
Abstract
Wheat improvement relies on genetic diversity associated with variation in target traits. While traditionally the main sources of novel genetic diversity for breeding are wheat varieties or various wild relatives of wheat, advances in gene mapping and genome editing technologies provide an opportunity for engineering new variants of genes that could have beneficial effect on agronomic traits. Here, we provide the overview of the genome editing technologies and their application to creating targeted variation in genes that could enhance wheat productivity. We discuss the potential utility of the genome editing technologies and CRISPR-Cas-induced variation incorporated into the pre-breeding pipelines for wheat improvement.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Learning Objectives
-
Understanding the basics of CRISPR-Cas-based technology and learning how to apply it in wheat improvement.
2 Introduction to the Development of Genome Editing (GE) Technologies
Compared to the conventional random mutagenesis, GE tools provide opportunity to modify specific genomic regions of interest. The GE tools share two major common features: (1) a programable DNA binding domain, and (2) a DNA nuclease capable of introducing double or single strand breaks (DSB or SSBs) into the targeted DNA sites. The introduced DNA breaks could then be repaired either through (1) error-prone non-homologous end joining (NHEJ) process resulting in insertions, deletions, or single base substitutions, or (2) homology-directed repair (HDR) process, or (3) be utilized as sites for sequence replacement using prime editing (PE) technology. The HDR could be used for precise sequence insertion or replacement through recombination with the exogenously supplied “donor DNA”.
Before the development of CRISPR-based GE technologies, targeted GE was performed using Zinc Finger Nuclease (ZFN) and Transcription Activator-Like Effector Nuclease (TALEN). ZFN, the first programable GE system, was based on a Zinc Finger Protein (ZFP) fused with the nuclease domain of restriction endonuclease FokI (Fig. 29.1a). ZFP comprised 3–6 tandemly repeated DNA binding units. Each unit contains 30 amino acids and recognizes a 3 base pair (bp) DNA sequence. The DNA targeting specificity of ZFNs is defined by the composition of DNA binding units. ZFNs are designed to work in pairs because the cleavage domain of FokI must dimerize to introduce DSBs (Fig. 29.1a). In the TALEN-based GE system, the DNA binding domain is composed of tandemly repeated DNA recognition units with additional N- and C- terminal domains derived from TALEs (Fig. 29.1b). Each unit contains 33–35 highly conserved amino acids except for the variable residues 12 and 13, which define the single nucleotide binding specificity. Both ZFN and TALEN systems have been broadly used for GE in multiple organisms. With the development of a more effective CRISPR-Cas-based technology, their application in research and biotechnology declined.

The schematic illustration of ZFN and TALEN GE systems. (a) ZFN is composed of tandemly repeated DNA binding units fused to a nuclease domain of restriction endonuclease FokI. Each DNA binding units recognize 3 bp of DNA. Left and right ZFN work in pairs to make DSBs. (b) TALEN is composed of TALE repeats, the N- and C-terminal domains, and the fused with FokI nuclease. Each unit of TALE repeats recognize 1 bp of DNA. TALENs work in pairs to introduce DSBs
The CRISPR-Cas system is composed of a CRISPR-associated protein (Cas) and a mature transcript originating from a Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) locus. In 2012, the groups led by two biochemists, Jennifer A. Doudna from USA and Emmanuelle Charpentier from France, demonstrated the first application of the CRISPR-Cas9 programmable endonuclease for in vitro DNA editing [1]. The group led by a Lithuanian biochemist, Virginijus Siksnys, also independently achieved the CRISPR-Cas9 mediated in vitro DNA cleavage [2]. It took only a few years for the newly developed CRISPR-Cas-based system [3, 4] to become a major GE tool for studying eukaryotic genomes. Because of the revolutionary changes brought into the basic genetic and genomic studies by the CRISPR-Cas-based technologies and new possibilities provided by this technology for curing diseases and improving crops, Jennifer A. Doudna and Emmanuelle Charpentier were awarded the 2020 Nobel Prize in Chemistry.
Since the first discovery of the CRISPR loci in 1987, it took scientists more than 20 years to understand that the CRISPR-Cas is a part of the bacteria/archaea immune system, which evolved to recognize and cleave invading DNA/RNA molecules [5]. The CRISPR-Cas complex is formed by Cas proteins and CRISPR RNAs (crRNAs) spliced from the RNA transcripts of the CRISPR loci. The CRISPR-Cas systems could be categorized into class 1 and class 2, which could adopt multiple and single Cas proteins as nucleases for target cleavage, respectively [6]. Class 2 is further divided into type II, V and VI. The two mostly widely used CRISRP-Cas-based genome editors are CRISPR-Cas9 and CRISPR-Cas12a, which belong to class 2 type II and V, respectively. The ease of target design, high GE efficiency and ability to simultaneously target multiple genomic regions made the CRISPR-Cas technology more popular than ZFN and TALEN. During the last decade, the power of CRISPR-Cas-based system has been harnessed to better understand function of genes underlying variation in major agronomic traits and to develop novel strategies for crop improvement.
3 CRISPR-Cas-Based GE Toolbox
3.1 CRISPR-Cas Variants and Their Basic Applications
The CRISPR-Cas9 from Streptococcus pyogenes (CRISPR-SpCas9) is one of the most commonly used CRISPR-Cas systems for GE. To induce targeted DSB, the SpCas9 needs a single-guide RNA (sgRNA) containing 20 nucleotide spacer complementary to the targeted DNA sequence, which is followed by the 3′-end NGG protospacer-adjacent motif (PAM) (Fig. 29.2a). The synthetic sgRNAs for CRISPR-Cas9 system are engineered based on the crRNAs and trans-activating crRNA (tracrRNA) [1]. In bacteria/archaea, the directed repeat sequences of the immature crRNA array form complexes with the tracrRNAs, which are processed into crRNA-tracrRNA duplexes by Cas9 and RNase III [5]. As part of the CRISPR-Cas9 complex, the sgRNA guides is responsible for guiding Cas9 to the target site. The two endonuclease domains of SpCas9, HNH and RuvC, will cleave the paired and non-paired DNA strands, respectively (Fig. 29.2a), and predominantly result in blunt end DSBs located 3 bp from the NGG PAM.

The schematic illustration of CRISPR-Cas9 and CIRSPR-Cas12a systems. (a) The CRISPR-Cas9 nuclease cuts the double stranded DNA 3 bp upstream of the NGG protospacer adjacent motif (PAM) and forms blunt end DSB. The DNA cuts catalyzed by the two nuclease domains of Cas9 are shown by the purple arrows. The NGG PAM is shown with the blue rectangle. Amino acid changing mutations in Cas9 (D10A in RucV domain and H840A in HNH domain) result in variants that are either capable of making cuts on only one DNA strand (nCas9) or incapable of cutting DNA (dCas9). (b) The CRISPR-Cas12a has one RuvC nuclease domain and one novel nuclease domain, which are shown as purple arrows. CRISPR-Cas12a has different PAM sequence (TTTV [V = A, C, G] or TTV) and makes 4–5 nucleotide long staggered double stranded DNA breaks at the distal end of the protospacer
By introducing either aspartate-to-alanine substitution (D10A) in the RuvC domain or Histidine-to-alanine substitution (H840A) in the HNH domain, SpCas9 could be converted to the DNA nickase (nCas9). The inactive form of Cas9, also called dead Cas9 or dCas9, could be created by introducing both D10A and H840A substitutions simultaneously. The nCas9 and dCas9 variants are currently used in a number of GE applications. The range of editable genomic targets was further expanded by creating the variants of Cas9 recognizing different PAMs. For example, the engineered Cas9NG [7], which predominantly recognizes ‘NG’ PAM, was shown to be effective for GE in wheat [8]. Additional information about the variants of Cas9 editors could be found in review by Alzalone et al. [9].
CRISPR-Cas12a, also known as Cpf1, is another broadly used CRIRSP-Cas system that has several features distinguishing it from CRISPR-Cas9 [10] (Fig. 29.2b). CRISPR-Cas12a (1) recognizes 5′-end T-rich PAMs (TTTV (V = A, C, G) for LbCas12a and AsCas12a, and TTV for FnCas12a); (2) generates staggered end DSBs with 4–5-nucleotide 5′-overhangs; (3) induces mutations on the distal end of protospacer, thereby preserving PAM and enabling multiple rounds of GE; (4) processes transcripts with tandem repeats into mature crRNAs and induces genome edits independent of tracrRNA, which simplifies the design of multiplex GE experiments. Similar to Cas9, a series of Cas12a variants recognizing various PAMs have been engineered [9]. Both the wild-type and engineered Cas12a have been shown to be effective in wheat [8].
The repair of DSBs created by Cas nucleases through the NHEJ process introduces stochastic mutations, mostly short DNA sequence insertions/deletions or base pair substitutions. This approach is commonly used to disrupt functional elements in genome, such as gene coding sequences or regulatory elements. In the earlier GE applications, the precise modifications in genome, including specific single-base mutations, gene replacements, targeted deletions or insertions could be achieved through HDR of DSBs by co-delivering a CRISPR-Cas reagent along with a donor DNA template. The donor DNA includes a DNA sequence with desired mutations flanked from both sides by sequences similar to the sequences around the CRISPR-Cas target site. This structure of a donor template promotes HDR and allows for replacing the original sequence with DNA carrying desired changes. Compared to NHEJ-mediated GE, the efficiency of HDR-based GE remains relatively low because it relies on DNA replication activity, which is initiated only at the S- and G2-phases of the cell cycle [3] One exception is precise DNA deletion, which could be achieved by the CRISPR-Cas targeting of a pair of sites flanking the region of interest [3].
3.2 Base-Editors and Prime-Editors
The low efficiency and precision of HDR-mediated DSB repair in the CRISPR-Cas applications aimed at sequence replacement necessitated the development of alternative approaches. A series of nonconventional GE systems, which do not rely on DSBs or donor DNA templates, greatly expanded the range of possible genome modifications [11,12,13].
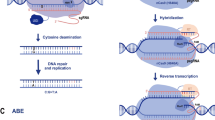
In base editors BE3 (cytosine base editor or CBE) and ABE7.10 (adenine base editor or ABE), single-stranded DNA deaminases are fused to the N-terminal domain of nCas9 (Fig. 29.3a) [12, 13]. During base editing, after the hybridization of sgRNA with its cognate target, nucleotides in the editing window on the PAM-containing DNA strand (non-targeted) are exposed to deaminase.

The mechanisms of Base Editing and Prime Editing. The (a) Cytosine Base Editor (CBE) BE3 and (b) Adenine Base Editor (ABE) ABE7.10 are based on nickase Cas9(D10A) fused with deaminases. (c) Prime Editor (PE). This figure shows how PE inserts sequence (shown in red) downstream of the targeted locus (shown in blue). PE is a fusion of nCas9 with Reverse Transcriptase (RT). pegRNA contains targeting spacer sequence, primer binding sequence and template used by RT to introduce desired changes into a target site
In BE3, the editing window of cytidine deaminase (APOBEC1 from rat) spans nucleotides 4–8 (Fig. 29.3a), in which CBE converts cytosine (C) to uracil (U). The U will be recognized as thymine (T) by DNA polymerase, and adenosine (A) will be added to the nCas9-nicked DNA strand when it is repaired using the modified strand as a template. Finally, during DNA replication U will be replaced with T, resulting in transition mutations C → T and G → A on the non-targeted and targeted strands, respectively (Fig. 29.3a). The U introduced by CBE could be rapidly removed by uracil DNA glycosylase and reduce the efficiency of editing. To address this issue, an uracil glycosylase inhibitor (UGI) is fused to the C-terminal of nCas9 (D10A) in CBE to increase the half-life of U. In ABE7.10, the editing window of the heterodimer including both the wild and engineered TadA deoxyadenosine deaminase spans nucleotides 4–7 (Fig. 29.3b). The engineered TadA in ABE7.10 converts A to inosine (I), which will be read as G by DNA polymerases. Similar to CBE, after two rounds of DNA repair, the transition mutations A → G and T → C will be formed on the non-targeted and targeted strands, respectively (Fig. 29.3b).
Base editing is restricted to the four types of transition mutations (C → T, G → A, A → G, and T → C), shows high off-target (non-specific) GE and induces mutations within the protospacer sequence at positions different from the targeted base (bystander mutations). These limitations were resolved in the recently developed prime editor (PE) that was demonstrated to be more versatile and precise, though its editing efficiency was lower than that of the BEs [9, 11]. The most commonly used PE2 system is based on the combination of (1) an engineered reverse transcriptase (RT) from Moloney Murine Leukemia Virus fused to the N-terminal domain of nCas9, and (2) prime editing gRNA (pegRNA), which includes both a primer binding site (PBS) and a template for RT carrying the desired genome edits (Fig. 29.3c). During prime editing, nCas9 cuts the unpaired (non-targeted) DNA strand 3 bases upstream of PAM. The 3′-end of cut DNA strand hybridizes with PBS and directs RT using the template in the pegRNA. This reverse transcription step copies the desired sequence from the pegRNA to target DNA. The prime editor could induce all possible types of DNA point mutations and deletions and insertions as long as 10 nucleotides, and as far as 30 nucleotides from the cut site. Once issues with reduced GE efficiency are resolved, these features will make PE a powerful tool for basic and applied studies in agricultural crops. Recently, PE was successfully used to edit the rice and wheat genomes [14].
3.3 Gene Suppressors and Activators, Epigenomic Modifiers, and Others
The level of gene expression is defined by regulatory elements, transcription factors (TFs), and the chromatin accessibility of genomic regions with regulatory function. Since dCas9 could be easily programmed to target any region of genome using sgRNAs, it was adopted to deliver transcription factors or proteins involved in chromatin remodeling, histone modification or epigenetic reprogramming to promoters or other regulatory regions of genome. The catalytically inactive dCas9 protein was used to engineer TFs capable of activating or suppressing the expression of any gene, or to develop a broad array of gene regulation and epigenome modification tools by fusing with various proteins domains [15]. For example, the dCas9 fused with protein domains SRDX and VP64 was successfully used for respectively suppressing and activating gene expression in plants. Thus, dCas9 provides a unique opportunity to reversibly modulate gene expression and investigate its role in the regulation of biological pathways underlying phenotypic diversity.
4 Recent Application of GE for Improving Major Agronomic Traits and Breeding Technologies
Since its invention, CRISPR-Cas-based GE technologies have been widely applied to modify agronomically important traits in crops. The GE has been applied to improve crop productivity, nutritional quality, storage life, abiotic and biotic stress resistance. The improved understanding of the genetic basis of trait variation in many crops combined with CRISRP-Cas GE enabled novel breeding strategies. These strategies include inducing targeted genetic variation in genes controlling agronomic traits, de-novo domestication of novel crops, development of herbicide resistant crop varieties and male sterile lines, manipulation of hybrid incompatibility, hybrid vigor fixation, and development of haploid induction (HI) lines and haploid induction editing technology (HI-Edit) [16].
5 Genome Editing in Wheat
Compared to other main crops (e.g. rice or maize), the adoption of CRISRP-Cas system for wheat improvement lags behind. Among factors that contributed to this trend are (1) the low efficiency of wheat transformation, (2) until recently, the lack of a high-quality reference genome, and (3) the complexity of allopolyploid wheat genetics that complicates comparative genomic analyses and require additional efforts for inferring the biological role of duplicated genes. The recent advances in wheat transformation methods [17] and release of the high-quality reference genomes of wheat [18, 19] hold great promise to broaden application of CRISRP-Cas technologies in wheat genetics and breeding. This section introduces the progress in CRISRP-Cas-based GE in wheat, and describe procedures utilized for modifying the wheat genome, which include CRISPR-Cas system optimization, target gene selection, GE strategy selection, gene target design and validation, genetic transformation, CRISPR-Cas mutant screening, phenotypic validation of CRISPR-Cas-based gene edits, and introgression of beneficial CRISPR-Cas-induced alleles into adapted wheat cultivars.
5.1 Optimization of the CRISRP-Cas System for Wheat Genome Editing
Since most of the CRISRP-Cas tools have originally been developed for human or mammalian cells, it was necessary to optimize these tools for crop genome editing. The optimization of CRISPR-Cas systems includes the selection of optimal codons for the effective Cas gene translation and promoters for the effective expression of Cas-encoding genes and sgRNAs. In the reported studies, both rice and maize codon-optimized Cas9 [20, 21] as well as the wheat codon-optimized Cas9 [22, 23] were successfully used for editing the wheat genome. The maize ubiquitin promoter is one of the most commonly used RNA polymerase II promoters to express the Cas genes in transgenic plants. The RNA polymerase III promoters, U3 or U6, are usually employed to drive the sgRNA expression. Recently, the ubiquitin promoter from switchgrass was shown could be used to express guide RNAs and support effective GE mediated by CRISPR-Cas12a [8].
The optimization of CRISPR-Cas systems for the multiplexed gene editing (multiplex GE or MGE) is also critical for the crop GE applications. In the MGE constructs, the expression of individual sgRNAs could be driven from independent promoters. Alternatively, multiple sgRNA units could be placed under the control of a single promoter and expressed as a precursor RNA molecule. The processing of this RNA into functional sgRNAs could be supported by the (1) self-cleaving ribozymes, (2) glycine tRNA, or (3) Csy4 recognition sites that separate the sgRNA units from each other [24].
5.2 Selection of Target Genes for CRISPR-Cas-Based GE
The application of GE for precision crop breeding depends on how well we understand the role of different genes and pathways in controlling phenotypic traits. Over the last decade, the development of novel sequence-based genotyping approaches and of new genetic and genomic resources including multiple annotated reference genomes, re-sequenced diversity panels, TILLING populations, gene expression atlases, and genetic mapping populations enabled quick validation of candidate genes underlying variation in important agronomic traits in wheat. These advances combined with the power of GE technologies opens unique opportunities to start improving wheat by introducing the new CRISPR-Cas-induced alleles of genes into the breeding process.
Using forward genetics approaches, the candidate gene(s) underlying quantitative trait loci (QTL) could be found by map-based cloning or genome-wide association mapping (GWAM). The function of a candidate gene could be validated by the CRISRP-Cas-induced knock-out. One such example is the CRISPR-Cas9-induced mutagenesis of the TaHRC gene, which was found to be a negative regulator of resistance to Fusarium head blight [25]. Thus, the CRISRP-Cas system could potentially be utilized for the large-scale functional screening of hundreds of genes identified by GWAM, comparative genomics or other genomic approaches.
The selection of candidate genes for GE could be guided by the bioinformatically or experimentally inferred gene interaction networks. For example, the experimental screening of protein-DNA interactions involving the promoter of the TaGlu-1Dy gene, which encodes high molecular weight glutenin contributing to the dough quality, helped to identify the TaNAC019 candidate gene [26]. The CRISPR-Cas9-induced knock-out mutants of this gene showed that TaNAC019 positively regulates the amount of seed storage protein and grain starch content by modulating the expression of grain quality genes.
Plant species showed parallel variation of their morphological and physiological traits, and the molecular mechanisms underlying these traits appear to be broadly conserved. The release of wheat genome references [18, 19] enabled identification of candidates genes based on the comparative analyses (e.g. using Ensembl Plants) involving closely and distantly related crop genomes. By integrating comparative genomics approach with the CRISPR-Cas-based GE, genes controlling grain number [27], grain size and weight [20, 23, 28], powdery mildew resistance [21], and herbicide resistance [29] have been functionally characterized. These findings indicate that comparative genomics is a powerful tool for the extrapolation of gene mapping information across related crop species.
5.3 Selection of GE Strategies
The choice of GE strategy and Cas Editors is primarily defined by the biological nature of alleles having positive effects on agronomic traits. The ease of the GE experiment design, and the genetic architecture and heritability of a trait are also taken into account. The loss of function mutations could be readily induced using the targeted random mutagenesis via the NHEJ process or point mutagenesis using BEs or PE. The DNA sequence insertions, deletions or replacements could be achieved by HDR-mediated GE or PE. However, among these strategies only random and point mutagenesis have successfully been used to create wheat lines with modified traits.
When a gene negatively regulates an agronomic trait, the most efficient GE strategy is the random mutagenesis of coding regions. For example, the loss-of-function mutants of the TaGW2 gene created by the CRISPR-Cas9-induced frame shift mutation in the coding region increased the grain size and weight (Fig. 29.4a) [20]. A point mutagenesis strategy was successfully used to develop herbicide-tolerant wheat [29]. Several plant herbicides target acetolactate synthase (ALS), which is essential for branched-chain amino acids synthesis and plant growth. A nonsense mutation at residue P174 of ALS was known to cause herbicide resistances in multiple plant species. In this case, the CBE-based GE strategy was adopted and successfully used to create herbicide resistant wheat with various ALS alleles (Fig. 29.4b).

Selection of GE strategies and target design. (a) An example of a gene negatively regulating agronomic trait. CRISPR-Cas9-induced frame shift mutations in the TaGW2 gene homoeologs increased the grain size and weight. (b) An example of CBE application to wheat GE. CBE was used to introduce point mutations in the TaALS gene and create herbicide resistant wheat lines [29]. The GE window of CBE and the targeted amino acid residue are shown in green, the CRISPR-Cas-induced mutations are shown in red
5.4 GE Target Selection and Plasmid Construction
The selection of the CRISPR-Cas targets is influenced by the availability of PAMs and the base composition of the targeted regions. A number of web-based CRISPR-Cas target design tools facilitate this task. When target selection is not restricted to a small region, the overlap between the top-ranked targets chosen by multiple tools is recommended to ensure effective GE. However, if target selection is restricted to a small region, a target could be selected manually. In addition, target selection in polyploid wheat should consider the presence of multiple target copies in distinct genomes. To modify all homoeologous copies of genes in the wheat genome, a target site should be located in the conserved region shared by all gene copies. To specifically modify only one homoeologous copy of a gene, a target should include nucleotide sites unique to that copy of a gene. The proximity of a genome-specific mutation to PAM correlates well with the specificity of GE. At least one unique mutation less than 10 bp away from the PAM would be needed to make the CRISPR-Cas target specific.
Commonly, the CRISPR-Cas constructs have the sgRNA promoter and scaffold connected by two back-to-back Type IIS restriction enzyme cut sites, which could create 4 nucleotide 5′ overhangs at the ends of the promoter and the sgRNA scaffold (Fig. 29.5). The spacers matching the target site are synthesized as two reverse complimentary oligonucleotides that after annealing generate 5′ overhangs matching the sticky ends of the restriction enzyme cut sites. The annealed oligos are then inserted into the construct via Golden Gate reaction [22]. For MGE, a tandem array of multiple sgRNA separated by tRNA, ribozyme or Cys4 recognition site sequences could be synthesized and inserted into the CRISPR-Cas construct.

Illustration of the CRISPR-Cas9 construct used for the wheat GE. The expression of sgRNA is driven by the U3 promoter from wheat (TaU3p) and terminated at the U3 terminator (U3t). Cas9 expressed from the maize ubiquitin promoter (zUbip) is terminated at the NOS terminator. A 3 x Flag tag, two nuclear location signals (NLSs), and the RuvC and HNH nuclease domains are marked. The sgRNA transcription start site is highlighted in green. The BsaI cut sites are used for inserting the spacer portion of sgRNA created by annealing the synthesized oligonucleotides. The Cas9 cut site on the target are shown with red triangles. The GE target site and the sequences of oligonucleotides complementary to the target site are shown in lowercase letter n
5.5 Validation of the Selected GE Targets
Though web-based tools substantially improved selection of optimal targets for GE, the efficiency of GE could still be influenced by factors whose effects are difficult to account for (e.g. high target duplication rate, chromosome state or epigenetic modifications at the targeted regions). As a result, the GE efficiency of selected targets could vary substantially, as was shown for the coding regions in the wheat genome (0.08%–8.33%) [23]. The transient expression of the CRISPR-Cas constructs in the wheat protoplasts was successfully used to experimentally assess the ability of sgRNAs to modify the selected targets. The GE efficiency could be estimated by restriction enzyme digestion (RED), if GE leads to mutation at the enzyme recognition site. Otherwise, the GE events could be validated by amplifying targeted regions followed by the Sanger sequencing of amplicons or by highly multiplexed next-generation sequencing (NGS) of pooled barcoded PCR products [23].
5.6 The Delivery of CRISPR-Cas Reagents and Regeneration of Genome-Edited Wheat Lines
The success of GE heavily relies on the efficiency of wheat transformation, which previously was performed using the biolistic transformation and limited only to certain wheat cultivars. However, the recently reported methods of wheat regeneration and Agrobacterium-mediated transformation [17, 30] broaden the range of wheat varieties amenable to transformation. The usage of transgenic plants for inducing CRISPR-Cas-mediated mutations remains one of the major concerns for general public. To eliminate the time-consuming backcrossing steps aimed at removing the transgenic constructs, biolistic transformations using a CRISPR-Cas-based RNA [28] or ribonucleoprotein complex [31] have been developed for wheat. However, these methods remain labor-intensive because of the lack of selectable markers at the tissue culture and plant regeneration steps.
Haploid induction editing technology (HI-Edit) uses pollen from a haploid inducer line (e.g. maize) expressing the CRISPR-Cas reagents to fertilize a recipient line. The genome of the recipient line is edited before the elimination of the haploid inducer genome [32] and the haploid embryo with the edited copy of a gene is then used to produce double haploid (DH) plants. This approach was applied to create the edited DH lines of wheat using the pollen of maize line carrying active CRISPR-Cas9 [32]. HI-Edit overcomes not only the drawbacks of time-consuming crossing in wheat breeding aimed at generating inbred lines and reducing linkage drag around beneficial alleles, but also the issues related to the genotype-specific regeneration efficiency in wheat transformation. However, the necessity to generate transgenic maize, the low editing efficiency, the need for DH production step and freedom-to-operate related to this technology will likely affect the adoption of HI-Edit in wheat breeding.
5.7 Screening Plants Carrying GE Events
The same methods used for mutation screening in the protoplasts, RED, Sanger sequencing and NGS, could be utilized to detect the GE events in plants. Among these methods, however, only NGS provides a throughput necessary for large-scale mutation screening, and also is capable of generating deep coverage sequence data for the targeted regions that would allow for assessing the types and allelic dosage of new GE mutations. This information is important for detecting the GE plants with homozygous and heterozygous mutations, or plants that show the evidence of mosaic mutations in somatic tissues, which are usually not heritable [33].
In some cases, when it is required to edit a genomic target showing the low GE efficiency or to obtain plants with multiple targets modified, it was found useful to screen the GE events across several generations of plants derived from the original transgenic event. This approach provides an alternative to regenerating a large number of plants from independent transgenic events, and relies on the CRISPR-Cas9 transgenerational activity to produce mutations in the next generation [23, 33]. In addition, this transgenerational activity allows for inducing the CRISPR-Cas9 mutations on the homologous chromosomes of other cultivars crossed with the transgenic lines expressing the CRISPR-Cas constructs.
5.8 Phenotypic Evaluation of GE Wheat Lines
The phenotypic effects of GE depend on the genetic architecture of traits. In allopolyploid wheat, the effects of GE are strongly influenced by polyploidy and were shown to be dosage-depend, with the strongest effects observed in lines carrying mutations in all copies of homoeologous genes [20]. The expansion of gene families in the wheat genome [18] represents another challenge for GE. The sub-functionalization or functional redundancy among duplicated genes would complicate estimating the phenotypic effects of GE, unless many gene family members are edited. The consequences of GE in genes selected based on the comparative genomics depend on the evolutionary conservation of gene function. For example, the GE of the wheat-rice orthologs TaGW7 and OsGW7 showed some differences in the phenotypic effects, although their main functions appear to be conserved [23].
One should be also cautious about the selection of a wild-type control for comparing with the GE lines. The regeneration of transgenic plants could induce epigenetic modifications in the genome, which by themselves could influence phenotypes. To reduce the effect of genetic background on phenotype, the best option is to use controls homozygous for the wild-type alleles, which are selected from a progeny of heterozygous mutants or cross between the wild-type genotype and the GE wheat line [20, 23].
5.9 Prospects of CRISRP-Cas Application in Wheat Improvement
Though the number of traits modified by GE in wheat is still limited and mostly based on the loss-of-function mutations in genes having a negative effect on agronomic traits, the CRISPR-Cas-based technologies has a great potential to accelerate wheat improvement. Combined with advances in wheat genome sequencing and the development of rich genetic resources for trait mapping, GE will accelerate identification and functional analyses of genes and pathways controlling major agronomic traits. The range of phenotypic variation for these traits could potentially be further expanded by applying GE to engineer the novel allelic variants of genes with modified regulatory regions or coding sequences. The development of herbicide resistant, haploid induction and male sterile lines using the GE is clear demonstration of how GE could be used to improve wheat breeding and production processes. The improvement of wheat transformation technologies holds promise to broaden the range of genotypes amenable to regeneration and in near future will likely allow for GE in a broad range of germplasm including wild relatives. The advent of HI-Edit technology is another step towards eliminating the dependence of GE from the genotype-specific variation in regeneration efficiency among different wheat cultivars. These advances will enable CRISPR-mediated induction of beneficial allelic diversity at multiple genes in diverse wheat cultivars, and identify the optimal combinations of CRISPR-induced and natural alleles with positive effects on agronomic traits by performing selection in the wheat breeding programs. In the future, the ability to transform wild relatives of wheat combined with the improved understanding of genes involved in domestication process will open possibilities for developing novel crops by the GE of domestication genes.
6 Key Concepts
The ability to engineer positive changes in agronomic traits using CRISPR-Cas genome editing technologies relies on decades of research performed to identify causal genes controlling phenotypic variation in agronomic crops. The CRISPR-Cas system uses guide RNAs designed to target specific regions of the genome to introduce precise modifications into coding or regulatory sequences of these causal genes. The genome editing projects start with the identification of these genes and choosing a CRISPR-Cas system and a genome editing strategy most suitable for performing desired modifications in the targeted genomic regions. The bioinformatically designed guide RNAs are experimentally validated and used to build plasmid constructs for genome editing. There are a number of methods exist for delivering the genome editing reagents into wheat plants including biolistic and Agrobacterium-mediated transformation, transformation with ribonucleoprotein complexes, and HI-Edit. The genome edited plants are screened for mutations in the targeted regions and phenotypically evaluated to assess the effect of newly engineered alleles on the traits of interest. These newly created variants of genes broaden the scope of genetic diversity available for selection in breeding programs and hold great potential to accelerate the improvement of agronomic traits.
7 Conclusions
CRISPR-Cas system is a powerful technology that could take full advantage of new genomic and genetic resources developed for wheat and related crops. Integration of this GE tool into the modern breeding practice may help to speed up the rate of genetic gain by accelerating the identification of novel agronomic genes, broadening genetic diversity of the identified genes, and reducing time required for transferring beneficial alleles into the adapted germplasm and evaluating their effects in the relevant environments. These advances will enable redesigning biological pathways underlying major agronomic traits in wheat by introducing the engineered genes into the breeding populations and selecting optimal allelic combinations to maximize target trait expression. The CRISPR-Cas-based tools will play important role in addressing many future wheat breeding challenges.
References
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. https://doi.org/10.1126/science.1225829
Gasiunas G, Barrangou R, Horvath P, Siksnys V (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci 109:E2579–E2586. https://doi.org/10.1073/pnas.1208507109
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. https://doi.org/10.1126/science.1231143
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826. https://doi.org/10.1126/science.1232033
Wiedenheft B, Sternberg SH, Doudna JA (2012) RNA-guided genetic silencing systems in bacteria and archaea. Nature 482:331–338. https://doi.org/10.1038/nature10886
Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, Charpentier E, Cheng D, Haft DH, Horvath P, Moineau S, Mojica FJM, Scott D, Shah SA, Siksnys V, Terns MP, Venclovas Č, White MF, Yakunin AF, Yan W, Zhang F, Garrett RA, Backofen R, van der Oost J, Barrangou R, Koonin EV (2020) Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol 18:67–83. https://doi.org/10.1038/s41579-019-0299-x
Nishimasu H, Shi X, Ishiguro S, Gao L, Hirano S, Okazaki S, Noda T, Abudayyeh OO, Gootenberg JS, Mori H, Oura S, Holmes B, Tanaka M, Seki M, Hirano H, Aburatani H, Ishitani R, Ikawa M, Yachie N, Zhang F, Nureki O (2018) Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361:1259–1262. https://doi.org/10.1126/science.aas9129
Wang W, Tian B, Pan Q, Chen Y, He F, Bai G, Akhunova A, Trick HN, Akhunov E (2020) Expanding the range of editable targets in the wheat genome using the variants of the Cas12a and Cas9 nucleases. bioRxiv. https://doi.org/10.1101/2020.12.09.418624
Anzalone AV, Koblan LW, Liu DR (2020) Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 38:824–844. https://doi.org/10.1038/s41587-020-0561-9
Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F (2015) Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163:759–771. https://doi.org/10.1016/j.cell.2015.09.038
Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, Liu DR (2019) Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576:149–157. https://doi.org/10.1038/s41586-019-1711-4
Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR (2017) Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551:464–471. https://doi.org/10.1038/nature24644
Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533:420–424. https://doi.org/10.1038/nature17946
Lin Q, Zong Y, Xue C, Wang S, Jin S, Zhu Z, Wang Y, Anzalone AV, Raguram A, Doman JL, Liu DR, Gao C (2020) Prime genome editing in rice and wheat. Nat Biotechnol 38:582–585. https://doi.org/10.1038/s41587-020-0455-x
Nakamura M, Gao Y, Dominguez AA, Qi LS (2021) CRISPR technologies for precise epigenome editing. Nat Cell Biol 23:11–22. https://doi.org/10.1038/s41556-020-00620-7
Gao C (2021) Genome engineering for crop improvement and future agriculture. Cell 184:1621–1635. https://doi.org/10.1016/j.cell.2021.01.005
Hayta S, Smedley MA, Demir SU, Blundell R, Hinchliffe A, Atkinson N, Harwood WA (2019) An efficient and reproducible Agrobacterium-mediated transformation method for hexaploid wheat (Triticum aestivum L.). Plant Methods 15(121) https://doi.org/10.1186/s13007-019-0503-z
Appels R, Eversole K, Stein N, Feuillet C, Keller B, Rogers J, Pozniak CJ, Choulet F, Distelfeld A, Poland J, Ronen G, Sharpe AG, Barad O, Baruch K, Keeble-Gagnère G, Mascher M, Ben-Zvi G, Josselin A-A, Himmelbach A, Balfourier F, Gutierrez-Gonzalez J, Hayden M, Koh C, Muehlbauer G, Pasam RK, Paux E, Rigault P, Tibbits J, Tiwari V, Spannagl M, Lang D, Gundlach H, Haberer G, Mayer KFX, Ormanbekova D, Prade V, Šimková H, Wicker T, Swarbreck D, Rimbert H, Felder M, Guilhot N, Kaithakottil G, Keilwagen J, Leroy P, Lux T, Twardziok S, Venturini L, Juhász A, Abrouk M, Fischer I, Uauy C, Borrill P, Ramirez-Gonzalez RH, Arnaud D, Chalabi S, Chalhoub B, Cory A, Datla R, Davey MW, Jacobs J, Robinson SJ, Steuernagel B, van Ex F, Wulff BBH, Benhamed M, Bendahmane A, Concia L, Latrasse D, Bartoš J, Bellec A, Berges H, Doležel J, Frenkel Z, Gill B, Korol A, Letellier T, Olsen O-A, Singh K, Valárik M, van der Vossen E, Vautrin S, Weining S, Fahima T, Glikson V, Raats D, Číhalíková J, Toegelová H, Vrána J, Sourdille P, Darrier B, Barabaschi D, Cattivelli L, Hernandez P, Galvez S, Budak H, JDG J, Witek K, Yu G, Small I, Melonek J, Zhou R, Belova T, Kanyuka K, King R, Nilsen K, Walkowiak S, Cuthbert R, Knox R, Wiebe K, Xiang D, Rohde A, Golds T, Čížková J, Akpinar BA, Biyiklioglu S, Gao L, N’Daiye A, Kubaláková M, Šafář J, Alfama F, Adam-Blondon A-F, Flores R, Guerche C, Loaec M, Quesneville H, Condie J, Ens J, Maclachlan R, Tan Y, Alberti A, Aury J-M, Barbe V, Couloux A, Cruaud C, Labadie K, Mangenot S, Wincker P, Kaur G, Luo M, Sehgal S, Chhuneja P, Gupta OP, Jindal S, Kaur P, Malik P, Sharma P, Yadav B, Singh NK, Khurana JP, Chaudhary C, Khurana P, Kumar V, Mahato A, Mathur S, Sevanthi A, Sharma N, Tomar RS, Holušová K, Plíhal O, Clark MD, Heavens D, Kettleborough G, Wright J, Balcárková B, Hu Y, Salina E, Ravin N, Skryabin K, Beletsky A, Kadnikov V, Mardanov A, Nesterov M, Rakitin A, Sergeeva E, Handa H, Kanamori H, Katagiri S, Kobayashi F, Nasuda S, Tanaka T, Wu J, Cattonaro F, Jiumeng M, Kugler K, Pfeifer M, Sandve S, Xun X, Zhan B, Batley J, Bayer PE, Edwards D, Hayashi S, Tulpová Z, Visendi P, Cui L, Du X, Feng K, Nie X, Tong W, Wang L (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361. https://doi.org/10.1126/science.aar7191
Walkowiak S, Gao L, Monat C, Haberer G, Kassa MT, Brinton J, Ramirez-Gonzalez RH, Kolodziej MC, Delorean E, Thambugala D, Klymiuk V, Byrns B, Gundlach H, Bandi V, Siri JN, Nilsen K, Aquino C, Himmelbach A, Copetti D, Ban T, Venturini L, Bevan M, Clavijo B, Koo D-H, Ens J, Wiebe K, N’Diaye A, Fritz AK, Gutwin C, Fiebig A, Fosker C, Fu BX, Accinelli GG, Gardner KA, Fradgley N, Gutierrez-Gonzalez J, Halstead-Nussloch G, Hatakeyama M, Koh CS, Deek J, Costamagna AC, Fobert P, Heavens D, Kanamori H, Kawaura K, Kobayashi F, Krasileva K, Kuo T, McKenzie N, Murata K, Nabeka Y, Paape T, Padmarasu S, Percival-Alwyn L, Kagale S, Scholz U, Sese J, Juliana P, Singh R, Shimizu-Inatsugi R, Swarbreck D, Cockram J, Budak H, Tameshige T, Tanaka T, Tsuji H, Wright J, Wu J, Steuernagel B, Small I, Cloutier S, Keeble-Gagnère G, Muehlbauer G, Tibbets J, Nasuda S, Melonek J, Hucl PJ, Sharpe AG, Clark M, Legg E, Bharti A, Langridge P, Hall A, Uauy C, Mascher M, Krattinger SG, Handa H, Shimizu KK, Distelfeld A, Chalmers K, Keller B, Mayer KFX, Poland J, Stein N, McCartney CA, Spannagl M, Wicker T, Pozniak CJ (2020) Multiple wheat genomes reveal global variation in modern breeding. Nature 2020:1–7
Wang W, Simmonds J, Pan Q, Davidson D, He F, Battal A, Akhunova A, Trick HN, Uauy C, Akhunov E (2018) Gene editing and mutagenesis reveal inter-cultivar differences and additivity in the contribution of TaGW2 homoeologues to grain size and weight in wheat. Theor Appl Genet 131:2463–2475. https://doi.org/10.1007/s00122-018-3166-7
Wang Y, Cheng Z, Shan W, Zhang Y, Liu J, Gao C, Qiu J (2014) Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol 32:947–951. https://doi.org/10.1038/nbt.2969
Wang W, Akhunova A, Chao S, Akhunov E (2016) Optimizing multiplex CRISPR/Cas9-based genome editing for wheat. bioRxiv. https://doi.org/10.1101/051342
Wang W, Pan Q, Tian B, He F, Chen Y, Bai G, Akhunova A, Trick HN, Akhunov E (2019) Gene editing of the wheat homologs of TONNEAU1-recruiting motif encoding gene affects grain shape and weight in wheat. Plant J 100:251–264. https://doi.org/10.1111/tpj.14440
Čermák T, Curtin SJ, Gil-Humanes J, Čegan R, Kono TJY, Konečná E, Belanto JJ, Starker CG, Mathre JW, Greenstein RL, Voytas DF (2017) A multipurpose toolkit to enable advanced genome engineering in plants. Plant Cell 29:1196–1217. https://doi.org/10.1105/tpc.16.00922
Su Z, Bernardo A, Tian B, Chen H, Wang S, Ma H, Cai S, Liu D, Zhang D, Li T, Trick H, St. Amand P, Yu J, Zhang Z, Bai G (2019) A deletion mutation in TaHRC confers Fhb1 resistance to Fusarium head blight in wheat. Nat Genet 51:1099–1105. https://doi.org/10.1038/s41588-019-0425-8
Gao Y, An K, Guo W, Chen Y, Zhang R, Zhang X, Chang S, Rossi V, Jin F, Cao X, Xin M, Peng H, Hu Z, Guo W, Du J, Ni Z, Sun Q, Yao Y (2021) The endosperm-specific transcription factor TaNAC019 regulates glutenin and starch accumulation and its elite allele improves wheat grain quality. Plant Cell. https://doi.org/10.1093/plcell/koaa040
Zhang Z, Hua L, Gupta A, Tricoli D, Edwards KJ, Yang B, Li W (2019) Development of an Agrobacterium-delivered CRISPR/Cas9 system for wheat genome editing. Plant Biotechnol J 17:1623–1635. https://doi.org/10.1111/pbi.13088
Zhang Y, Liang Z, Zong Y, Wang Y, Liu J, Chen K, Qiu J-L, Gao C (2016) Efficient and transgene-free genome editing in wheat through transient expression of CRISPR/Cas9 DNA or RNA. Nat Commun 7:12617. https://doi.org/10.1038/ncomms12617
Zhang R, Liu J, Chai Z, Chen S, Bai Y, Zong Y, Chen K, Li J, Jiang L, Gao C (2019) Generation of herbicide tolerance traits and a new selectable marker in wheat using base editing. Nat Plants 5:480–485. https://doi.org/10.1038/s41477-019-0405-0
Debernardi JM, Tricoli DM, Ercoli MF, Hayta S, Ronald P, Palatnik JF, Dubcovsky J (2020) A GRF–GIF chimeric protein improves the regeneration efficiency of transgenic plants. Nat Biotechnol 38:1274–1279. https://doi.org/10.1038/s41587-020-0703-0
Liang Z, Chen K, Li T, Zhang Y, Wang Y, Zhao Q, Liu J, Zhang H, Liu C, Ran Y, Gao C (2017) Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nat Commun 8:14261. https://doi.org/10.1038/ncomms14261
Kelliher T, Starr D, Su X, Tang G, Chen Z, Carter J, Wittich PE, Dong S, Green J, Burch E, McCuiston J, Gu W, Sun Y, Strebe T, Roberts J, Bate NJ, Que Q (2019) One-step genome editing of elite crop germplasm during haploid induction. Nat Biotechnol 37:287–292. https://doi.org/10.1038/s41587-019-0038-x
Wang W, Pan Q, He F, Akhunova A, Chao S, Trick H, Akhunov E (2018) Transgenerational CRISPR-Cas9 activity facilitates multiplex gene editing in allopolyploid wheat. CRISPR J 1:65–74. https://doi.org/10.1089/crispr.2017.0010
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this chapter

Cite this chapter
Wang, W., Akhunov, E. (2022). Application of CRISPR-Cas-Based Genome Editing for Precision Breeding in Wheat. In: Reynolds, M.P., Braun, HJ. (eds) Wheat Improvement. Springer, Cham. https://doi.org/10.1007/978-3-030-90673-3_29
Download citation
DOI: https://doi.org/10.1007/978-3-030-90673-3_29
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-90672-6
Online ISBN: 978-3-030-90673-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)