Chem. Lett. 9, 2703 (2018)
L. Xin-Ying, Cao xue, Phys. Rev. A 77, 022508 (2008)
P. Martini et al., J. Phys. Chem. A 123, 4599 (2019)
A. Mauracher et al., J. Chem. Phys. 142, 104306 (2015)
P. Khare et al., Rev. Geophys. 37, 227 (1999)
B. Zuckerman, J.A. Ball, C.A. Gottlieb, Astrophys. J. Lett. 163, L41 (1971)
S.-Y. Liu, D.M. Mehringer, L.E. Snyder, ApJ 552, 654 (2001)
P. Redondo, A. Largo, C. Barrientos, A&A 579, A125 (2015)
C. Rice et al., J. Power Sourc. 111, 83 (2002)
Y. Zhu, S.Y. Ha, R.I. Masel, J. Power Sourc. 130, 8 (2004)
X. Wang et al., J. Power Sourc. 175, 784 (2008)
Z. Bai et al., J. Phys. Chem. C 113, 10568 (2009)
T. Gunji, F. Matsumoto, Inorganics 7, 36 (2019)
Z. Bai et al., Int. J. Electrochem. Sci. 8, 12 (2013)
F. Nitze et al., Electrochim. Acta 63, 323 (2012)
M. Meksi et al., Chem. Lett. 44, 1774 (2015)
M. Mahmoodi-Darian et al., J. Am. Soc. Mass Spectrom. 30, 787 (2019)
M. Mahmoodi-Darian et al., Int. J. Mass Spectrom. 450, 116293 (2020)
M. Goulart et al., Phys. Chem. Chem. Phys. 15, 3577 (2013)
S. Heinbuch et al., J. Chem. Phys. 126, 244301 (2007)
D.P.P. Andrade et al., J. Phys. Chem. C 112, 11954 (2008)
D.P.P. Andrade et al., J. Electron Spectrosc. Relat. Phenom. 155, 124 (2007)
I. Martin et al., Phys. Chem. Chem. Phys. 7, 2212 (2005)
L. Ziemczonek, T. Wróblewski, Eur. Phys. J. Spec. Top. 144, 251 (2007)
Y. Valadbeigi, H. Farrokhpour, Int. J. Quantum Chem. 113, 1717 (2013)
W.Y. Feng, C. Lifshitz, J. Phys. Chem. 98, 6075 (1994)
Y. Inokuchi, N. Nishi, J. Phys. Chem. A 106, 4529 (2002)
V. Aviyente et al., Int. J. Mass Spectrom. Ion Processes 161, 123 (1997)
R. Zhang, C. Lifshitz, J. Phys. Chem. 100, 960 (1996)
L. Baptista et al., J. Phys. Chem. A 112, 13382 (2008)
F.F. da Silva et al., Phys. Chem. Chem. Phys. 11, 11631 (2009)
M. Šala et al., J. Phys. Chem. A 113, 3223 (2009)
H. Schöbel et al., Phys. Chem. Chem. Phys. 13, 1092 (2010)
H. Schwertfeger, P.R. Schreiner, Chem. unserer Zeit 44, 248 (2010)
H. Schwertfeger, A.A. Fokin, P.R. Schreiner, Angew. Chem. Int. Ed. 47, 1022 (2008)
K.-W. Yeung et al., Nanotechnol. Rev. 9, 650 (2020)
A.M. Schrand, S.A.C. Hens, O.A. Shenderova, Crit. Rev. Solid State Mater. Sci. 34, 18 (2009)
M. Goulart et al., J. Phys. Chem. C 121, 10767 (2017)
R.M. Sehgal, D. Maroudas, Langmuir 31, 11428 (2015)
L. Kranabetter et al., Phys. Chem. Chem. Phys. 20, 21573 (2018)
B.A. Buffett, Annu. Rev. Earth Planet. Sci. 28, 477 (2000)
Yu.A. Dyadin et al., J Struct Chem 40, 645 (1999)
P. Englezos, Ind. Eng. Chem. Res. 32, 1251 (1993)
F. Laimer et al., Chem. A Eur. J. n/a, (2020)
D. Mateo, J. Eloranta, J. Phys. Chem. A 118, 6407 (2014)
T. Jiang, C. Kim, J.A. Northby, Phys. Rev. Lett. 71, 700 (1993)
J.A. Northby, C. Kim, Physica B 194–196, 1229 (1994)
M. Farnık et al., 6 (1996)
W. Saunders, Phys. Rev. A 46, 7028 (1992)
O. Echt et al., Phys. Rev. A 38, 3236 (1988)
I. Mähr et al., Phys. Rev. Lett. 98, 023401 (2007)
F. Laimer, F. Zappa, P. Scheier, Size and Velocity Distribution of Negatively Charged Helium Nanodroplets. J. Phys. Chem. A. 125(35), 7662–7669 (2021)
U. Henne, Ph.D. Dissertation, available as Bericht 5/1996, Max-Planck-Instiut für Stömungsforschung, Göttingen, Germany, 1996
R. M. P. Tanyag et al., J. Chem. Phys. 152, 234306 (2020).
M. Hartmann et al., Phys. Rev. Lett. 75, 1566 (1995)
S. Yang et al., Nanoscale 5, 11545 (2013)
J.P. Toennies, A.F. Vilesov, Angew. Chem. Int. Ed. 43, 2622 (2004)
J. Mansikka-Aho, M. Manninen, E. Hammarén, Z. Phys. D Atoms Mol. Clust. 21, 271 (1991)
Y. Dong, M. Springborg, J. Phys. Chem. C 111, 12528 (2007)
J. Roithová et al., Faraday Discuss. 217, 98 (2019)
A. Günther et al., J. Mol. Spectrosc. 332, 8 (2017)
E.K. Campbell, J.P. Maier, ApJ 850, 69 (2017)
P. Bartl et al., J Phys Chem A 118, 8050 (2014)
A. Kramida, Y. Ralchenko, (1999)
S. Denifl et al., J Chem Phys 124, 054320 (2006)
R.J. Beuhler et al., Biochemistry 13, 5060 (1974)
M.P. Ziemkiewicz, D.M. Neumark, O. Gessner, Int. Rev. Phys. Chem. 34, 239 (2015)
J.P. Maier, E.K. Campbell, Int. J. Mass Spectrom. 434, 116 (2018)
M. Töpfer et al., Mol. Phys. 117, 1481 (2019)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this chapter

Cite this chapter
Schiller, A., Laimer, F., Tiefenthaler, L. (2022). Helium Droplet Mass Spectrometry. In: Slenczka, A., Toennies, J.P. (eds) Molecules in Superfluid Helium Nanodroplets. Topics in Applied Physics, vol 145. Springer, Cham. https://doi.org/10.1007/978-3-030-94896-2_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-94896-2_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-94895-5
Online ISBN: 978-3-030-94896-2
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)