
Abstract
Lipoprotein(a) [Lp(a)] is an atherogenic lipoprotein with a strong genetic regulation. Up to 90% of the concentrations are explained by a single gene, the LPA gene. The concentrations show a several-hundred-fold interindividual variability ranging from less than 0.1 mg/dL to more than 300 mg/dL. Lp(a) plasma concentrations above 30 mg/dL and even more above 50 mg/dL are associated with an increased risk for cardiovascular disease including myocardial infarction, stroke, aortic valve stenosis, heart failure, peripheral arterial disease, and all-cause mortality. Since concentrations above 50 mg/dL are observed in roughly 20% of the Caucasian population and in an even higher frequency in African-American and Asian-Indian ethnicities, it can be assumed that Lp(a) is one of the most important genetically determined risk factors for cardiovascular disease.
Carriers of genetic variants that are associated with high Lp(a) concentrations have a markedly increased risk for cardiovascular events. Studies that used these genetic variants as a genetic instrument to support a causal role for Lp(a) as a cardiovascular risk factor are called Mendelian randomization studies. The principle of this type of studies has been introduced and tested for the first time ever with Lp(a) and its genetic determinants.
There are currently no approved pharmacologic therapies that specifically target Lp(a) concentrations. However, some therapies that target primarily LDL cholesterol have also an influence on Lp(a) concentrations. These are mainly PCSK9 inhibitors that lower LDL cholesterol by 60% and Lp(a) by 25–30%. Furthermore, lipoprotein apheresis lowers both, Lp(a) and LDL cholesterol, by about 60–70%. Some sophisticated study designs and statistical analyses provided support that lowering Lp(a) by these therapies also lowers cardiovascular events on top of the effect caused by lowering LDL cholesterol, although this was not the main target of the therapy. Currently, new therapies targeting RNA such as antisense oligonucleotides (ASO) or small interfering RNA (siRNA) against apolipoprotein(a), the main protein of the Lp(a) particle, are under examination and lower Lp(a) concentrations up to 90%. Since these therapies specifically lower Lp(a) concentrations without influencing other lipoproteins, they will serve the last piece of the puzzle whether a decrease of Lp(a) results also in a decrease of cardiovascular events.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Apolipoprotein(a)
- Association study
- Cardiovascular disease
- Copy number variation
- Lipoprotein(a)
- Lp(a)
- Mendelian randomization
- Therapy
1 Introduction
Lipoprotein(a) [Lp(a)] is one of the strongest genetically determined risk factors for cardiovascular disease (CVD) (Kronenberg and Utermann 2013). It contains besides an LDL particle an additional apolipoprotein that is called apolipoprotein(a) [apo(a)]. This apolipoprotein shows a high homology with plasminogen. After more than 50 years of research, the physiological function of Lp(a) is still unexplained. An astonishing characteristic of Lp(a) is the more than 1,000-fold range of concentrations between individuals from almost zero to more than 300 mg/dL (Kronenberg and Utermann 2013). The distribution of Lp(a) is very skewed in most populations: for example, roughly 50% of the Europeans have concentrations below 10 mg/dL and about 25% have concentrations above 30 mg/dL.
2 Sites of Production and Catabolism of Lp(a)
Apo(a) is synthesized in the liver (Tomlinson et al. 1989; Kraft et al. 1989). Variation in Lp(a) concentrations among individuals is determined by the rate of production rather than by differences in the catalytic rate (Rader et al. 1994). After secretion of apo(a), it binds to LDL-apoB lysine-binding residues by its lysine-binding sites with subsequent forming of a disulfide bond. Whether the assembly occurs in the circulation at the hepatic surface or intracellularly is a matter of debate (Koschinsky and Marcovina 2004).
The site and mechanism of catabolism is discussed controversially. Studies in mice revealed that the liver is the main route of Lp(a) catabolism (Cain et al. 2005). The finding of an arteriovenous difference of Lp(a) concentrations in the renal circulation (Kronenberg et al. 1997), of apo(a) fragments in urine (Mooser et al. 1996), and of a disturbed Lp(a) metabolism in kidney disease (Frischmann et al. 2007) has suggested also a role of the kidney in Lp(a) catabolism. The receptors involved in the catabolic process are a matter of debate as recently comprehensively reviewed (McCormick and Schneider 2019) and include lipoprotein receptors such as the LDL receptor, VLDL receptor, LDL receptor related proteins (LRP1 and LRP2), toll-like and scavenger receptors (e.g., CD36 and SR-BI), carbohydrate receptors or lectins, and plasminogen receptors.
3 Physiology and Pathophysiology of Lp(a)
The physiological function of Lp(a) is still in the dark. It is believed that Lp(a) has proatherogenic and prothrombotic properties (Fig. 1). The apo(a) glycoprotein has a high degree of homology to plasminogen (McLean et al. 1987) suggesting that Lp(a) might not only be a link between the cholesterol transport system in plasma and the fibrinolytic system but may also act as a modulator of the balance between blood clotting and fibrinolysis. At least in vitro, Lp(a) indeed interferes with the blood clotting/fibrinolytic cascades at several steps (Boffa and Koschinsky 2016). As reviewed extensively (Koschinsky and Marcovina 2004; Boffa and Koschinsky 2016), this includes the inhibition of streptokinase and urokinase-mediated activation of plasminogen by the tissue-type plasminogen activator (t-PA), inhibition of t-PA in solution, fibrin and fibrinogen binding, competition with plasminogen and t-PA binding for soluble fibrinogen, competition with plasminogen for binding to cellular receptors, and enhancement of the plasminogen-activator-inhibitor PAI-1 activity. Considering that many individuals have low Lp(a) concentrations and some of them even almost no Lp(a) and others have concentrations far above 100 mg/dL, one might expect major effects on the balance between clotting and fibrinolysis which, however, have not convincingly been described by in vivo data. Epidemiologic and genetic studies did not provide support for a thrombogenic role and if there is any, it can only be found at very high Lp(a) values above the 95th percentile (Nordestgaard and Langsted 2016). Interestingly, there are reports that the situation might be different in childhood where high Lp(a) concentrations were found to be accompanied with venous thromboembolism (Nowak-Gottl et al. 1999).

Proatherogenic and prothrombotic properties of Lp(a)
A further property of Lp(a)/apo(a) is the interaction with components of the extracellular matrix including fibrin, fibronectin, tetranectin, proteoglycans, and β2-glycoprotein (Köchl et al. 1997). The domains in apo(a), which mediate binding to fibrin/fibrinogen, are lysine-binding sites in KIV-8 and KIV-10 (Koschinsky and Marcovina 2004). Binding of Lp(a) to fibrin has been proposed as a mechanism to deliver cholesterol to sites of injury and wound healing of the vascular wall with the negative side effect that Lp(a) also deposits cholesterol in growing atherosclerotic plaques and inhibits fibrinolysis at the plaque surface. Furthermore, high Lp(a) levels impair activation of transforming growth factor-β by downregulation of plasmin generation, thereby contributing to smooth muscle cell proliferation (Grainger et al. 1993). The published data provide clear evidence that Lp(a) can interfere with many key reactions of clotting/fibrinolysis in vitro and is deposited in atherosclerotic plaques. Moreover, effects on monocytes/macrophages result in foam cell formation (Poon et al. 1997). Lp(a) induces chemoattractant activity of monocytes and induces macrophage expression of interleucin-8 (IL-8) (Klezovitch et al. 2001).
The connection between innate immune system and Lp(a) has been further strengthened by the identification of Lp(a) as the major plasma carrier of oxidized phospholipids (OxPLs) (Bergmark et al. 2008) which can stimulate many pro-inflammatory pathways in the arterial wall. The binding-site for oxidized phospholipids has been identified in the protein moiety of Lp(a), specifically in the KIV type 10 domain of apo(a) (Leibundgut et al. 2013). Levels of Lp(a) and OxPL in human plasma are highly correlated and it is therefore not unexpected that this also results in an association of OxPL levels with cardiovascular disease (Tsimikas et al. 2005, 2010; Kiechl et al. 2007). These oxidized lipid species are recognized by pattern recognition receptors of innate immune cells and trigger the whole cascade of inflammatory processes that can finally lead to plaque destabilization (Boffa and Koschinsky 2019). During recent years a series of publications (Fig. 2) from the group around Eric Stroes investigated the impact of Lp(a) and LDL-C on arterial wall inflammation and found that individuals with elevated Lp(a) concentrations have increased arterial inflammation and enhanced peripheral blood mononuclear cell trafficking to the arterial wall compared with subjects with normal Lp(a). Monocytes isolated from subjects with high Lp(a) concentrations showed an increased capacity to transmigrate the endothelium and produce pro-inflammatory cytokines. This was mediated by OxPLs and subsequent blocking of the OxPLs by specific antibodies also reduced the pro-inflammatory responsiveness of the monocytes (Fig. 2a) (van der Valk et al. 2016). The subsequent publications revealed that a pronounced reduction of LDLC by PCSK9 inhibitors is not sufficient to substantially reduce the arterial wall inflammation (Fig. 2b) (Stiekema et al. 2019). In a further investigation they examined whether patients with cardiovascular disease and elevated Lp(a) concentrations experience anti-inflammatory effects following large reductions of Lp(a) by apo(a) antisense therapy. They observed in a first step that circulating monocytes of healthy individuals and patients, both having high Lp(a) concentrations, are characterized by a markedly pro-inflammatory gene expression profile with several pathways of the innate immune system being upregulated. In an intervention with apo(a) antisense therapy the authors showed that the resulting 47% lowering of Lp(a) concentrations was indeed capable of reversing the pro-inflammatory gene expression signature to levels close to that of controls with normal Lp(a) concentrations. This was accompanied with a 22% functional reduction in transendothelial migration capacity of monocytes ex vivo (Fig. 2c) (Stiekema et al. 2020; Coassin and Kronenberg 2020). These findings proposed a further mechanism how Lp(a) might mediate cardiovascular disease and added a new layer to our still shallow understanding of the exact pathophysiological mechanisms by which high Lp(a) causes cardiovascular disease.

Impact of Lp(a) on arterial wall inflammation. Panel (a) Individuals with high Lp(a) (n = 30, median 108 mg/dL) present significantly increased arterial wall inflammation as assessed by magnetic resonance imaging, 18-F fluorodeoxyglucose (18F-FDG) uptake PET/CT and SPECT/CT, and activated monocytes assessed by transendothelial migration and monocyte priming and challenge assays (van der Valk et al. 2016). Panel (b) 420 mg of the PCSK9 inhibitor evolocumab vs. placebo over 16 weeks reduced LDL-C consistently but did not improve arterial wall inflammation in individuals with high Lp(a) (n = 129, median ≈80 mg/dL), despite modest concomitant Lp(a) reduction (−13.9%) (Stiekema et al. 2019). Panel (c) High Lp(a) was associated with a pro-inflammatory transcriptome in monocytes. Treatment with different regimes of AKCEA-APO(a)-LRx resulted in a 47% reduction of Lp(a) and in reductions of the pro-inflammatory signatures in the transcriptome, the transendothelial migration capacity, and the expression of chemokines and toll-like receptors on the monocytes surface (Stiekema et al. 2020; Coassin and Kronenberg 2020)
4 Genetic Control of Lp(a) Concentrations
Lp(a) concentrations are not much influenced by age, sex, fasting state, inflammation (Kronenberg 2014a; Langsted et al. 2014) and lifestyle factors such as diet or physical activity. However, the concentrations are under strict genetic control. Family studies revealed a heritability estimate of Lp(a) concentrations of about 90% (Austin et al. 1992; Lamon-Fava et al. 1991; Utermann 1989). Lp(a) is therefore the lipoprotein with the strongest genetic control. The discovery of the size polymorphism of apo(a) in serum (Utermann et al. 1987) which is based on a variable number of the so-called kringle-IV (K-IV) repeats in the LPA gene (Utermann 1989; Kraft et al. 1992; Lackner et al. 1991, 1993) resulted in the identification of the LPA gene as the major gene for Lp(a) levels. Each of these up to more than 40 repeats has a size of 5.6 kB which results in a highly polymorphic and informative copy number variation (CNV).
There exists a pronounced inverse correlation between the number of K-IV repeats and Lp(a) concentrations. Individuals expressing a low number of K-IV repeats resulting in the so-called small apo(a) isoforms (up to 22K-IV repeats) have on average markedly higher Lp(a) concentrations than individuals carrying only large apo(a) isoforms (more than 22K-IV repeats) (Kronenberg and Utermann 2013). This K-IV size polymorphism of apo(a) explains about 20–80% of the variability of Lp(a) concentrations depending on the ethnicity (Utermann et al. 1987).
Interestingly, Lp(a) levels of unrelated individuals carrying the same isoform size combination can still vary by up to 200-fold (Cohen et al. 1993; Perombelon et al. 1994). On the other hand, same-sized alleles within families which are identical-by-descent show typically a less than 2.5-fold variation in Lp(a) concentrations (Perombelon et al. 1994). This is a clear indication that further genetic variants exist that regulate the Lp(a) concentrations in addition to the isoform size (Cohen et al. 1993; Coassin et al. 2017). The search for single nucleotide polymorphisms in the LPA gene region had a first peak in 2009 with the description of the two SNPs rs10455872 and rs3798220 which since then are regularly used in hundreds of association studies (Clarke et al. 2009). However, there are many more SNPs in the wider LPA gene region which are associated with Lp(a) concentrations and genome-wide association studies (GWAS) have explored this in a systematic way (Li et al. 2015; Mack et al. 2017; Ober et al. 2009). In a recent GWAS meta-analysis in 13,781 individuals from 5 populations we identified 2001 SNPs in a 1.76 MB large region around the LPA gene. 48 of these SNPs were associated independently of each other with Lp(a) concentrations (p-values below 5 × 10−8) (Mack et al. 2017). However, that does not mean that each of these SNPs is causally (functionally) related to Lp(a) concentrations. Many of these SNPs are probably in linkage disequilibrium with another SNP that is causally influencing Lp(a) concentrations. A typical example is rs75692336 which showed the strongest association with Lp(a) concentrations in a statistical model in our GWAS which adjusted besides age and sex also for the apo(a) isoform size. This SNP turned out to be a proxy SNP for the real culprit, a splice-site variant (G4925A) in the kringle-IV type 2 which is not easily accessible for genotyping (Coassin et al. 2017) and therefore not found in GWAS analyses (Mack et al. 2017).
Besides the LPA gene region, the APOE gene was found to be independently associated with Lp(a) concentrations. Especially the SNP which is responsible for the APO E2 allele (rs7412) decreased Lp(a) concentrations by 3.34 mg/dL or ≈15% of the mean values of the population (Mack et al. 2017). The recent GWAS in 293,274 White British individuals revealed two further loci, the CETP locus as well as the APOH locus, the latter encodes for beta2-glycoprotein I (Hoekstra et al. 2021).
The major part of the LPA gene is a so-called camouflaged gene since up to 70% of the coding sequence harboring the K-IV type 2 with up to more than 40 repeats is not easily accessible to modern sequencing technologies. Each copy of the K-IV type 2 has a size of 5.6 kb. Until recently almost no sequence information was available for that region. Our group recently developed an ultra-deep sequencing strategy for this region and a variant analysis pipeline and reported the first map of genetic variation in the KIV-2 region. By sequencing 123 Central-European individuals and reanalyzing public data of 2,504 individuals from 26 populations, we found 14 different loss-of-function and splice-site mutations, as well as >100 partially even common missense variants (Coassin et al. 2019). One of these variants is the above-mentioned splice-site variant (G4925A) which has a frequency of about 22% and tremendously decreases Lp(a) concentrations by more than 30 mg/dL (Coassin et al. 2019). Another variant is the R21X in the KIV-2 region which was not easy to analyze until recently. It is a likely causal SNP resulting in a nonsense mutation, which leads to a truncated protein that is rapidly degraded (Parson et al. 2004). We recently developed a highly sensitive allele-specific qPCR assay and genotyped R21X in 10,910 individuals from three populations and showed that R21X carriers have significantly lower (−11.7 mg/dL) mean Lp(a) concentrations (Di Maio et al. 2020). A study by Morgan and colleagues investigated two very rare variants, R990Q (rs41259144) located in the KIV type 4 and R1771C (rs139145675) located in the kringle V which were present in four null Lp(a) individuals. These two variants are hypothesized to impair the ability of the protein folding and thereby circumventing its processing to maturity for secretion (Morgan et al. 2020). Other detected mutations are currently under investigation to explain the functional consequences of these variants. It might very well be that some of these variants explain the large ethnic and interindividual differences in Lp(a) concentrations.
5 Lp(a) Concentrations and Risk for CVD
5.1 Searching for Lp(a) Thresholds Associated with an Increased Coronary Artery Disease Risk
The evidence is quite strong that high Lp(a) concentrations are associated with an increasing risk for cardiovascular disease (Kronenberg and Utermann 2013; Cegla et al. 2019). The Copenhagen City Heart Study observed for individuals from a general population with concentrations between 30 and 76 mg/dL (corresponding to the 67th–90th percentile) a 1.60-fold increased risk for incident myocardial infarction compared to individuals with Lp(a) concentrations below 5 mg/dL (corresponding to the lower 22% of the population). This risk increased to 1.90 for persons with Lp(a) concentrations between 77 and 117 mg/dL (90th to 95th percentile) and to 2.60 for individuals with Lp(a) concentrations above 117 mg/dL (>95th percentile) (Kamstrup et al. 2009) (Fig. 3, panel (a)). The concentration threshold for an increased risk has been discussed controversially and an European Atherosclerosis Society (EAS) consensus statement proposed 50 mg/dL (Nordestgaard et al. 2010). Most importantly, such a threshold corresponds to the 80th percentile of the concentration distribution in a Caucasian population and means that 20% of the population have probably an increased risk for CVD due to elevated Lp(a) concentrations. From a standpoint of public health relevance, this makes Lp(a) a very important risk factor for CVD.

Mendelian randomization approach to demonstrate a causal association between Lp(a) concentrations and cardiovascular disease. Panel (a) shows the association between elevated Lp(a) concentrations and cardiovascular disease (CVD) as shown in the Copenhagen City Heart Study (Kamstrup et al. 2009). Panel (b) shows the association between the number of K-IV repeats in the LPA gene and Lp(a) concentrations: individuals with small apo(a) isoform have markedly higher median Lp(a) concentrations than individuals with large apo(a) isoforms (Laschkolnig et al. 2014). Panel (c) shows the preponderance of small apo(a) isoforms in patients with CVD when compared to controls (Sandholzer et al. 1992). Since a low number of K-IV copies (11–22 copies) is associated with high Lp(a) levels and high Lp(a) levels are associated with CVD, it follows that a low number of K-IV copies has to be associated with CVD if the association of Lp(a) with CVD is causal. Figure is taken and adapted with permission from reference (Kronenberg 2016b)
The most recent data from the UK Biobank with more than 460,000 study participants followed for more than 11 years are in line with a rather linear increase in risk for CVD with increasing Lp(a) concentrations. In that sufficiently powered study the risk started already to increase above the median of Lp(a) which was 19.6 nmol/L (corresponds to roughly 8 mg/dL) (Patel et al. 2021). Of course, this raises the question what is a clinically meaningful increase in risk? The same study revealed also that Lp(a) is not only a risk factor in non-White populations but also in Black or Asian people (Patel et al. 2021), a matter of major discussions over the past. This is in line with earlier results from the ARIC study (Virani et al. 2012), the MESA Study (Guan et al. 2015), or the Dallas Heart Study (Lee et al. 2017), but in contrast to NHANES III or the INTERHEART Study which did not found Lp(a) to be associated with CVD in non-Hispanic Blacks (Brandt et al. 2020) or Africans (Pare et al. 2019), respectively.
The recent statement by the new ESC/EAS guidelines for the management of dyslipidemia might have introduced more confusion than clarification by stating that “Lp(a) measurement should be considered at least once in each adult person’s lifetime to identify those with very high inherited Lp(a) levels >180 mg/dL (>430 nmol/L) who may have a lifetime risk of ASCVD equivalent to the risk associated with heterozygous familial hypercholesterolaemia.” (Mach et al. 2019) The first part of this sentence to measure Lp(a) at least once in each individual is indeed a forward-looking advice important for risk stratification and in the near future maybe also for therapeutic interventions. However, some interpret the second part of this statement as the introduction of a new threshold for risk stratification. However, this is not the case since they mention that these values are associated with “very high inherited risk.” The risk is already markedly increased at lower levels as discussed above. These very high levels are only observed in far less than 1% of White people and should not be used as an excuse either not to screen or to decline any therapeutic options to lower Lp(a) concentrations with currently available therapies such as lipid apheresis or with therapies that will become available in the future. This part of the guidelines and the cited literature did not show the data and the calculations which resulted in this threshold in a convincing and reproducible way (Mach et al. 2019). It has therefore to be considered with caution and needs further clarification.
5.2 Lp(a) and Other Vascular Diseases
Besides numerous studies on coronary artery disease, during recent years several studies found even a strong association between high Lp(a) concentrations and stroke (Erqou et al. 2009; Langsted et al. 2019a; Zhang et al. 2019), aortic valve calcification stenosis (Arsenault et al. 2014; Cairns et al. 2017; Capoulade et al. 2018; Chen et al. 2018; Kamstrup et al. 2014; Thanassoulis et al. 2013; Vongpromek et al. 2015; Vuorio et al. 2019; Perrot et al. 2019), heart failure (Kamstrup and Nordestgaard 2016) as well as peripheral arterial disease (Dieplinger et al. 2007; Gurdasani et al. 2012; Laschkolnig et al. 2014). A recent GWAS for peripheral arterial disease identified for the LPA gene region the strongest signal which was even more pronounced that for the CDKN2B region on chromosome 9p21 or for the LDL receptor (Klarin et al. 2019). This makes Lp(a) a risk factor for many CVD endpoints although the strength of the association might differ between the various entities of endpoints. Studies that investigated the association of the LPA gene region with all-cause mortality (Langsted et al. 2019b) demonstrated also a significant association in case they were sufficiently powered. A sufficient study size and number of endpoints is in this context a prerequisite since the finding of an association with all-cause mortality is mainly driven by death in the wider context of CVD.
5.3 Differences Between Primary and Secondary Prevention Studies
There is strong evidence for an association between high Lp(a) concentrations and future CVD events in studies in whom participants were free of CVD at baseline (primary prevention studies). However, findings are inconsistent for study populations with preexisting CVD at baseline (secondary prevention studies). A meta-analysis of 11 secondary prevention studies found that elevated Lp(a) predicted major adverse cardiovascular events with an odds ratio of 1.40 (95% CI 1.15–1.71, p < 0.001) with a considerable heterogeneity between studies. When the authors stratified the studies into those with high (≥130 mg/dL) versus low (<130 mg/dL) average on-treatment LDL-C, the association for Lp(a) was still significant for the studies with high average LDL-C (OR = 1.46, 95% CI 1.23–1.73) but not for those with low average LDL-C (OR = 1.20, 95% CI 0.90–1.60 (O’Donoghue et al. 2014). This has raised many discussions since especially first intervention studies to specifically lower Lp(a) would target patients with established CVD. A seminal review on secondary prevention studies elucidating the reasons why some of these studies did not find an association has recently been published by Boffa and colleagues (2018). The authors distinguished between major potential general “confounders” and confounders related to Lp(a) which might have influenced the findings of some of the secondary prevention studies. As general confounders they discussed the restrictive inclusion and exclusion criteria of randomized controlled trials, the lack of statistical power, differences in clinical management between primary and secondary prevention trials and finally an index event bias. Index event bias may underlie paradoxical findings whereby risk factors that are well established to contribute to pathological events do not appear to predict recurrence of these events (Dahabreh and Kent 2011). A famous example is hypertension which increases the risk for a first stroke about fourfold but is not associated with stroke recurrence (Smits et al. 2013). As confounders related to Lp(a) the authors mentioned the use of log-transformed Lp(a) levels, secondary changes in Lp(a) concentrations due to the events (consider that Lp(a) is thought to be an acute-phase protein), and issues related to the measurement of Lp(a) (e.g., standardization of the assays, isoform-dependency of the assays, or effects of sample handling and storage on Lp(a) measurement) (Boffa et al. 2018).
5.4 Is Lp(a) an Independent Risk Factor for CVD?
There is ample evidence that Lp(a) is an independent risk factor for CVD (Kronenberg and Utermann 2013). That means that high Lp(a) can increase the risk for CVD even if other classical risk factors are not present. And if combined with other risk factors the entire risk of a particular person increases further. Whether this risk increase is linear or exponential is much easier to predict on the population level than for a given person. Risk prediction is associated with uncertainties as long as we do not know or measure all factors that contribute to the risk of a disease. This has recently been demonstrated quite convincingly in the Malmö Diet and Cancer Study with almost 30,000 study participants and more than 4,122 incident coronary artery disease cases during a long median observation period of 21.3 years. The authors calculated polygenic risk scores for each individual using 6.24 million SNPs and observed that the lifetime risk for coronary artery disease increased from about 16% in those 10% of the participants with the lowest polygenic risk scores to more than 45% in the 10% of the population with the highest score. Most interestingly, this was independent from traditional risk factors meaning that in each traditional risk factor category (low, medium, borderline, and high) the risk increased two- to threefold for those with a high genetic risk score category compared to a low genetic risk score category (Hindy et al. 2020). This clearly demonstrates that a single risk factor including Lp(a) should never be seen “isolated” without considering the other risk factors as good as possible. This can be seen best when looking at the distribution of CVD risk contributors such as systolic blood pressure or LDL cholesterol in individuals who remain free and those who develop CVD over the upcoming years: there is always a substantial overlap in the distribution of these factors as already shown in the ancient studies from the Framingham cohort (Kannel et al. 1964) and lately by cohorts from Finland and Sweden (Ripatti et al. 2010). This explains also why not everybody with high Lp(a) will develop CVD.
With Lp(a) and LDL-C a very special situation is present since almost each method of LDL-C measurement (or calculation by formulas) includes also the cholesterol content present in in the LDL particle of Lp(a). It is estimated that the proportion of cholesterol in the Lp(a) particle is about 30% of the Lp(a) mass (Kinpara et al. 2011), although some discuss an even higher value of 45% (Kronenberg et al. 2004) which also depends on the apo(a) isoform size. In some individuals with high Lp(a) concentrations the amount of cholesterol derived from Lp(a) can be quite substantial as in the roughly 5% of the general population with an Lp(a) concentration above 100 mg/dL. In these persons the cholesterol content included in the LDL-C measurement but originating from Lp(a) would be 30 to 45 mg/dL. In persons with 200 mg/dL this would contribute 60 to 90 mg/dL to the LDL-C measurement. This additional amount of LDL-C can even result in a diagnosis (or misclassification) of hypercholesterolemia which is actually caused by high Lp(a) concentrations (Langsted et al. 2016). A very recent study, however, reported that the percentage of Lp(a) cholesterol relative to Lp(a) mass varied from 5.8% to 57.3% (Yeang et al. 2021). Especially this low percentage of cholesterol might require further investigation and validation.
A further consequence from this measurement issue might become of clinical relevance, especially when a patient is given a statin and shows no response or a low response to this LDL-C-targeting treatment. A reason for this might be a high Lp(a) concentration: statins do not lower Lp(a) concentrations. If Lp(a) is very high, LDL-C might be misclassified as high which could result in a “wrong” indication for a statin under particular circumstances. When the targeted “true” LDL-C (the LDL-C without the Lp(a) cholesterol) is already in the target range, a treatment with a statin might result in a lower response than expected from the uncorrected LDL-C value (Scanu and Hinman 2002; Miltiadous et al. 2006).
6 What Evidence Do We Have for a Causal Association of High Lp(a) with CVD?
When a biomarker is changed in diseased patients, the important discussion starts whether this biomarker is a risk factor or a risk marker as illustrated recently (Kronenberg 2019a). In case of a risk factor, this parameter is causally related to disease and it might become an interesting drug target. If it is a biomarker, this parameter might be interesting for diagnostic purposes because it is changed secondarily to the disease and points the physician to the disease. It would not make sense to develop drugs which influence that parameter (see recent discussion on this issue in reference (Kronenberg 2016a). Mendelian randomization studies illustrated in Fig. 3 provide a strong support for causality. Genetic variants that are strongly associated with high Lp(a) concentrations (Fig. 3, panel (b)) show also an increased risk with CVD (Fig. 3, panel (c)) which underscores the causal link between high Lp(a) concentrations and CVD. Actually Lp(a) was the first practical example applying this approach almost a decade before the term “Mendelian randomization” has been coined: at the beginning of the 1990s data on the small apo(a) isoforms were used as genetic instrument: small isoforms with up to 22 K-IV repeats were associated with a significantly increased risk for coronary heart disease in six different populations (Sandholzer et al. 1992). A later meta-analysis including 7,382 coronary heart disease cases and 8,514 controls identified a 2.08-fold increased risk for carriers of small apo(a) isoforms (Erqou et al. 2010). Later approaches that investigated LPA variants on the DNA level either by the number of KIV repeats identified by pulsed-field gel electrophoresis or by the sum of KIV repeats of the two alleles by quantitative PCR or by the investigation of SNPs that are associated with high Lp(a) concentrations revealed the same finding (Clarke et al. 2009; Kamstrup et al. 2009; Kraft et al. 1996). On the other hand, genetic variants that are associated with low Lp(a) concentrations are obviously protective from CVD (Coassin et al. 2017; Di Maio et al. 2020; Lim et al. 2014). These strong associations make Lp(a) probably the most important genetic risk factor for CVD if we keep in mind the high frequency of small apo(a) isoforms or variants which go along with high Lp(a) concentrations in the population (Kronenberg 2016b).
7 RNA-Targeting Therapies to Specifically Lower Lp(a)
Based on the epidemiological and genetic studies the next and most consequent logical step is the therapeutic lowering of Lp(a) and the investigation whether such a therapy also decreases the number of CVD events (Fig. 4). To specifically accomplish an Lp(a)-lowering without influencing other lipoproteins, two main approaches are currently available and both selectively reduce the synthesis of apo(a) in the liver (Landmesser et al. 2020) and are therefore the optimal approach not only to lower Lp(a) but also to serve the last piece of the puzzle to demonstrate that a specific lowering of Lp(a) will also reduce CVD events.

Effect of therapeutic interventions on lipoproteins and clinical outcomes
7.1 Antisense Oligonucleotides (ASO) Against Apolipoprotein(a)
In principle, antisense oligonucleotides are 13–20 nucleic acid long and bind to the target RNA. As soon as the single-stranded ASOs are taken up by the cell, they bind directly to the mRNA creating a duplex that forms a complex with the intracellularly available RNAse H1. This finally mediates the target mRNA cleavage preventing the production of the targeted protein (Landmesser et al. 2020).
Using this principle to target apo(a), already the first phase I trial with IONIS-APO(a)Rx given subcutaneously in various amounts and numbers of dosages revealed a potent Lp(a)-lowering effect up to almost 80% (Tsimikas et al. 2015). In further trials it has been shown that this therapy results also in a reduction of oxidized phospholipids and a reduced monocyte inflammatory activation that returned close to baseline levels after stopping the medication. Furthermore, a new chemistry was used in this next trial in which a modified IONIS-APO(a)Rx antisense oligonucleotide is conjugated with a GalNAc3 complex (IONIS-APO(a)-LRx). This formulation targets the drug to the hepatocyte via the asialoglycoprotein receptor, making it 30 times more potent than the parent antisense oligonucleotide. This enabled the administered dose to be reduced 10-fold, thereby improving its tolerability. The highest dose administered resulted in a 92% mean reduction of Lp(a) with no serious side effects (Viney et al. 2016). The most recent trial included 286 patients with established CVD and Lp(a) concentrations of at least 60 mg/dL (Tsimikas et al. 2020a). Administration of APO(a)-LRx resulted in dose-dependent decreases in Lp(a) concentrations with up to 72% at 60 mg every 4 weeks, and 80% at 20 mg every week, as compared with 6% with placebo. There were no significant differences between any APO(a)-LRx dose and placebo with respect to platelet counts, liver and renal measures, or influenza-like symptoms. The most common adverse events were injection-site reactions (Tsimikas et al. 2020a).
Based on those data the Lp(a)HORIZON trial (NCT04023552: https://clinicaltrials.gov/ct2/show/study/NCT04023552) started recently and will recruit 7,680 patients with the key inclusion criteria of Lp(a) ≥70 mg/dL at the screening visit, an optimal LDL cholesterol lowering treatment, an optimal treatment of other CV risk factors, and a myocardial infarction or an ischemic stroke ≥3 months to ≤10 years prior to the screening visit or a clinically significant symptomatic peripheral artery disease. Patients will be injected monthly 80 mg of the drug (now called TQJ230 or Pelacarsen) or placebo subcutaneously. The estimated completion date is April 2024.
7.2 Short Interfering RNA (siRNA) to Target Apo(a)
The siRNAs are chemically synthesized and a GalNAc-siRNA conjugate selectively enters the hepatocyte via receptor-mediated endocytosis using the asialoglycoprotein receptor (similar as for ASO). After this, the double-stranded siRNA is released from the endosome and the two RNA stands dissociate into the sense and antisense strand. The antisense strand forms a highly stable complex with the RNA-induced silencing complex (RISC) which induces the cleavage of the target mRNA, degradation by exonucleases, and reduced synthesis of the protein of interest. The complex of siRNA with RISC is highly stable which results in a long-term cleavage of the targeted transcripts with a suppression of the protein production lasting more than 6 months (Landmesser et al. 2020).
Currently, two drugs under Phase I and II trials are using the siRNA principle: the first one is AMG890 (also called olpasiran) with a phase 1 (NCT03626662) and a phase 2 trial (NCT04270760). Obviously the phase 1 trial showed an Lp(a) reduction of more than 90% that persisted for 3–6 months (Abstract by Koren MJ et al.; Circulation 2020;142:A13951). The second one is SLN360: a phase 1 trial with 88 patients is currently under way and is expected to be completed in November 2022.
8 Other Lipid-Lowering Drugs and Therapies with Possible Influence on Lp(a) Concentrations and Clinical Outcomes
Before reviewing the various other approaches of lowering Lp(a) (Fig. 4), there are two issues which should be stressed when considering the results from the trials which investigated the Lp(a) lowering effects of various interventions:
-
Most previous therapeutic options were not developed and were not aiming for a specific Lp(a)-lowering but reached out for other lipoproteins and the Lp(a)-lowering is a concomitant observation. Therefore, it is hard to disentangle whether an effect observed in an intervention group is an effect of Lp(a)-lowering or an effect of the influence on other lipoproteins or risk factors which were the primary target of that intervention.
-
In many cases, studies were not designed to reach out for patients with high or solely high Lp(a) concentrations but targeted other lipoproteins such as high LDL-C or low HDL-C. Therefore in many cases the median Lp(a) levels were often as low as the median in general populations and these studies do not necessarily reflect the study population one would reach out to study the therapeutic effect in a high Lp(a) group.
8.1 Lipoprotein Apheresis
The data available on lipoprotein apheresis are coming mostly from Germany and are very impressive in terms of a 60–70% lowering of LDL-C and Lp(a) concentrations as well as the massive reduction of CHD outcomes. Of course, it is a sawtooth picture of the Lp(a) changes that can be observed with a decrease of Lp(a) from before to immediately after apheresis of up to more than 70%. After the apheresis session the Lp(a) concentrations increase again and the interval mean values were calculated to be roughly 35% lower compared to the values before the session (Julius et al. 2019).
In one of the first studies patients were included whose Lp(a) concentrations were above the 90th percentile. Lipoprotein apheresis not only reduced Lp(a) by 73%, but also coronary events by 86% (Jaeger et al. 2009). Interesting was the observation that in the subgroup of patients who had a very low Lp(a)-corrected LDL-C already before the onset of apheresis, the reduction in coronary heart disease events was about the same as in the other group in which LDL-C could additionally be lowered by apheresis. This means that in the first group, the reduction of coronary heart disease events is more likely due to the decrease in Lp(a) concentrations (Jaeger et al. 2009). Similar results for reducing Lp(a) concentrations and coronary heart disease events were also shown in other investigations (Leebmann et al. 2013; Roeseler et al. 2016). A significant decline of the mean annual cardiovascular event rate was observed from 0.58 ± 0.53 in the period 2 years before regular lipoprotein apheresis to 0.11 ± 0.15 thereafter (Roeseler et al. 2016). Similar observations have been made in an US-American study (Moriarty et al. 2019).
Lipoprotein apheresis studies have been criticized since they were hard to control due to the lack of randomization and blinding. A recent small study has tried to overcome this problem in 20 patients with refractory angina pectoris and mean Lp(a) concentrations at 110 mg/dL. These patients underwent 3 months of blinded weekly lipoprotein apheresis or sham, followed by crossover. Despite the relatively short-term therapy, this resulted in a significant decrease in angina pectoris frequency, an increase in myocardial perfusion reserve, an increase in exercise capacity, and a decrease in total carotid wall volume (Khan et al. 2017).
A specific approach to study the effect of Lp(a)-lowering came from a small study applying a specific Lp(a) apheresis with sheep polyclonal monospecific antibodies against human apo(a). This intervention decreased Lp(a) on average by 73% without significant changes in true LDL-C and other risk factors. The mean percent diameter stenosis of the coronary arteries after 18 months decreased by 5% in the Lp(a) intervention group and increased by 5% in the control group that received only statins (Safarova et al. 2013).
8.2 PCSK9 Inhibitors
PCSK9 inhibitors primarily target LDL-C and lower this atherogenic lipoprotein by roughly 60%. However, Lp(a) concentrations are also lowered by 25–30% (Table 1). There are two therapeutic principles: (1) a monoclonal antibody against PCSK9 given every 2 weeks or monthly as used in evolocumab and alirocumab or (2) a small interfering RNA (siRNA) that targets the mRNA of PCSK9 as used in inclisiran which will be given twice yearly.
In a post-hoc analysis of the FOURIER Trial including 25,096 patients with established atherosclerotic CVD, the PCSK9 inhibitor evolocumab reduced the risk of CVD outcomes by 23% in patients with a baseline Lp(a) > median of 37 nmol/L (≈15 mg/dL), and by 7% in those ≤median (p-value interaction = 0.07). The conclusion was the same when 120 nmol/L (≈50 mg/dL) instead of the median was used as grouping threshold. The study observed a significant relationship with a 15% lower risk (95% CI, 2–26%; p = 0.0199) per 25 nmol/L (≈10 mg/dL) reduction in Lp(a) after adjusting for the change in LDL-C (Table 1) (O’Donoghue et al. 2019). This would mean that even a relatively small lowering of Lp(a) should result in a clinical benefit.
Recently published data from pre-specified analysis of the ODYSSEY Outcomes trial showed that Lp(a)-lowering by alirocumab contributes to a reduction of major adverse cardiovascular events (MACE) independently of LDL-C-lowering in 18,924 patients with recent acute coronary syndrome and LDL-C ≥ 70 mg/dL despite intensive or maximum tolerated statin treatment (Bittner et al. 2020). Of course, the major contribution to the risk reductions comes from the reduction of Lp(a)-corrected LDL-C which is the primary target of the PCSK9 inhibitors. However, alirocumab additionally reduced Lp(a) by a median of 23%. The LDL-independent contribution to the proportion of MACE reduction attributable to changes in Lp(a) caused by alirocumab treatment increased from 4% to 11% to 25% for Lp(a) levels at the 25th, 50th, and 75th percentiles, respectively (Table 1) (Bittner et al. 2020).
Furthermore, pooled data from 10 controlled phase 3 ODYSSEY trials have been analyzed. The authors observed a 12% relative risk reduction in MACE per 25% reduction in Lp(a) which was no longer significant after adjustment for LDL-C changes. In subgroup analysis, the association between Lp(a) reduction and MACE remained significant in a fully adjusted model among participants with baseline Lp(a) ≥50 mg/dL (Table 1) (Ray et al. 2019).
The siRNA therapy inclisiran has been studied in 501 patients using different dosages and revealed median Lp(a) reductions from baseline to day 180 from −14% to −18% in the single-dose groups and −15% to −26% in the 2-dose groups. The Lp(a) reduction did not reach statistical significance in any of the dosing groups which was probably caused by the very wide interindividual variability in the Lp(a) reduction (Ray et al. 2018). It needs to be seen how pronounced the Lp(a) reductions will be in the phase III trials.
8.3 Statins
There is currently a major discussion whether statins increase Lp(a) concentrations or not. Due to the structure of Lp(a) containing an LDL particle, the LDL receptor was always an interesting candidate for Lp(a) removal. This idea was supported by the observations that Lp(a) levels are markedly elevated in patients with familial hypercholesterolemia (Utermann 1989; Utermann et al. 1989). It was therefore very surprising when first studies by Kostner et al. (1989) and later by O’Donoghue et al. (2014) observed an increase in Lp(a) concentrations after successful lowering of LDL cholesterol by statins. This is in line with the results of the JUPITER Study: patients under rosuvastatin treatment showed even a statistically significant positive shift in the overall Lp(a) distribution that was not observed in the placebo group (Khera et al. 2014; Kronenberg 2014b). Recently, Willeit and colleagues collated patient-level data from seven randomized, placebo-controlled, statin outcome trials including 29,069 patients with repeated Lp(a) measurements. 14,536 patients were randomly allocated statin treatment. The effect of statin therapy on Lp(a) concentrations was heterogeneous across studies: the pooled percentage change was −0.4% with three trials showing a mean increase (between 2 and 15%) and four trials showing a mean decrease (between −1 and −13%) in Lp(a) concentrations (Willeit et al. 2018). A further subject-level meta-analysis included 5,256 patients (1,371 on placebo and 3,885 on statin) from six randomized trials. All six trials used the same Lp(a) assay. The mean percent change from baseline ranged from 8.5% to 19.6% in the statin groups and −0.4% to −2.3% in the placebo groups. When various statins were compared, the mean percent change from baseline ranged from 11.6% to 20.4% in the pravastatin group and 18.7% to 24.2% in the atorvastatin group (Tsimikas et al. 2020b). A study in patients with dyslipidemia included 39 patients who first initiated statin treatment and a control group of 42 patients who were already on stable statin treatment for at least 4 months. Overall, Lp(a) concentrations did not increase significantly in both groups. However, when the analysis was stratified for the apo(a) isoforms, it was found that Lp(a) levels increased significantly from 66.4 to 97.4 mg/dL in patients with small apo(a) isoforms in the initiation group, but not in the control group and not in patients having only large apo(a) isoforms (Yahya et al. 2019). This interesting observation needs confirmation in larger studies.
The findings that statins might increase Lp(a) concentrations triggered discussions whether the benefit of statins by lowering LDL-C might be diminished or outweighed by the increase in Lp(a) concentrations especially in patients who have already increased Lp(a) concentrations before statin therapy is started. The JUPITER trial observed for the extended endpoint that patients with Lp(a) concentrations above the median reduced their risk for events by 28% (95% CI 3–48%) whereas those with baseline Lp(a) concentration below the median reduced the risk by 54% (95% CI 31–68%) (p-value for interaction = 0.10) (Khera et al. 2014). The meta-analysis by Willeit et al. found for those patients on statins a 47% higher risk when the Lp(a) levels were above compared to below 50 mg/dL (HR = 1.48, 95% CI 1.23–1.78). The risk was only 23% higher in the controls when Lp(a) was above compared to below 50 mg/dL (HR = 1.23, 95% CI 1.04–1.45%). This has been interpreted that the Lp(a)-associated risk becomes an even stronger predictor of residual risk when LDL-attributable risk is reduced with statin treatment (Willeit et al. 2018). Since this analysis did not use one of the groups as reference group for all comparisons (e.g., the controls with Lp(a) levels below 50 mg/dL), the risk estimates cannot simply be compared and might therefore be misinterpreted resulting in the idea that statins should be stopped in case of high Lp(a) concentrations. However, the probable risk benefit from lowering LDL-C by a statin outweighs the moderate increase in Lp(a) levels in most patients. This can be extrapolated from the study by Willeit et al. (see Fig. 2 of that publication): the cumulative risk was 21.7% in the placebo group (3,148 cases within 14,533 patients) which is higher compared to 17.9% in the statin-treated group (2,603 cases within 14,536 patients). This corresponds to a risk reduction to 0.83 by statins. Ethical considerations would not allow to test in a randomized controlled trial whether the avoidance of statins in patients with high Lp(a) concentrations would result in a benefit. In any case, the recent observations need careful attention and Lp(a) concentrations might better be monitored especially in patients who start statin therapy and having elevated Lp(a) concentrations since the observed increase under statin therapy in single patients can be sometimes very significant (Tsimikas et al. 2020c). In such patients a change to PCSK9 inhibitors might be an option to consider.
8.4 Drugs That Are Probably No Longer Followed for Lp(a)-Lowering Potential
There are some drugs that were not specifically designed to lower Lp(a) but showed an interesting Lp(a)-lowering effect. However, various reasons often related to the primary target and the observed side effects or efficacy revealed that they were stopped after clinical trials or they are no longer followed in extended clinical trials.
The CETP inhibitors anacetrapib (HPS3/TIMI55–REVEAL Collaborative Group et al. 2017; Thomas et al. 2017), evacetrapib (Nicholls et al. 2016), and TA-8995 (Ford et al. 2014) have in common that they not only increase HDL-C tremendously, but they also decrease Lp(a) between 25 and 40%. However, outcome studies using these drugs did not show a benefit or were even harmful in terms of CVD events. Both the thyroid analogue eprotirome (Ladenson et al. 2010) and the MTP inhibitor lomitapide (Samaha et al. 2008) lower Lp(a). Mipomersen, an antisense oligonucleotide targeting apoB mRNA lowers Lp(a) by roughly 30% but is restricted to very specific patient groups and countries due to its side effects (Raal et al. 2010).
In earlier times, niacin was thought to be a good candidate in treating Lp(a) elevation. However, in patients with optimally low levels of LDL-C, and despite favorable effects on HDL-C, triglycerides, and Lp(a), two clinical outcome trials failed to show any incremental clinical benefit on cardiovascular events with niacin when added to simvastatin (Boden et al. 2011; Landray et al. 2014). Following these trials, niacin is no longer available in most countries. However, besides other limitations these trials were not designed to examine the clinical effect in the patients with elevated Lp(a).
9 Therapeutic Lowering of Lipoprotein(a): How Much Is Enough?
On the eve of the introduction of specific Lp(a)-lowering therapies, the question has been raised, how much Lp(a) should be lowered to result in a clinical benefit. As recently discussed (Kronenberg 2019b), there are two approaches to get a rough estimate, which are a genetic estimation by using a Mendelian randomization approach or post-hoc analyses from clinical trials which target primarily LDL-C and as a “collateral yield” also lower Lp(a).
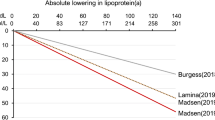
The Mendelian randomization approach uses genetic data from association studies on Lp(a) and LDL-C concentrations as well as association studies on CHD to estimate the required Lp(a)-lowering effect size to show the same association with CHD risk-lowering as a 38.67-mg/dL (1 mmol/L) therapeutic reduction in LDL-C. The latter is only used for benchmarking reasons. This intriguing idea used by Burgess and colleagues revealed that Lp(a) would have to be lowered by 101.5 mg/dL to show the same effect as lowering LDL-C by 38.67 mg/dL (Burgess et al. 2018). This number was probably markedly overestimated since the main study on which these results are based on had a median Lp(a) concentration twofold to threefold higher compared to other studies of the same ethnicity. We therefore repeated these calculations using the same approach and the same data basis except for the estimates of various SNPs on Lp(a) concentrations. For these we used our own data from almost 14.000 individuals in whom Lp(a) was measured within one laboratory and with median Lp(a) levels in the range of expectations for typical Caucasian populations (Mack et al. 2017). We calculated that an Lp(a)-lowering of roughly 65 mg/dL would be required instead of more than 100 mg/dL (Lamina and Kronenberg 2019). These data were mainly based on population-based data and it was not clear whether these hold also true for secondary prevention patients. Exactly this situation has very recently been tested in a Danish study (Madsen et al. 2020). They used population-based data from individuals with a history of CVD who were followed after their initial event. Since this equals a secondary prevention setting, they calculated that plasma Lp(a) should be lowered by 50 and 99 mg/dL for 5 years to achieve 20 and 40% MACE risk reduction in secondary prevention. Accordingly, for a 22% MACE reduction, a reduction of Lp(a) by 55 mg/dL would be required. Interestingly, when the authors used genetic data for short-term risk reduction, they found that an absolute Lp(a) reduction of 66 mg/dL would be needed to obtain a MACE reduction of 22%. This is exactly the same number as calculated by our group but using primary prevention studies (Lamina and Kronenberg 2019).
For all these studies it has to be added that the comparison with an LDL-lowering of 38.67 mg/dL was only used as a benchmark which results for LDL-C in a 22% lowering of MACE (Baigent et al. 2010). Of course, even smaller lowering might be beneficial as has been demonstrated for LDL-C.
The data from Mendelian randomization studies gave the impression that Lp(a) would have to be lowered by a higher extent than LDL-C to have the same clinical benefit. The ratio would be 2.6 to 1.0 according to Burgess and colleagues (2018) or 1.7 to 1.0 according to two other studies (Lamina and Kronenberg 2019; Madsen et al. 2020). This would mean that LDL-C would have markedly higher atherogenic properties than Lp(a) which is not necessarily supported by data. The post-hoc analyses of interventional studies with PCSK9 inhibitors provide some major hope that even a smaller Lp(a)-lowering might already show a clinical benefit (Table 1). As discussed above, the FOURIER study described a 15% lower risk per 25 nmol/L (≈10 mg/dL) reduction in Lp(a) after adjusting for the change in LDL-C (O’Donoghue et al. 2019). From the recently published ODYSSEY Outcomes trial it can be calculated that a lowering of Lp(a) by 42 mg/dL would be required to lower the MACE rate by 22% (Bittner et al. 2020) which brings the above-mentioned ratio of required Lp(a)/LDL-C-lowering closer to 1.0. Furthermore, the pooled data from 10 controlled phase 3 ODYSSEY trials also showed that the relative risk reduction per 25% reduction of Lp(a) adjusted for LDL-C changes was higher for the group of patients with Lp(a) ≥50 mg/dL with an HR = 0.60 (95% CI 0.39–0.92, p = 0.0201) (Ray et al. 2019).
Limitations for this Mendelian randomization approach come from various assumptions which have to be considered with cautions: first, it is unclear whether Lp(a) and LDL-C particles have a similar cumulative effect on CVD over time, or with other words, whether they have the same atherogenic potential? This is not necessarily the case since there is some evidence that even Lp(a) particles of different apo(a) isoforms have different atherogenic potential (Kronenberg et al. 1999; Saleheen et al. 2017). Second, besides an atherogenic nature of Lp(a) some data point also to a thrombogenic nature (Boffa and Koschinsky 2016; Romagnuolo et al. 2018). If this is also the case in vivo, the pathogenic mechanism of Lp(a) might even be stronger than for LDL-C and the lowering of Lp(a) would have an additional benefit on the thrombogenic axis.
All the available observations from post-hoc analysis of intervention studies and from Mendelian randomization studies are helpful for the planning of future trials. These trials will have to consider the uncertainties connected with these data such as more or less wide confidence intervals of the estimates. One of these uncertainties are introduced by the insufficiently standardized Lp(a) assays used in the various studies. This became obvious when looking at the data presented by Burgess et al. with the above-mentioned very high Lp(a) concentrations in their main cohort (Burgess et al. 2018). An overestimation of the Lp(a) concentration by an insufficiently standardized assay might result in a misclassification of patients to be at high risk and an enrollment of patients which would otherwise not be appropriate for the study. Therefore, major efforts should be invested in the screening phase for suitable study patients to avoid misclassification of patients by inappropriate Lp(a) assays (Kronenberg and Tsimikas 2019; Scharnagl et al. 2019).
10 Conclusions
The causal association between high Lp(a) concentrations and CVD is strongly supported by genetic studies. Post-hoc analyses following intervention studies with PCSK9-inhibitors that targeted primarily LDL-C but additionally lower also Lp(a) provide some evidence that an additional lowering of Lp(a) besides LDL-C is beneficial. However, these studies were not designed to target patients with high Lp(a) levels and are therefore limited. Mendelian randomization studies provide further strong support that lowering Lp(a) might be beneficial.
References
Arsenault BJ, Boekholdt SM, Dube MP, Rheaume E, Wareham NJ, Khaw KT, Sandhu MS, Tardif JC (2014) Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: a prospective Mendelian randomization study and replication in a case-control cohort. Circ Cardiovasc Genet 7:304–310
Austin MA, Sandholzer C, Selby JV, Newman B, Krauss RM, Utermann G (1992) Lipoprotein(a) in women twins: heritability and relationship to apolipoprotein(a) phenotypes. Am J Hum Genet 51:829–840
Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R (2010) Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 376:1670–1681
Bergmark C, Dewan A, Orsoni A, Merki E, Miller ER, Shin MJ, Binder CJ, Horkko S, Krauss RM, Chapman MJ, Witztum JL, Tsimikas S (2008) A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J Lipid Res 49:2230–2239
Bittner VA, Szarek M, Aylward PE, Bhatt DL, Diaz R, Edelberg JM, Fras Z, Goodman SG, Halvorsen S, Hanotin C, Harrington RA, Jukema JW, Loizeau V, Moriarty PM, Moryusef A, Pordy R, Roe MT, Sinnaeve P, Tsimikas S, Vogel R, White HD, Zahger D, Zeiher AM, Steg PG, Schwartz GG (2020) Effect of Alirocumab on lipoprotein(a) and cardiovascular risk after acute coronary syndrome. J Am Coll Cardiol 75:133–144
Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W (2011) Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 365:2255–2267
Boffa MB, Koschinsky ML (2016) Lipoprotein (a): truly a direct prothrombotic factor in cardiovascular disease? J Lipid Res 57:745–757
Boffa MB, Koschinsky ML (2019) Oxidized phospholipids as a unifying theory for lipoprotein(a) and cardiovascular disease. Nat Rev Cardiol 16:305–318
Boffa MB, Stranges S, Klar N, Moriarty PM, Watts GF, Koschinsky ML (2018) Lipoprotein(a) and secondary prevention of atherothrombotic events: a critical appraisal. J Clin Lipidol 12:1358–1366
Brandt EJ, Mani A, Spatz ES, Desai NR, Nasir K (2020) Lipoprotein(a) levels and association with myocardial infarction and stroke in a nationally representative cross-sectional US cohort. J Clin Lipidol 14:695–706.e694
Burgess S, Ference BA, Staley JR, Freitag DF, Mason AM, Nielsen SF, Willeit P, Young R, Surendran P, Karthikeyan S, Bolton TR, Peters JE, Kamstrup PR, Tybjaerg-Hansen A, Benn M, Langsted A, Schnohr P, Vedel-Krogh S, Kobylecki CJ, Ford I, Packard C, Trompet S, Jukema JW, Sattar N, Di Angelantonio E, Saleheen D, Howson JMM, Nordestgaard BG, Butterworth AS, Danesh J (2018) Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a Mendelian randomization analysis. JAMA Cardiol 3:619–627
Cain WJ, Millar JS, Himebauch AS, Tietge UJ, Maugeais C, Usher D, Rader DJ (2005) Lipoprotein [a] is cleared from the plasma primarily by the liver in a process mediated by apolipoprotein [a]. J Lipid Res 46:2681–2691
Cairns BJ, Coffey S, Travis RC, Prendergast B, Green J, Engert JC, Lathrop M, Thanassoulis G, Clarke R (2017) A replicated, genome-wide significant association of aortic stenosis with a genetic variant for lipoprotein(a): meta-analysis of published and novel data. Circulation 135:1181–1183
Capoulade R, Yeang C, Chan KL, Pibarot P, Tsimikas S (2018) Association of mild to moderate aortic valve stenosis progression with higher lipoprotein(a) and oxidized phospholipid levels: secondary analysis of a randomized clinical trial. JAMA Cardiol 3:1212–1217
Cegla J, Neely RDG, France M, Ferns G, Byrne CD, Halcox J, Datta D, Capps N, Shoulders C, Qureshi N, Rees A, Main L, Cramb R, Viljoen A, Payne J, Soran H (2019) HEART UK consensus statement on lipoprotein(a): a call to action. Atherosclerosis 291:62–70
Chen HY, Dufresne L, Burr H, Ambikkumar A, Yasui N, Luk K, Ranatunga DK, Whitmer RA, Lathrop M, Engert JC, Thanassoulis G (2018) Association of LPA variants with aortic stenosis: a large-scale study using diagnostic and procedural codes from electronic health records. JAMA Cardiol 3:18–23
Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, Parish S, Barlera S, Franzosi MG, Rust S, Bennett D, Silveira A, Malarstig A, Green FR, Lathrop M, Gigante B, Leander K, de Faire U, Seedorf U, Hamsten A, Collins R, Watkins H, Farrall M (2009) Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med 361:2518–2528
Coassin S, Kronenberg F (2020) Mechanistic insights into lipoprotein(a): from infamous to ‘inflammous’. Eur Heart J 41:2272–2274
Coassin S, Erhart G, Weissensteiner H, de Eca Guimaraes AM, Lamina C, Schönherr S, Forer L, Haun M, Losso JL, Köttgen A, Schmidt K, Utermann G, Peters A, Gieger C, Strauch K, Finkenstedt A, Bale R, Zoller H, Paulweber B, Eckardt KU, Hüttenhofer A, Huber LA, Kronenberg F (2017) A novel but frequent variant in LPA KIV-2 is associated with a pronounced Lp(a) and cardiovascular risk reduction. Eur Heart J 38:1823–1831
Coassin S, Schoenherr S, Weissensteiner H, Erhart G, Forer L, Losso JL, Lamina C, Haun M, Utermann G, Paulweber B, Specht G, Kronenberg F (2019) A comprehensive map of single base polymorphisms in the hypervariable LPA Kringle IV-2 copy number variation region. J Lipid Res 60:186–199
Cohen JC, Chiesa G, Hobbs HH (1993) Sequence polymorphisms in the apolipoprotein (a) gene. Evidence for dissociation between apolipoprotein(a) size and plasma lipoprotein(a) levels. J Clin Invest 91:1630–1636
Dahabreh IJ, Kent DM (2011) Index event bias as an explanation for the paradoxes of recurrence risk research. JAMA 305:822–823
Di Maio S, Grüneis R, Streiter G, Lamina C, Maglione M, Schoenherr S, Öfner D, Thorand B, Peters A, Eckardt K-U, Köttgen A, Kronenberg F, Coassin S (2020) Investigation of a nonsense mutation located in the complex KIV-2 copy number variation region of apolipoprotein(a) in 10,910 individuals. Genome Med 12:74
Dieplinger B, Lingenhel A, Baumgartner N, Poelz W, Dieplinger H, Haltmayer M, Kronenberg F, Mueller T (2007) Increased serum lipoprotein(a) concentrations and low molecular weight phenotypes of apolipoprotein(a) are associated with symptomatic peripheral arterial disease. Clin Chem 53:1298–1305
Erqou S, Kaptoge S, Perry PL, Di AE, Thompson A, White IR, Marcovina SM, Collins R, Thompson SG, Danesh J (2009) Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA 302:412–423
Erqou S, Thompson A, Di AE, Saleheen D, Kaptoge S, Marcovina S, Danesh J (2010) Apolipoprotein(a) isoforms and the risk of vascular disease: systematic review of 40 studies involving 58,000 participants. J Am Coll Cardiol 55:2160–2167
Ford J, Lawson M, Fowler D, Maruyama N, Mito S, Tomiyasu K, Kinoshita S, Suzuki C, Kawaguchi A, Round P, Boyce M, Warrington S, Weber W, van Deventer S, Kastelein JJ (2014) Tolerability, pharmacokinetics and pharmacodynamics of TA-8995, a selective cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Br J Clin Pharmacol 78:498–508
Frischmann ME, Kronenberg F, Trenkwalder E, Schaefer J, Schweer H, Dieplinger B, König P, Ikewaki K, Dieplinger H (2007) In vivo turnover study demonstrates diminished clearance of lipoprotein(a) in hemodialysis patients. Kidney Int 71:1036–1043
Grainger DJ, Kirschenlohr HL, Metcalfe JC, Weissberg PL, Wade DP, Lawn RM (1993) Proliferation of human smooth muscle cells promoted by lipoprotein(a). Science 260:1655–1658
Guan W, Cao J, Steffen BT, Post WS, Stein JH, Tattersall MC, Kaufman JD, McConnell JP, Hoefner DM, Warnick R, Tsai MY (2015) Race is a key variable in assigning lipoprotein(a) cutoff values for coronary heart disease risk assessment: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol 35:996–1001
Gurdasani D, Sjouke B, Tsimikas S, Hovingh GK, Luben RN, Wainwright NW, Pomilla C, Wareham NJ, Khaw KT, Boekholdt SM, Sandhu MS (2012) Lipoprotein(a) and risk of coronary, cerebrovascular, and peripheral artery disease: the EPIC-Norfolk prospective population study. Arterioscler Thromb Vasc Biol 32:3058–3065
Hindy G, Aragam KG, Ng K, Chaffin M, Lotta LA, Baras A, Regeneron Genetics C, Drake I, Orho-Melander M, Melander O, Kathiresan S, Khera AV (2020) Genome-wide polygenic score, clinical risk factors, and long-term trajectories of coronary artery disease. Arterioscler Thromb Vasc Biol 40:2738–2746
Hoekstra M, Chen HY, Rong J, Dufresne L, Yao J, Guo X, Tsai MY, Tsimikas S, Post WS, Vasan RS, Rotter JI, Larson MG, Thanassoulis G, Engert JC (2021) Genome-wide association study highlights APOH as a novel locus for lipoprotein(a) levels. Arterioscler Thromb Vasc Biol 41:ATVBAHA120314965
HPS3/TIMI55–REVEAL Collaborative Group, Bowman L, Hopewell JC, Chen F, Wallendszus K, Stevens W, Collins R, Wiviott SD, Cannon CP, Braunwald E, Sammons E, Landray MJ (2017) Effects of anacetrapib in patients with atherosclerotic vascular disease. N Engl J Med 377:1217–1227
Jaeger BR, Richter Y, Nagel D, Heigl F, Vogt A, Roeseler E, Parhofer K, Ramlow W, Koch M, Utermann G, Labarrere CA, Seidel D (2009) Longitudinal cohort study on the effectiveness of lipid apheresis treatment to reduce high lipoprotein(a) levels and prevent major adverse coronary events. Nat Clin Pract Cardiovasc Med 6:229–239
Julius U, Tselmin S, Schatz U, Fischer S, Birkenfeld AL, Bornstein SR (2019) Actual situation of lipoprotein apheresis in patients with elevated lipoprotein(a) levels. Atheroscler Suppl 40:1–7
Kamstrup PR, Nordestgaard BG (2016) Elevated lipoprotein(a) levels, LPA risk genotypes, and increased risk of heart failure in the general population. J Am Coll Cardiol Heart Fail 4:78–87
Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG (2009) Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA 301:2331–2339
Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG (2014) Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol 63:470–477
Kannel WB, Dawber TR, Friedman GD, GLENNON WE, McNamara PM (1964) Risk factors in coronary heart disease.An evaluation of several serum lipids as predictors of coronary heart disease. The Framingham study. Ann Intern Med 61:888–899
Khan TZ, Hsu LY, Arai AE, Rhodes S, Pottle A, Wage R, Banya W, Gatehouse PD, Giri S, Collins P, Pennell DJ, Barbir M (2017) Apheresis as novel treatment for refractory angina with raised lipoprotein(a): a randomized controlled cross-over trial. Eur Heart J 38:1561–1569
Khera AV, Everett BM, Caulfield MP, Hantash FM, Wohlgemuth J, Ridker PM, Mora S (2014) Lipoprotein(a) concentrations, Rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER trial (justification for the use of statins in prevention: an intervention trial evaluating Rosuvastatin). Circulation 129:635–642
Kiechl S, Willeit J, Mayr M, Viehweider B, Oberhollenzer F, Kronenberg F, Wiedermann CJ, Oberthaler S, Xu Q, Witztum JL, Tsimikas S (2007) Oxidized phospholipids, lipoprotein(a), lipoprotein-associated phospholipase A2 activity and 10-year cardiovascular outcomes: prospective results from the Bruneck study. Arterioscler Thromb Vasc Biol 27:1788–1795
Kinpara K, Okada H, Yoneyama A, Okubo M, Murase T (2011) Lipoprotein(a)-cholesterol: a significant component of serum cholesterol. Clin Chim Acta 412:1783–1787
Klarin D, Lynch J, Aragam K, Chaffin M, Assimes TL, Huang J, Lee KM, Shao Q, Huffman JE, Natarajan P, Arya S, Small A, Sun YV, Vujkovic M, Freiberg MS, Wang L, Chen J, Saleheen D, Lee JS, Miller DR, Reaven P, Alba PR, Patterson OV, DuVall SL, Boden WE, Beckman JA, Gaziano JM, Concato J, Rader DJ, Cho K, Chang KM, Wilson PWF, O'Donnell CJ, Kathiresan S, Tsao PS, Damrauer SM (2019) Genome-wide association study of peripheral artery disease in the million veteran program. Nat Med 25:1274–1279
Klezovitch O, Edelstein C, Scanu AM (2001) Stimulation of interleukin-8 production in human THP-1 macrophages by apolipoprotein (a): evidence for a critical involvement of elements in its C-terminal domain. J Biol Chem 276:46864–46869
Köchl S, Fresser F, Lobentanz E, Baier G, Utermann G (1997) Novel interaction of apolipoprotein(a) with b-2 glycoprotein I mediated by the kringle IV domain. Blood 90:1482–1489
Koschinsky ML, Marcovina SM (2004) Structure-function relationships in apolipoprotein(a): insights into lipoprotein(a) assembly and pathogenicity. Curr Opin Lipidol 15:167–174
Kostner GM, Gavish D, Leopold B, Bolzano K, Weintraub MS, Breslow JL (1989) HMG CoA reductase inhibitors lower LDL cholesterol without reducing Lp(a) levels. Circulation 80:1313–1319
Kraft HG, Menzel HJ, Hoppichler F, Vogel W, Utermann G (1989) Changes of genetic apolipoprotein phenotypes caused by liver transplantation. Implications for apolipoprotein synthesis. J Clin Invest 83:137–142
Kraft HG, Köchl S, Menzel HJ, Sandholzer C, Utermann G (1992) The apolipoprotein(a) gene: a transcribed hypervariable locus controlling plasma lipoprotein(a) concentration. Hum Genet 90:220–230
Kraft HG, Lingenhel A, Köchl S, Hoppichler F, Kronenberg F, Abe A, Mühlberger V, Schönitzer D, Utermann G (1996) Apolipoprotein(a) Kringle IV repeat number predicts risk for coronary heart disease. Arterioscler Thromb Vasc Biol 16:713–719
Kronenberg F (2014a) Lipoprotein(a) in various conditions: to keep a sense of proportions. Atherosclerosis 234:249–251
Kronenberg F (2014b) Lipoprotein(a): there's life in the old dog yet. Circulation 129:619–621
Kronenberg F (2016a) High-density lipoprotein cholesterol on a roller coaster: where will the ride end? Kidney Int 89:747–749
Kronenberg F (2016b) Human genetics and the causal role of lipoprotein(a) for various diseases. Cardiovasc Drugs Ther 30:87–100
Kronenberg F (2019a) Prediction of cardiovascular risk by Lp(a) concentrations or genetic variants within the LPA gene region. Clin Res Cardiol Suppl 14:5–12
Kronenberg F (2019b) Therapeutic lowering of lipoprotein(a): how much is enough? Atherosclerosis 288:163–165
Kronenberg F, Tsimikas S (2019) The challenges of measuring Lp(a): a fight against Hydra? Atherosclerosis 289:181–183
Kronenberg F, Utermann G (2013) Lipoprotein(a) – resurrected by genetics. J Intern Med 273:6–30
Kronenberg F, Trenkwalder E, Lingenhel A, Friedrich G, Lhotta K, Schober M, Moes N, König P, Utermann G, Dieplinger H (1997) Renovascular arteriovenous differences in Lp(a) plasma concentrations suggest removal of Lp(a) from the renal circulation. J Lipid Res 38:1755–1763
Kronenberg F, Kronenberg MF, Kiechl S, Trenkwalder E, Santer P, Oberhollenzer F, Egger G, Utermann G, Willeit J (1999) Role of lipoprotein(a) and apolipoprotein(a) phenotype in atherogenesis: prospective results from the Bruneck study. Circulation 100:1154–1160
Kronenberg F, Lingenhel A, Lhotta K, Rantner B, Kronenberg MF, König P, Thiery J, Koch M, Von Eckardstein A, Dieplinger H (2004) Lipoprotein(a)- and low-density lipoprotein-derived cholesterol in nephrotic syndrome: impact on lipid-lowering therapy? Kidney Int 66:348–354
Lackner C, Boerwinkle E, Leffert CC, Rahmig T, Hobbs HH (1991) Molecular basis of apolipoprotein (a) isoform size heterogeneity as revealed by pulsed-field gel electrophoresis. J Clin Invest 87:2153–2161
Lackner C, Cohen JC, Hobbs HH (1993) Molecular definition of the extreme size polymorphism in apolipoprotein(a). Hum Mol Genet 2:933–940
Ladenson PW, Kristensen JD, Ridgway EC, Olsson AG, Carlsson B, Klein I, Baxter JD, Angelin B (2010) Use of the thyroid hormone analogue eprotirome in statin-treated dyslipidemia. N Engl J Med 362:906–916
Lamina C, Kronenberg F (2019) Estimation of the required lipoprotein(a)-lowering therapeutic effect size for reduction in coronary heart disease outcomes: a Mendelian randomization analysis. JAMA Cardiol 4:575–579
Lamon-Fava S, Jimenez D, Christian JC, Fabsitz RR, Reed T, Carmelli D, Castelli WP, Ordovas JM, Wilson PWF, Schaefer EJ (1991) The NHLBI twin study: heritability of apolipoprotein A-I and B, and low density lipoprotein subclasses and concordance for lipoprotein(a). Atherosclerosis 91:97–106
Landmesser U, Poller W, Tsimikas S, Most P, Paneni F, Luscher TF (2020) From traditional pharmacological towards nucleic acid-based therapies for cardiovascular diseases. Eur Heart J 41:3884–3899
Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, Tomson J, Wallendszus K, Craig M, Jiang L, Collins R, Armitage J (2014) Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med 371:203–212
Langsted A, Kamstrup PR, Nordestgaard BG (2014) Lipoprotein(a): fasting and nonfasting levels, inflammation, and cardiovascular risk. Atherosclerosis 234:95–101
Langsted A, Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG (2016) High lipoprotein(a) as a possible cause of clinical familial hypercholesterolaemia: a prospective cohort study. Lancet Diabetes Endocrinol 4:577–587
Langsted A, Nordestgaard BG, Kamstrup PR (2019a) Elevated lipoprotein(a) and risk of ischemic stroke. J Am Coll Cardiol 74:54–66
Langsted A, Kamstrup PR, Nordestgaard BG (2019b) High lipoprotein(a) and high risk of mortality. Eur Heart J 40:2760–2770
Laschkolnig A, Kollerits B, Lamina C, Meisinger C, Rantner B, Stadler M, Peters A, Koenig W, Stöckl A, Dähnhardt D, Böger CA, Krämer BK, Fraedrich G, Strauch K, Kronenberg F (2014) Lipoprotein(a) concentrations, apolipoprotein(a) phenotypes and peripheral arterial disease in three independent cohorts. Cardiovasc Res 103:28–36
Lee SR, Prasad A, Choi YS, Xing C, Clopton P, Witztum JL, Tsimikas S (2017) LPA gene, ethnicity, and cardiovascular events. Circulation 135:251–263
Leebmann J, Roseler E, Julius U, Heigl F, Spitthoever R, Heutling D, Breitenberger P, Maerz W, Lehmacher W, Heibges A, Klingel R (2013) Lipoprotein apheresis in patients with maximally tolerated lipid lowering therapy, Lp(a)-hyperlipoproteinemia and progressive cardiovascular disease: prospective observational multicenter study. Circulation 128:2567–2576
Leibundgut G, Scipione C, Yin H, Schneider M, Boffa MB, Green S, Yang X, Dennis E, Witztum JL, Koschinsky ML, Tsimikas S (2013) Determinants of binding of oxidized phospholipids on apolipoprotein (a) and lipoprotein (a). J Lipid Res 54:2815–2830
Li J, Lange LA, Sabourin J, Duan Q, Valdar W, Willis MS, Li Y, Wilson JG, Lange EM (2015) Genome- and exome-wide association study of serum lipoprotein (a) in the Jackson heart study. J Hum Genet 60:755–761
Lim ET, Wurtz P, Havulinna AS, Palta P, Tukiainen T, Rehnstrom K, Esko T, Magi R, Inouye M, Lappalainen T, Chan Y, Salem RM, Lek M, Flannick J, Sim X, Manning A, Ladenvall C, Bumpstead S, Hamalainen E, Aalto K, Maksimow M, Salmi M, Blankenberg S, Ardissino D, Shah S, Horne B, McPherson R, Hovingh GK, Reilly MP, Watkins H, Goel A, Farrall M, Girelli D, Reiner AP, Stitziel NO, Kathiresan S, Gabriel S, Barrett JC, Lehtimaki T, Laakso M, Groop L, Kaprio J, Perola M, McCarthy MI, Boehnke M, Altshuler DM, Lindgren CM, Hirschhorn JN, Metspalu A, Freimer NB, Zeller T, Jalkanen S, Koskinen S, Raitakari O, Durbin R, MacArthur DG, Salomaa V, Ripatti S, Daly MJ, Palotie A (2014) Distribution and medical impact of loss-of-function variants in the finnish founder population. PLoS Genet 10:e1004494
Mach F, Baigent C, Catapano AL, Koskina KC, Casula M, Badimon L, Chapman MJ, De Backer GG, Delgado V, Ference BA, Graham IM, Halliday A, Landmesser U, Mihaylova B, Pedersen TR, Riccardi G, Richter DJ, Sabatine MS, Taskinen MR, Tokgozoglu L, Wiklund O, Windecker S, Aboyans V, Collet JP, Dean V, Fitzsimons D, Gale CP, Grobbee D, Halvorsen S, Hindricks G, Iung B, Jüni P, Katus HA, Leclercq C, Lettino M, Lewis BS, Merkely B, Mueller C, Petersen S, Petronio AS, Roffi M, Shlyakhto E, Simpson IA, Sousa-Uva M, Touyz RM, Nibouche D, Zelveian PH, Siostrzonek P, Najafov R, van de Borne P, Pojskic B, Postadzhiyan A, Kypris L, Špinar J, Larsen ML, Eldin HS, Viigimaa M, Strandberg TE, Ferrières J, Agladze R, Laufs U, Rallidis L, Bajnok L, Gudjónsson T, Maher V, Henkin Y, Gulizia MM, Mussagaliyeva A, Bajraktari G, Kerimkulova A, Latkovskis G, Hamoui O, Slapikas R, Visser L, Dingli P, Ivanov V, Boskovic A, Nazzi M, Visseren F, Mitevska I, Retterstøl K, Jankowski P, Fontes-Carvalho R, Gaita D, Ezhov M, Foscoli M, Giga V, Pella D, Fras Z, Perez de Isla L, Hagström E, Lehmann R, Abid L, Ozdogan O, Mitchenko O, Patel RS (2019) ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Atherosclerosis 290:140–205
Mack S, Coassin S, Rueedi R, Yousri NA, Seppala I, Gieger C, Schoenherr S, Forer L, Erhart G, Marques-Vidal P, Ried JS, Waeber G, Bergmann S, Daehnhardt D, Stoeckl A, Raitakari OT, Khahonen M, Peters A, Meitinger T, Strauch K, Kedenko L, Paulweber B, Lehtimaki T, Hunt SC, Vollenweider P, Lamina C, Kronenberg F, Grp K-S (2017) A genome-wide association meta-analysis on lipoprotein (a) concentrations adjusted for apolipoprotein (a) isoforms. J Lipid Res 58:1834–1844
Madsen CM, Kamstrup PR, Langsted A, Varbo A, Nordestgaard BG (2020) Lp(a) (lipoprotein[a])-lowering by 50 mg/dL (105 nmol/L) may be needed to reduce cardiovascular disease 20% in secondary prevention: a population-based study. Arterioscler Thromb Vasc Biol 40:255–266
McCormick SPA, Schneider WJ (2019) Lipoprotein(a) catabolism: a case of multiple receptors. Pathology 51:155–164
McLean JW, Tomlinson JE, Kuang W-J, Eaton DL, Chen EY, Fless GM, Scanu AM, Lawn RM (1987) cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature 330:132–137
Miltiadous G, Saougos V, Cariolou M, Elisaf MS (2006) Plasma lipoprotein(a) levels and LDL-cholesterol lowering response to statin therapy in patients with heterozygous familial hypercholesterolemia. Ann Clin Lab Sci 36:353–355
Mooser V, Marcovina SM, White AL, Hobbs HH (1996) Kringle-containing fragments of apolipoprotein(a) circulate in human plasma and are excreted into the urine. J Clin Invest 98:2414–2424
Morgan BM, Brown AN, Deo N, Harrop TWR, Taiaroa G, Mace PD, Wilbanks SM, Merriman TR, Williams MJA, McCormick SPA (2020) Nonsynonymous SNPs in LPA homologous to plasminogen deficiency mutants represent novel null apo(a) alleles. J Lipid Res 61:432–444
Moriarty PM, Gray JV, Gorby LK (2019) Lipoprotein apheresis for lipoprotein(a) and cardiovascular disease. J Clin Lipidol 13:894–900
Nicholls SJ, Ruotolo G, Brewer HB, Wang MD, Liu L, Willey MB, Deeg MA, Krueger KA, Nissen SE (2016) Evacetrapib alone or in combination with statins lowers lipoprotein(a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J Clin Lipidol 10:519–527
Nordestgaard BG, Langsted A (2016) Lipoprotein (a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res 57:1953–1975
Nordestgaard BG, Chapman MJ, Ray K, Boren J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Reiner Z, Taskinen MR, Tokgozoglu L, Tybjaerg-Hansen A (2010) Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J 31:2844–2853
Nowak-Gottl U, Junker R, Hartmeier M, Koch HG, Munchow N, Assmann G, Von Eckardstein A (1999) Increased lipoprotein(a) is an important risk factor for venous thromboembolism in childhood. Circulation 100:743–748
O’Donoghue ML, Fazio S, Giugliano RP, Stroes ESG, Kanevsky E, Gouni-Berthold I, Im K, Lira PA, Wasserman SM, Ceska R, Ezhov MV, Jukema JW, Jensen HK, Tokgozoglu SL, Mach F, Huber K, Sever PS, Keech AC, Pedersen TR, Sabatine MS (2019) Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation 139:1483–1492
Ober C, Nord AS, Thompson EE, Pan L, Tan Z, Cusanovich D, Sun Y, Nicolae R, Edelstein C, Schneider DH, Billstrand C, Pfaffinger D, Phillips N, Anderson RL, Philips B, Rajagopalan R, Hatsukami TS, Rieder MJ, Heagerty PJ, Nickerson DA, Abney M, Marcovina S, Jarvik GP, Scanu AM, Nicolae DL (2009) Genome-wide association study of plasma lipoprotein(a) levels identifies multiple genes on chromosome 6q. J Lipid Res 50:798–806
O'Donoghue ML, Morrow DA, Tsimikas S, Sloan S, Ren AF, Hoffman EB, Desai NR, Solomon SD, Domanski M, Arai K, Chiuve SE, Cannon CP, Sacks FM, Sabatine MS (2014) Lipoprotein(a) for risk assessment in patients with established coronary artery disease. J Am Coll Cardiol 63:520–527
Pare G, Caku A, McQueen M, Anand SS, Enas E, Clarke R, Boffa MB, Koschinsky M, Wang X, Yusuf S (2019) Lipoprotein(a) levels and the risk of myocardial infarction among 7 ethnic groups. Circulation 139:1472–1482
Parson W, Kraft HG, Niederstatter H, Lingenhel AW, Kochl S, Fresser F, Utermann G (2004) A common nonsense mutation in the repetitive Kringle IV-2 domain of human apolipoprotein(a) results in a truncated protein and low plasma Lp(a). Hum Mutat 24:474–480
Patel AP, Wang M, Pirruccello JP, Ellinor PT, Ng K, Kathiresan S, Khera AV (2021) Lp(a) (lipoprotein[a]) concentrations and incident atherosclerotic cardiovascular disease: new insights from a large national biobank. Arterioscler Thromb Vasc Biol 41:465–474
Perombelon YFN, Soutar AK, Knight BL (1994) Variation in lipoprotein(a) concentration associated with different apolipoprotein(a) alleles. J Clin Invest 93:1481–1492
Perrot N, Theriault S, Dina C, Chen HY, Boekholdt SM, Rigade S, Despres AA, Poulin A, Capoulade R, Le TT, Messika-Zeitoun D, Trottier M, Tessier M, Guimond J, Nadeau M, Engert JC, Khaw KT, Wareham NJ, Dweck MR, Mathieu P, Pibarot P, Schott JJ, Thanassoulis G, Clavel MA, Bosse Y, Arsenault BJ (2019) Genetic variation in LPA, calcific aortic valve stenosis in patients undergoing cardiac surgery, and familial risk of aortic valve microcalcification. JAMA Cardiol 4:620–627
Poon M, Zhang XX, Dunsky KG, Taubman MB, Harpel PC (1997) Apolipoprotein(a) induces monocyte chemotactic activity in human vascular endothelial cells. Circulation 96:2514–2519
Raal FJ, Santos RD, Blom DJ, Marais AD, Charng MJ, Cromwell WC, Lachmann RH, Gaudet D, Tan JL, Chasan-Taber S, Tribble DL, Flaim JD, Crooke ST (2010) Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet 375:998–1006
Rader DJ, Cain W, Ikewaki K, Talley G, Zech LA, Usher D, Brewer HB (1994) The inverse association of plasma lipoprotein(a) concentrations with apolipoprotein(a) isoform size is not due to differences in Lp(a) catabolism but to differences in production rate. J Clin Invest 93:2758–2763
Ray KK, Stoekenbroek RM, Kallend D, Leiter LA, Landmesser U, Scott Wright R, Wijngaard P, Kastelein JJP (2018) Effect of an siRNA therapeutic targeting PCSK9 on atherogenic lipoproteins: prespecified secondary end points in Orion 1. Circulation 138:1304–1316
Ray KK, Vallejo-Vaz AJ, Ginsberg HN, Davidson MH, Louie MJ, Bujas-Bobanovic M, Minini P, Eckel RH, Cannon CP (2019) Lipoprotein(a) reductions from PCSK9 inhibition and major adverse cardiovascular events: pooled analysis of alirocumab phase 3 trials. Atherosclerosis 288:194–202
Ripatti S, Tikkanen E, Orho-Melander M, Havulinna AS, Silander K, Sharma A, Guiducci C, Perola M, Jula A, Sinisalo J, Lokki ML, Nieminen MS, Melander O, Salomaa V, Peltonen L, Kathiresan S (2010) A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet 376:1393–1400
Roeseler E, Julius U, Heigl F, Spitthoever R, Heutling D, Breitenberger P, Leebmann J, Lehmacher W, Kamstrup PR, Nordestgaard BG, Maerz W, Noureen A, Schmidt K, Kronenberg F, Heibges A, Klingel R (2016) Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of follow-up and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol 36:2019–2027
Romagnuolo R, Scipione CA, Bazzi ZA, Boffa MB, Koschinsky ML (2018) Inhibition of pericellular plasminogen activation by apolipoprotein(a): roles of urokinase plasminogen activator receptor and integrins aMb2 and aVb3. Atherosclerosis 275:11–21
Safarova MS, Ezhov MV, Afanasieva OI, Matchin YG, Atanesyan RV, Adamova IY, Utkina EA, Konovalov GA, Pokrovsky SN (2013) Effect of specific lipoprotein(a) apheresis on coronary atherosclerosis regression assessed by quantitative coronary angiography. Atheroscler Suppl 14:93–99
Saleheen D, Haycock PC, Zhao W, Rasheed A, Taleb A, Imran A, Abbas S, Majeed F, Akhtar S, Qamar N, Zaman KS, Yaqoob Z, Saghir T, Rizvi SNH, Memon A, Mallick NH, Ishaq M, Rasheed SZ, Memon FU, Mahmood K, Ahmed N, Frossard P, Tsimikas S, Witztum JL, Marcovina S, Sandhu M, Rader DJ, Danesh J (2017) Apolipoprotein(a) isoform size, lipoprotein(a) concentration, and coronary artery disease: a Mendelian randomisation analysis. Lancet Diabetes Endocrinol 5:524–533
Samaha FF, McKenney J, Bloedon LT, Sasiela WJ, Rader DJ (2008) Inhibition of microsomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. Nat Clin Pract Cardiovasc Med 5:497–505
Sandholzer C, Saha N, Kark JD, Rees A, Jaross W, Dieplinger H, Hoppichler F, Boerwinkle E, Utermann G (1992) Apo(a) isoforms predict risk for coronary heart disease: a study in six populations. Arterioscler Thromb 12:1214–1226
Scanu AM, Hinman J (2002) Issues concerning the monitoring of statin therapy in hypercholesterolemic subjects with high plasma lipoprotein(a) levels. Lipids 37:439–444
Scharnagl H, Stojakovic T, Dieplinger B, Dieplinger H, Erhart G, Kostner GM, Hermann M, März W, Grammer TB (2019) Comparison of lipoprotein(a) serum concentrations measured by six commercially available immunoassays. Atherosclerosis 289:206–213
Smits LJ, van Kuijk SM, Leffers P, Peeters LL, Prins MH, Sep SJ (2013) Index event bias-a numerical example. J Clin Epidemiol 66:192–196
Stiekema LCA, Stroes ESG, Verweij SL, Kassahun H, Chen L, Wasserman SM, Sabatine MS, Mani V, Fayad ZA (2019) Persistent arterial wall inflammation in patients with elevated lipoprotein(a) despite strong low-density lipoprotein cholesterol reduction by proprotein convertase subtilisin/kexin type 9 antibody treatment. Eur Heart J 40:2775–2781
Stiekema LCA, Prange KHM, Hoogeveen RM, Verweij SL, Kroon J, Schnitzler JG, Dzobo KE, Cupido AJ, Tsimikas S, Stroes ESG, de Winther MPJ, Bahjat M (2020) Potent lipoprotein(a) lowering following apolipoprotein(a) antisense treatment reduces the pro-inflammatory activation of circulating monocytes in patients with elevated lipoprotein(a). Eur Heart J 41:2262–2271
Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, Kerr KF, Pechlivanis S, Budoff MJ, Harris TB, Malhotra R, O’Brien KD, Kamstrup PR, Nordestgaard BG, Tybjaerg-Hansen A, Allison MA, Aspelund T, Criqui MH, Heckbert SR, Hwang SJ, Liu Y, Sjogren M, van der Pals J, Kalsch H, Muhleisen TW, Nothen MM, Cupples LA, Caslake M, Di AE, Danesh J, Rotter JI, Sigurdsson S, Wong Q, Erbel R, Kathiresan S, Melander O, Gudnason V, O’Donnell CJ, Post WS (2013) Genetic associations with valvular calcification and aortic stenosis. N Engl J Med 368:503–512
Thomas T, Zhou H, Karmally W, Ramakrishnan R, Holleran S, Liu Y, Jumes P, Wagner JA, Hubbard B, Previs SF, Roddy T, Johnson-Levonas AO, Gutstein DE, Marcovina SM, Rader DJ, Ginsberg HN, Millar JS, Reyes-Soffer G (2017) CETP (cholesteryl ester transfer protein) inhibition with anacetrapib decreases production of lipoprotein(a) in mildly hypercholesterolemic subjects. Arterioscler Thromb Vasc Biol 37:1770–1775
Tomlinson JE, McLean JW, Lawn RM (1989) Rhesus monkey apolipoprotein(a). Sequence, evolution, and sites of synthesis. J Biol Chem 264:5957–5965
Tsimikas S, Brilakis ES, Miller ER, McConnell JP, Lennon RJ, Kornman KS, Witztum JL, Berger PB (2005) Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med 353:46–57
Tsimikas S, Mallat Z, Talmud PJ, Kastelein JJ, Wareham NJ, Sandhu MS, Miller ER, Benessiano J, Tedgui A, Witztum JL, Khaw KT, Boekholdt SM (2010) Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol 56:946–955
Tsimikas S, Viney NJ, Hughes SG, Singleton W, Graham MJ, Baker BF, Burkey JL, Yang Q, Marcovina SM, Geary RS, Crooke RM, Witztum JL (2015) Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. Lancet 386:1472–1483
Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, Tardif JC, Baum SJ, Steinhagen-Thiessen E, Shapiro MD, Stroes ES, Moriarty PM, Nordestgaard BG, Xia S, Guerriero J, Viney NJ, O'Dea L, Witztum JL (2020a) Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med 382:244–255
Tsimikas S, Gordts PLSM, Nora C, Yeang C, Witztum JL (2020b) Statin therapy increases lipoprotein(a) levels. Eur Heart J 41:2275–2284
Tsimikas S, Gordts PLSM, Nora C, Yeang C, Witztum JL (2020c) Statins and increases in Lp(a): an inconvenient truth that needs attention. Eur Heart J 41:192–193
Utermann G (1989) The mysteries of lipoprotein(a). Science 246:904–910
Utermann G, Menzel HJ, Kraft HG, Duba HC, Kemmler HG, Seitz C (1987) Lp(a) glycoprotein phenotypes: inheritance and relation to Lp(a)-lipoprotein concentrations in plasma. J Clin Invest 80:458–465
Utermann G, Hoppichler F, Dieplinger H, Seed M, Thompson G, Boerwinkle E (1989) Defects in the low density lipoprotein receptor gene affect lipoprotein (a) levels: multiplicative interaction of two gene loci associated with premature atherosclerosis. Proc Natl Acad Sci U S A 86:4171–4174
van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, Ravandi A, Nederveen AJ, Verberne HJ, Scipione C, Nieuwdorp M, Joosten LA, Netea MG, Koschinsky ML, Witztum JL, Tsimikas S, Riksen NP, Stroes ES (2016) Oxidized phospholipids on lipoprotein(a) elicit Arterial Wall inflammation and an inflammatory monocyte response in humans. Circulation 134:611–624
Viney NJ, van Capelleveen JC, Geary RS, Xia S, Tami JA, Yu RZ, Marcovina SM, Hughes SG, Graham MJ, Crooke RM, Crooke ST, Witztum JL, Stroes ES, Tsimikas S (2016) Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 388:2239–2253
Virani SS, Brautbar A, Davis BC, Nambi V, Hoogeveen RC, Sharrett AR, Coresh J, Mosley TH, Morrisett JD, Catellier DJ, Folsom AR, Boerwinkle E, Ballantyne CM (2012) Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the atherosclerosis risk in communities (ARIC) study. Circulation 125:241–249
Vongpromek R, Bos S, Ten Kate GJ, Yahya R, Verhoeven AJ, De Feyter PJ, Kronenberg F, van Lennep JE, Sijbrands EJ, Mulder MT (2015) Lipoprotein(a) levels are associated with aortic valve calcification in asymptomatic patients with familial hypercholesterolaemia. J Intern Med 278:166–173
Vuorio A, Watts GF, Kovanen PT (2019) Lipoprotein(a) as a risk factor for calcific aortic valvulopathy in heterozygous familial hypercholesterolemia. Atherosclerosis 281:25–30
Willeit P, Ridker PM, Nestel PJ, Simes J, Tonkin AM, Pedersen TR, Schwartz GG, Olsson AG, Colhoun HM, Kronenberg F, Drechsler C, Wanner C, Mora S, Lesogor A, Tsimikas S (2018) Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: individual patient-data meta-analysis of statin outcome trials. Lancet 392:1311–1320
Yahya R, Berk K, Verhoeven A, Bos S, van der Zee L, Touw J, Erhart G, Kronenberg F, Timman R, Sijbrands E, van Roeters LJ, Mulder M (2019) Statin treatment increases lipoprotein(a) levels in subjects with low molecular weight apolipoprotein(a) phenotype. Atherosclerosis 289:201–205
Yeang C, Witztum JL, Tsimikas S (2021) Novel method for quantification of lipoprotein(a)-cholesterol: implications for improving accuracy of LDL-C measurements. J Lipid Res 62:100053. https://doi.org/10.1016/j.jlr.2021.100053
Zhang J, Du R, Peng K, Wu X, Hu C, Li M, Xu Y, Xu M, Wang S, Bi Y, Wang W, Lu J, Chen Y (2019) Serum lipoprotein (a) is associated with increased risk of stroke in Chinese adults: a prospective study. Atherosclerosis 289:8–13
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Kronenberg, F. (2021). Lipoprotein(a). In: von Eckardstein, A., Binder, C.J. (eds) Prevention and Treatment of Atherosclerosis . Handbook of Experimental Pharmacology, vol 270. Springer, Cham. https://doi.org/10.1007/164_2021_504
Download citation
DOI: https://doi.org/10.1007/164_2021_504
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-86075-2
Online ISBN: 978-3-030-86076-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)