
Abstract
Ilumatobacter coccineum YM16-304T (=NBRC 103263T) is a novel marine actinobacterium isolated from a sand sample collected at a beach in Shimane Prefecture, Japan. Strain YM16-304T is the type strain of the species. Phylogenetically, strain YM16-304T is close to Ilumatobacter nonamiense YM16-303T (=NBRC 109120T), Ilumatobacter fluminis YM22-133T and some uncultured bacteria including putative marine sponge symbionts. Whole genome sequence of these species has not been reported. Here we report the complete genome sequence of strain YM16-304T. The 4,830,181 bp chromosome was predicted to encode a total of 4,291 protein-coding genes.
Similar content being viewed by others


Introduction
Strain YM16-304T (=NBRC 103263T) is the type strain of Ilumatobacter coccineum Matsumoto et al. 2013 [1]. I. coccineum YM16-304T and Ilumatobacter nonamiense YM16-303T (=NBRC 109120T) were isolated from a sand sample collected at Nonami Beach in Shimane Prefecture in Japan, and represent the second and the third species of the genus Ilumatobacter [2]. Phylogenetic analysis showed that genus Ilumatobacter branches near the presumed root of the class Actinobacteria (Figure 1), and thus may represent a new taxon outside the known family Acidimicrobiaceae, although the family accommodating this genus has not been decided yet [1,2]. Iamia majanohamensis is also located outside the family Acidimicrobiaceae, and is the sole genus and species in family Iamiaceae [4]. Among the organisms for which whole genome sequences have been reported, the most closely related to YM16-304T is Acidimicrobium ferrooxidans DSM 10331 [5], which is phylogenetically distant from I. coccineum with 16S rRNA gene sequence similarity of 86%. No complete or draft genome information is currently available for the genera Ilumatobacter and Iamia. The taxon contains a number of uncultured bacteria including putative marine sponge symbionts, and the complete genome sequence of strain YM16-304T would provide a basis of technological developments for the isolation and better understanding of related uncultured actinobacteria.

Phylogenetic tree highlighting the position of I. coccineum strain YM16-304T relative to other representative type strains. The tree was constructed by the neighbor-joining method [3] based on a 1,326 bp alignment of 16S rRNA gene sequences. Corresponding INSDC accession numbers are shown in parentheses. Numbers at nodes indicate support values obtained from 1,000 bootstrap replications. Species for which complete genome sequences are available are labeled with two asterisks.
Classification and features
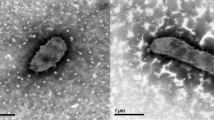
Strain YM16-304T is a mesophilic, neutrophilic, aerobic bacterium with features as summarized in Table 1. Growth occurs at 12–36 °C and at pH 7–8. Cells are rods and non-motile [Figure 2]. Gram staining was positive. Electron microscope observation demonstrated no flagella and pili formation [1]. In agreement with this observation, the genome encodes no gene necessary for flagella, chemotaxis and pili.

Scanning electron micrograph of I. coccineum strain YM16-304T
Strain YM16-304T grows poorly even in artificial seawater medium supplemented with 0.5% peptone and 0.1% yeast extract under optimum growth conditions [1]. From the genome sequence, strain YM16-304T seems to possess either deficient or unusual pathways for the synthesis of some amino acids and other essential cellular components as outlined in the later section (Primary metabolism).
Genome sequencing information
Genome project history
I. coccineum YM16-304T was selected for sequencing because of its isolated phylogenetic position and characteristics which distinguish this strain from other described actinobacterial species. Table 2 presents the project information and its association with MIGS version 2.0 compliance [15].
Growth conditions and DNA isolation
I. coccineum YM16-304T cells were grown in a 20 L volume at 27°C in DifcoTM Marine broth 2216 (Beckton Dickinson). DNA was isolated from 0.5 g of wet cells by manual extraction after lysis with lysozyme and SDS.
Genome sequencing and assembly
The genome of I. coccineum YM16-304T was sequenced using the conventional whole-genome shotgun sequencing method. Plasmid libraries with average insert sizes of 1.5 kb and 6.0 kb were generated in pTS1 (Nippon Gene) and pUC118 (TaKaRa) vectors, respectively, while a fosmid library with average insert size of 38 kb was constructed in pCC1FOS (EPICENTRE) as described previously [16]. A total of 26,592 clones (18,432, 5,376 and 2,784 clones from libraries with 1.5 kb, 6.0 kb and 38 kb inserts, respectively) were subjected to sequencing from both ends of the inserts on a ABI 3730xl DNA Analyzer (Applied Biosystems). Sequence reads were trimmed at a threshold of 20 in Phred score and assembled by using Phrap and CONSED assembly tools [12,13]. Gaps between contigs were closed by sequencing PCR products which bridge two neighboring contigs. Finally, each base of the genome was ensured to be sequenced from multiple clones either from both directions with Phrap quality score ≥ 70 or from one direction with Phrap quality score ≥40.
Genome annotation
The complete sequence of the chromosome was analyzed using Glimmer3 [14] for predicting protein-coding genes, tRNAscan-SE [17] and ARAGORN [18] for tRNA genes, and RNAmmer [19] for rRNA genes. The functions of predicted protein-coding genes were assigned manually, using the in-house genome annotation system OCSS (unpublished), in comparison with Uniprot [20], Interpro [21], HAMAP [22] and KEGG [23] databases.
Genome properties
The genome of I. coccineum YM16-304T consisted of a circular chromosome of 4,830,181 bp (Figure 3). The chromosome was predicted to contain 4,291 protein-coding genes, 46 tRNA genes, two copies of rRNA operons. Protein functions were manually assigned based on UniProt and InterPro searches, and specific functions were predicted for 1,824 genes (42.5% of the protein-coding genes). Among the remaining predicted proteins, 520 (12.1%) were assigned to proteins belonging to specific protein families, 1,535 (35.8%) were assigned to hypothetical proteins (showing sequence similarity to published proteins without known function), and 409 (9.5%) were assigned to hypothetical proteins (prediction only) (lacking sequence similarity to published proteins). Average G+C content was 67.29%. The properties and the statistics of the genome are summarized in Tables 3–4.

Circular representation of the I. coccineum YM16-304T chromosome From outside to the center: circles 1 and 2, predicted protein coding genes on the forward and reverse strands, respectively; circle 3, tRNA genes; circle 4, rRNA operons; circle 5, G+C content; circle 6, GC skew. Predicted protein coding genes are colored according to their assigned COG functional categories (see Table 4).
Primary metabolism
Strain YM16-304T lacks the dapE gene for succinyl-diaminopimelate desuccinylase (EC:3.5.1.18) in the biosynthesis pathway of lysine and diaminopimelic acids (DAPs). Instead, two candidate genes (YM304_26990 and YM304_19190) for LL-DAP aminotransferase (EC:2.6.1.83, dapL), that constitutes an alternative DAP-lysine biosynthesis pathway (DAP aminotransferase pathway [24,25]), were identified. The dapL gene is found in discrete lineages of Bacteria and Archaea, and is known to complement Escherichia coli dapD and dapE mutants, although purified proteins favor the reverse reaction rather than the synthesis of LL-DAP [25].
Among the genes of serine biosynthesis pathway, the serB gene for phosphoserine phosphatase (EC:3.1.3.3) was not identified by similarity searches. On the other hand, the thrH gene for phosphoserine / homoserine phosphotransferase [26] (EC:3.1.3.3, 2.7.1.39) was identified (YM304_28950). The possibility of using thrH gene product for serine biosynthesis instead of serB gene product was suggested.
Strain YM16-304Tseems to possess an alternative form of histidine biosynthesis pathway in which hisB gene for the synthesis of L-histidinol was replaced with the hisN gene (YM304_12240) as typically found in Corynebacterium glutamicum ATCC13032 [27] and other actinomycetes. However, the hisE gene for phosphoribosyl-ATP pyrophosphohydrolase (EC:3.6.1.31), which is responsible for the second step in histidine biosynthesis pathway, was not identified by similarity searches.
Metabolic reconstruction based on the annotation suggested that strain YM16-304T possesses the enzymes required for the biosynthesis of saturated fatty acids, unsaturated fatty acids, branched-chain fatty acids and carotenoids. The putative carotenoid biosynthesis pathway comprises crtE (YM304_37400), crtB (YM304_37420), crtI (YM304_37410) and crtLm (YM304_23780) gene homologs, which most probably synthesizes γ-carotene from isopentenyl pyrophosphate derived from non-mevalonate pathway [28–30]. Strain YM16-304T also possesses genes homologous to crtO (YM304_25370) and crtZ (YM304_38780), which were suggested to be involved in the synthesis of ketolated carotenoid such as canthaxanthin and astaxanthin [30]. Actual products of this pathway need to be experimentally verified.
The annotation also suggests that strain YM16-304T possesses the enzymes required for the biosynthesis of menaquinone (vitamin K), vitamin B6, nicotinate and nicotinamide, pantothenate and CoA, lipoic acid, protoheme, mycothiol and coenzyme F420, while biosynthetic pathways for folate, thiamine, riboflavin, biotin and adenosylcobalamin (coenzyme B12) are either missing or incomplete.
Secondary metabolism
The phylogenetic analysis based on 16S rRNA gene sequences showed that three species in the genus Ilumatobacter were closely related to some uncultured actinobacteria including marine sponge symbionts [31]. Marine sponges are noted as a rich source of biologically active secondary metabolites, true producers of such compound being suspected to be symbiotic bacteria [32–34]. However, only a small percentage of these symbiotic microorganisms are culturable [35,36], and genes involved in the synthesis of bioactive compounds such as polyketide synthases have often been isolated by metagenomic approaches [37,38].
The strain YM16-304T genome seemed to encode only a limited number of secondary metabolic enzymes, i.e., two type I polyketide synthases (PKS). The genome does not contain genes for type II and type III PKS nor a gene for nonribosomal peptide synthetase.
The type I PKS genes of the strain YM16-304T (YM304_13420, YM304_13410), together with the adjacent pfaD homolog (YM304_13430), most probably encode omega-3 polyunsaturated fatty acid (PUFA) synthase gene cluster. In some Gammaproteobacteria from marine sources such as Photobacterium profundum strain SS9, omega-3 polyunsaturated fatty acids such as eicosapentaenoic acid (20:5n-3; EPA) and docosahexaenoic acid (22:6n-3; DHA) are known to be synthesized by a PKS system consisting of pfaA, pfaB, pfaC and pfaD genes [39–41]. The domain organization of YM304_13420 was identical to that of the pfaA gene of P. profundum SS9. The N-terminal ketosynthase domain and the C-terminal dehydratase domains of YM304_13410 were similar to those of the pfaC gene of P. profundum, while the internal acyltransferase domain of YM304_13410 was moderately similar to that of the pfaB gene of P. profundum, representing a presumed chimeric form of PKS. As PfaB is the key enzyme determining the final product in EPA or DHA biosynthesis [42], the actual product of this PKS system may need to be clarified experimentally. Some PUFA-producing bacteria such as Moritella marina MP-1 [39,43] were reported to require an additional gene, pfaE, encoding a phosphopantheteinyl transferase. However, the pfaE gene was not identified in strain YM16-304T. Other classes of phosphopantheteinyl transferase (e.g. YM304_08850) may substitute the function of PfaE, similar to the case suggested in P. profundum SS9 [44].
Cell surface
Strain YM16-304 seemed to possess 13 ORFs containing LPXTG motif (InterPro ID: IPR001899), the presumed sorting signal of cell surface proteins in Gram-positive bacteria [45]. It was reported that several cell surface proteins containing LPXTG motif act as an adhesion factor known as microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) [46]. The genome of strain YM16-304 contained extracellular polysaccharide gene cluster (YM304_29910-YM304_30490), including gene cluster for the synthesis of sialic acids (YM304_30300-YM304_30320), which are also crucial for cell adhesion [47]. These extracellular components might serve for the bacterium to adhere to host tissues such as marine sponges.
Many marine bacteria use the Na+ cycle and require Na+ for their growth [48]. In these bacteria, Na+ is often used in the respiratory chain, ATP synthase, flagellar rotation and solute uptake instead of H+ [49]. Some bacteria can use both Na+ and H+ to expand the range of environments in which the bacteria can grow [50]. Strain YM16-304 was isolated from a sand sample collected at a beach and grows optimally in marine broth media, suggesting its marine origin. However, the gene products for the respiratory chain and ATP synthase were predicted to be of the H+-dependent type by similarity search. The Na+-dependent amino acid symporters were also not identified, nor was the H+-dependent symporters.
References
Matsumoto A, Kasai H, Matsuo Y, Shizuri Y, Ichikawa N, Fujita N, Omura S, Takahashi Y. Ilumatobacter nonamiense sp. nov. and Ilumatobacter coccineum sp. nov. isolated from seashore sand. Int J Syst Evol Microbiol 2013. PubMed http://dx.doi.org/10.1099/ijs.0.047316-0
Matsumoto A, Kasai H, Matsuo Y, Omura S, Shizuri Y, Takahashi Y. Ilumatobacter fluminis gen. nov., sp. nov., a novel actinobacterium isolated from the sediment of an estuary. J Gen Appl Microbiol 2009; 55:201–205. PubMed http://dx.doi.org/10.2323/jgam.55.201
Saitou N, Nei M. The neighbor-joining methods: a new method for reconstructing phylogenetic trees. Mol Biol Evol 1987; 4:406–425. PubMed
Kurahashi M, Fukunaga Y, Sakiyama Y, Harayama S, Yokota A. Iamia majanohamensis gen. nov., sp. nov., an actinobacterium isolated from sea cucumber Holothuria edulis, and proposal of Iamiaceae fam. nov. Int J Syst Evol Microbiol 2009; 59:869–873. PubMed http://dx.doi.org/10.1099/ijs.0.005611-0
Clum A, Nolan M, Lang E, Del Rio TG, Tice H, Copeland A, Cheng JF, Lucas S, Chen F, Bruce D, et al. Complete genome sequence of Acidimicrobium ferrooxidans type strain (ICPT). Stand Genomic Sci 2009; 1:38–45. PubMed http://dx.doi.org/10.4056/sigs.1463
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed http://dx.doi.org/10.1073/pnas.87.12.4576
Garrity GM, Holt JG. The Road Map to the Manual. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 119–169.
Stackebrandt E, Rainey FA, Ward-Rainey NL. Proposal for a new hierarchic classification system. Actinobacteria classis nov. Int J Syst Bacteriol 1997; 47:479–491. http://dx.doi.org/10.1099/00207713-47-2-479
Zhi XY, Li WJ, Stackebrandt E. An update of the structure and 16S rRNA gene sequence-based definition of higher ranks of the class Actinobacteria, with the proposal of two new suborders and four new families and emended descriptions of the existing higher taxa. Int J Syst Evol Microbiol 2009; 59:589–608. PubMed http://dx.doi.org/10.1099/ijs.0.65780-0
List of new names and new combinations previously effectively, but not validly, published. Validation List n. 129. Int J Syst Evol Microbiol 2009; 59:2129–2130. http://dx.doi.org/10.1099/ijs.0.017988-0
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25–29. PubMed http://dx.doi.org/10.1038/75556
Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 1998; 8:175–185. PubMed http://dx.doi.org/10.1101/gr.8.3.175
Ewing B, Green P. Basecalling of automated sequencer traces using phred. II. Error probabilities. Genome Res 1998; 8:175–185. PubMed http://dx.doi.org/10.1101/gr.8.3.175
Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007; 23:673–679. PubMed http://dx.doi.org/10.1093/bioinformatics/btm009
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed http://dx.doi.org/10.1038/nbt1360
Takarada H, Sekine M, Kosugi H, Matsuo Y, Fujisawa T, Omata S, Kishi E, Shimizu A, Tsukatani N, Tanikawa S, et al. Complete genome sequence of the soil actinomycete Kocuria rhizophila. J Bacteriol 2008; 190:4139–4146. PubMed http://dx.doi.org/10.1128/JB.01853-07
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997; 25:955–964. PubMed
Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res 2004; 32:11–16. PubMed http://dx.doi.org/10.1093/nar/gkh152
Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 2007; 35:3100–3108. PubMed http://dx.doi.org/10.1093/nar/gkm160
UniProt Consortium. The universal protein resource (UniProt) in 2010. Nucleic Acids Res 2010; 38:D142–D148. PubMed http://dx.doi.org/10.1093/nar/gkp846
Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Buillard V, Cerutti L, Copley R, et al. New developments in the InterPro database. Nucleic Acids Res 2007; 35:D224–D228. PubMed http://dx.doi.org/10.1093/nar/gkl841
Lima T, Auchincloss AH, Coudert E, Keller G, Michoud K, Rivoire C, Bulliard V, de Castro E, Lachaize C, Baratin D, et al. HAMAP: a database of completely sequenced microbial proteome sets and manually curated microbial protein families in UniProtKB/Swiss-Prot. Nucleic Acids Res 2009; 37:D471–D478. PubMed http://dx.doi.org/10.1093/nar/gkn661
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. KEGG for linking genomes to life and the environment. Nucleic Acids Res 2007; 36:D480–D484. PubMed http://dx.doi.org/10.1093/nar/gkm882
McCoy AJ, Adams NE, Hudson AO, Gilvarg C, Leustek T, Maurelli AT. L,L-diaminopimelate aminotransferase, a trans-kingdom enzyme shared by Chlamydia and plants for synthesis of diaminopimelate/lysine. Proc Natl Acad Sci USA 2006; 103:17909–17914. PubMed http://dx.doi.org/10.1073/pnas.0608643103
Hudson AO, Gilvarg C, Leustek T. Biochemical and phylogenetic characterization of a novel diaminopimelate biosynthesis pathway in prokaryotes identifies a diverged form of LL-diaminopimelate aminotransferase. J Bacteriol 2008; 190:3256–3263. PubMed http://dx.doi.org/10.1128/JB.01381-07
Singh SK, Yang K, Karthikeyan S, Huynh T, Zhang X, Phillips MA, Zhang H. The thrH gene product of Pseudomonas aeruginosa is a dual activity enzyme with a novel phosphoserine:homoserine phosphotransferase activity. J Biol Chem 2004; 279:13166–13173. PubMed http://dx.doi.org/10.1074/jbc.M311393200
Mormann S, Lömker A, Rückert C, Gaigalat L, Tauch A, Pühler A, Kalinowski J. Random mutagenesis in Corynebacterium glutamicum ATCC 13032 using an IS6100-based transposon vector identified the last unknown gene in the histidine biosynthesis pathway. BMC Genomics 2006; 7:205. PubMed http://dx.doi.org/10.1186/1471-2164-7-205
Tao L, Cheng Q. Novel beta-carotene ketolases from non-photosynthetic bacteria for canthaxanthin synthesis. Mol Genet Genomics 2004; 272:530–537. PubMed http://dx.doi.org/10.1007/s00438-004-1083-8
Tao L, Rouvière PE, Cheng Q. A carotenoid synthesis gene cluster from a non-marine Brevundimonas that synthesizes hydroxylated astaxanthin. Gene 2006; 379:101–108. PubMed http://dx.doi.org/10.1016/j.gene.2006.04.017
Cheng Q. Structural diversity and functional novelty of new carotenoid biosynthesis genes. J Ind Microbiol Biotechnol 2006; 33:552–559. PubMed http://dx.doi.org/10.1007/s10295-006-0121-4
Montalvo NF, Mohamed NM, Enticknap JJ, Hill RT. Novel actinobacteria from marine sponges. Antonie van Leeuwenhoek 2005; 87:29–36. PubMed http://dx.doi.org/10.1007/s10482-004-6536-x
Thomas TR, Kavlekar DP. LokaBharathi PA. Marine drugs from sponge-microbe association—a review. Mar Drugs 2010; 8:1417–1468. PubMed http://dx.doi.org/10.3390/md8041417
Taylor MW, Radax R, Steger D, Wagner M. Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol Mol Biol Rev 2007; 71:295–347. PubMed http://dx.doi.org/10.1128/MMBR.00040-06
Radwan M, Hanora A, Zan J, Mohamed NM, Abo-Elmatty DM, Abou-El-Ela SH, Hill RT. Bacterial community analyses of two red sea sponges. [NY]. Mar Biotechnol (NY) 2010; 12:350–360. PubMed http://dx.doi.org/10.1007/s10126-009-9239-5
Devi P, Wahidullah S, Rodrigues C, Souza LD. The sponge-associated bacterium Bacillus licheniformis SAB1: a source of antimicrobial compounds. Mar Drugs 2010; 8:1203–1212. PubMed http://dx.doi.org/10.3390/md8041203
Khan ST, Tamura T, Takagi M, Shin-Ya K. Streptomyces tateyamensis sp. nov., Streptomyces marinus sp. nov. and Streptomyces haliclonae sp. nov., three novel species of Streptomyces isolated from marine sponge Haliclona sp. Int J Syst Evol Microbiol 2010; 60:2775–2779. PubMed http://dx.doi.org/10.1099/ijs.0.019869-0
Piel J, Hui D, Wen G, Butzke D, Platzer M, Fusetani N, Matsunaga S. Antitumor polyketide biosynthesis by an uncultivated bacterial symbiont of the marine sponge Theonella swinhoei. Proc Natl Acad Sci USA 2004; 101:16222–16227. PubMed http://dx.doi.org/10.1073/pnas.0405976101
Hochmuth T, Piel J. Polyketide synthases of bacterial symbionts in sponges—evolution-based applications in natural products research. Phytochemistry 2009; 70:1841–1849. PubMed http://dx.doi.org/10.1016/j.phytochem.2009.04.0 10
Okuyama H, Orikasa Y, Nishida T, Watanabe K, Morita N. Bacterial genes responsible for the biosynthesis of eicosapentaenoic and docosahexaenoic acids and their heterologous expression. Appl Environ Microbiol 2007; 73:665–670. PubMed http://dx.doi.org/10.1128/AEM.02270-06
Metz JG, Roessler P, Facciotti D, Levering C, Dittrich F, Lassner M, Valentine R, Lardizabal K, Domergue F, Yamada A, et al. Production of polyunsaturated fatty acids by polyketide synthases in both prokaryotes and eukaryotes. Science 2001; 293:290–293. PubMed http://dx.doi.org/10.1126/science.1059593
Allen EE, Bartlett DH. Structure and regulation of the omega-3 polyunsaturated fatty acid synthase genes from the deep-sea bacterium Photobacterium profundum strain SS9. Microbiology 2002; 148:1903–1913. PubMed
Orikasa Y, Tanaka M, Sugihara S, Hori R, Nishida T, Ueno A, Morita N, Yano Y, Yamamoto K, Shibahara A, et al. pfaB products determine the molecular species produced in bacterial polyunsaturated fatty acid biosynthesis. FEMS Microbiol Lett 2009; 295:170–176. PubMed http://dx.doi.org/10.1111/j.1574-6968.2009.01582.x
Orikasa Y, Nishida T, Hase A, Watanabe K, Morita N, Okuyama H. A phosphopantetheinyl transferase gene essential for biosynthesis of n-3 polyunsaturated fatty acids from Moritella marina strain MP-1. FEBS Lett 2006; 580:4423–4429. PubMed http://dx.doi.org/10.1016/j.febslet.2006.07.008
Sugihara S, Orikasa Y, Okuyama H. An EntD-like phosphopantetheinyl transferase gene from Photobacterium profundum SS9 complements pfa genes of Moritella marina strain MP-1 involved in biosynthesis of docosahexaenoic acid. Biotechnol Lett 2008; 30:411–414. PubMed http://dx.doi.org/10.1007/s10529-007-9579-z
Marraffini LA, Dedent AC, Schneewind O. Sortases and the art of anchoring proteins to the envelopes of gram-positive bacteria. Microbiol Mol Biol Rev 2006; 70:192–221. PubMed http://dx.doi.org/10.1128/MMBR.70.1.192-221.2006
Patti JM, Allen BL, McGavin MJ, Höök M. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu Rev Microbiol 1994; 48:585–617. PubMed http://dx.doi.org/10.1146/annurev.mi.48.100194. 003101
Mizanur RM, Pohl NL. Bacterial CMP-sialic acid synthetases: production, properties, and applications. Appl Microbiol Biotechnol 2008; 80:757–765. PubMed http://dx.doi.org/10.1007/s00253-008-1643-7
Kogure K. Bioenergetics of marine bacteria. Curr Opin Biotechnol 1998; 9:278–282. PubMed http://dx.doi.org/10.1016/S0958-1669(98)80059-1
Mulkidjanian AY, Dibrov P, Galperin MY. The past and present of sodium energetics: may the sodium-motive force be with you. Biochim Biophys Acta 2008; 1777:985–992. PubMed http://dx.doi.org/10.1016/j.bbabio.2008.04.028
Häse CC, Fedorova ND, Galperin MY, Dibrov PA. Sodium ion cycle in bacterial pathogens: evidence from cross-genome comparisons. Microbiol Mol Biol Rev 2001; 65:353–370. PubMed http://dx.doi.org/10.1128/MMBR.65.3.353-370.2001
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Fujinami, S., Takarada, H., Kasai, H. et al. Complete genome sequence of Ilumatobacter coccineum YM16-304T. Stand in Genomic Sci 8, 430–440 (2013). https://doi.org/10.4056/sigs.4007734
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.4007734