
Abstract
Background
Corynebacterium glutamicum, a Gram-positive bacterium of the class Actinobacteria, is an industrially relevant producer of amino acids. Several methods for the targeted genetic manipulation of this organism and rational strain improvement have been developed. An efficient transposon mutagenesis system for the completely sequenced type strain ATCC 13032 would significantly advance functional genome analysis in this bacterium.
Results
A comprehensive transposon mutant library comprising 10,080 independent clones was constructed by electrotransformation of the restriction-deficient derivative of strain ATCC 13032, C. glutamicum RES167, with an IS6100-containing non-replicative plasmid. Transposon mutants had stable cointegrates between the transposon vector and the chromosome. Altogether 172 transposon integration sites have been determined by sequencing of the chromosomal inserts, revealing that each integration occurred at a different locus. Statistical target site analyses revealed an apparent absence of a target site preference. From the library, auxotrophic mutants were obtained with a frequency of 2.9%. By auxanography analyses nearly two thirds of the auxotrophs were further characterized, including mutants with single, double and alternative nutritional requirements. In most cases the nutritional requirement observed could be correlated to the annotation of the mutated gene involved in the biosynthesis of an amino acid, a nucleotide or a vitamin. One notable exception was a clone mutagenized by transposition into the gene cg0910, which exhibited an auxotrophy for histidine. The protein sequence deduced from cg0910 showed high sequence similarities to inositol-1(or 4)-monophosphatases (EC 3.1.3.25). Subsequent genetic deletion of cg0910 delivered the same histidine-auxotrophic phenotype. Genetic complementation of the mutants as well as supplementation by histidinol suggests that cg0910 encodes the hitherto unknown essential L-histidinol-phosphate phosphatase (EC 3.1.3.15) in C. glutamicum. The cg0910 gene, renamed hisN, and its encoded enzyme have putative orthologs in almost all Actinobacteria, including mycobacteria and streptomycetes.
Conclusion
The absence of regional and sequence preferences of IS6100-transposition demonstrate that the established system is suitable for efficient genome-scale random mutagenesis in the sequenced type strain C.glutamicum ATCC 13032. The identification of the hisN gene encoding histidinol-phosphate phosphatase in C. glutamicum closed the last gap in histidine synthesis in the Actinobacteria. The system might be a valuable genetic tool also in other bacteria due to the broad host-spectrum of IS6100.
Similar content being viewed by others


Background
Corynebacterium glutamicum is a non-sporulating soil bacterium, belonging to the high G+C Gram-positive Actinobacteria. The bacterium, originally isolated as a natural glutamic acid excreter, is nowadays widely used as an industrial relevant producer of several L-amino acids and vitamins [1, 2]. The common method to isolate amino acid-producing strains of C. glutamicum uses the iterative approach of general mutagenesis and subsequent screening for mutants with a high capacity to secrete the amino acid of interest [3]. With the development of genetic engineering methods, like gene disruption and replacement [4, 5] or over-expression and deregulation of single genes [6, 7], tools for more rational strain improvement are provided and successfully applied [8, 9]. The availability of the complete genome sequence of C. glutamicum ATCC 13032 in combination with automatic annotation tools [10] enhanced the potential of corynebacterial research greatly. However, despite single gene manipulation techniques and the acquired knowledge about genes or the function and regulation of their corresponding enzymes in the metabolic context of the cell, there are still gaps in some metabolic pathways and a large amount of genes for which functions could not yet be assigned. A practicable strategy to analyse genes of unknown function is to mutate them, e.g. by generating a genomic insertion mutant library, and subsequently characterize the phenotypic properties of individual clones within the library. An ideal resource for this purpose would be an ordered or random collection of insertion mutants, each containing a defined insertion in a particular non-essential gene, with which a large number of clones could be analysed simultaneously [11, 12].
In response to this demand, transposon mutagenesis systems became of increasing interest for genome-scale manipulation, analyses and single-gene studies in bacteria. Transposon systems are efficient mutagenesis tools in a wide variety of bacterial species [13]. The advantages of a mutagenesis system employing antibiotic resistance conferring transposons are the ability of a random integration into the chromosome, a positive selection for transposon mutants and the physical marker introduced at the site of mutation [14].
Transposons and insertion sequences (IS) are defined segments of DNA that can relocate as a unit between genomic regions [15]. Transposons range from class I composite transposons consisting of a pair of IS elements that enclose additional genetic information for antibiotic resistance or other properties, to complex class II transposons, to conjugative transposons that combine hybrid features of transposons, plasmids and bacteriophages [16].
Insertion sequences (IS) are the simplest form of mobile genetic elements. These elements generally possess one or two open reading frames encoding the protein that causes its mobility, the transposase. The majority of IS elements is flanked by terminal inverted-repeat sequences (IR). As a result of the integration mechanism IS generate directly repeated sequences (DR) of the target DNA flanking the element of a fixed length, which are characteristic for a given element.
A large number of native insertion sequences and their isoforms as part of transposons could be isolated from chromosomes and plasmids of coryneform bacteria and assigned to different members of mobile element families [17]. Furthermore, insertion sequences can be used to construct artificial transposons [18] and artificial transposon vectors [19] to circumvent, for instance, the natural limitation of missing antibiotic resistance markers. Several active mobile IS have been published for C. glutamicum. Most of these appear to transpose randomly, but are of limited usability for random mutagenesis due to a more or less distinctive target site preference. The IS elements IS1249 and IS1513 as part of the transposons Tn5432 and Tn5564, prefer triple A/T or a central palindromic tetranucleotide (CTAG) as target sequences [20, 21]. Recent studies with IS31831, as part of Tn14751 or IS14999 revealed A/T-rich regions [22] or an 8-bp palindromic sequence as preferred target sites [23].
In 2003, Bonamy et al. [24] reported about an IS1207-based transposon Tn5531 with an apparent low target site-specificity in the C. glutamicum strain ATCC 14752. However, the endogenous occurrence of seven copies of the highly similar insertion sequences (ISCg1a – ISCg1d, ISCg7, ISCg10 and ISCg17) in the strain ATCC 13032 impede the application of this transposon there since integrations would mainly be a result of RecA-mediated homologous recombination events.
The insertion element IS6100 was initially isolated as part of the composite transposon Tn610 of Mycobacterium fortuitum [25]. Later studies revealed its presence in a wide spectrum of host organisms from different bacterial lineages, e.g. Arthrobacter sp. [26], Pseudomonas aeruginosa [27], Xanthomonas campestris [28], or Aeromonas salmonicida [29]. It is 880-bp in size and is bordered by 14-bp perfect terminal inverted repeats. In common with other members of the IS6 family it translocates by replicative transposition which results in both the formation of a cointegration complex with an additional copy of the element itself as an end-product and the occurrence of an 8-bp direct repeat at the target site [17, 30]. Tauch et al. [31] isolated IS6100 from the C. glutamicum antibiotic resistance-plasmid pTET3 present in the strain C. glutamicum LP-6 and showed that the element is transpositionally active in the strain ATCC 13032. Subsequent Southern hybridization analyses of a small number of clones suggested that IS6100 inserts in a random manner into the C. glutamicum chromosome.
In this study a transposon library of the completely sequenced type strain ATCC 13032 with a statistically representative size was constructed using an transposon vector based on the IS6100 element. By auxanography analyses several auxotrophic mutants could be obtained. Plasmid rescue and sequencing of the insertion sites revealed a number of known biosynthesis genes as well hitherto unknowns. The additional development of a PCR-based screening strategy permitted the rapid detection of insertion mutants in any chromosomal region of interest. Taken together, the transposon used and the library created represent novel tools with high impact on functional genome analyses in C. glutamicum ATCC 13032.
Results
Transposon mutagenesis of Corynebacterium glutamicum ATCC 13032 with an IS6100-based transposon vector
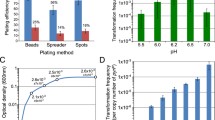
In vivo mutagenesis of the C. glutamicum ATCC 13032 genome was performed with the IS6100- based artificial transposon vector named pAT6100. The vector is a construct resulting from cloning IS6100 into the vector pK18mob2 [31]. The pAT6100 plasmid DNA extracted from Escherichia coli DH5αMCR was transformed into electrocompetent cells of C. glutamicum RES167, a restriction-deficient strain derived from the wild-type, by electroporation. Since the vector shares no homologous sequences with the host genome and it is not able to replicate autonomously in C. glutamicum the kanamycin resistance phenotype is the result of a transposition event into the chromosome. The transposition efficiencies ranged from five to ten c.f.u. per μg of desalted plasmid DNA.
An ordered clone library was constructed comprising 10,080 independent mutant clones. With a coverage more than three-fold with respect to the 3,002 coding regions determined for C. glutamicum ATCC 13032 [10] we expect that nearly all non-essential genes contain insertions. In order to estimate the theoretical quality of the library, statistical analyses were made: Given the number of clones in the transposon library (10,080), the estimated number of non-essential genes (2,400) [32], and the average gene length (952 bp), the average probability for any given gene to be disrupted by at least one transposon insertion is 97%.
In order to perform experimental analyses of the library, first a potential target site preference of the transposon was tested. Therefore, 26 clones were randomly selected and plasmid rescue cloning was carried out by digesting chromosomal DNA of the selected clones with EcoRI or XbaI, respectively, religating of the rescue constructs, transferring to E. coli and subsequent sequencing of the genomic insert with IS6100-specific primers. The sequence data obtained was analysed for sequence homologies with BLASTN searches against the C. glutamicum ATCC 13032 whole genome sequence [10]. The BLAST results revealed the common 14-bp inverted repeat of either left and right flank of the cointegrate followed by the individual 8-bp direct repeat target site for each clone, indicating the position of the transposon integration (Table 1). As the target sites of the clones were each different, with no duplicates with the same or similar sequences, it can be assumed that these 26 transposition events occurred independently. The position of the transposon in every mutant, as well those selected randomly (Fig. 1, red bars) and those investigated in auxanographic analyses (Fig. 1, black bars) were plotted on a circular map representing the C. glutamicum ATCC 13032 chromosome. The picture indicates a random distribution throughout the genome without regions of significant accumulation of insertions. Both, intragenic and intergenic insertions were detected. The G+C content was calculated from the eight nucleotides of the target site and ten flanking nucleotides upstream and downstream. It showed a high variance ranging from 39.3% to 78.5%, whereas the medium G+C content for the chromosome of the strain ATCC 13032 is 53.8%. The two possible orientations of the cointegrate with respect to the directions of replication or transcription are present in nearly equal proportions indicating that neither orientation is favoured.

Distribution of transposon insertions on a circular plot of the C. glutamicum ATCC 13032 genome [GenBank:BX927147]. Coloured bars of the outer circle pointing inward and outward show the orientation of the cointegrates with respect to the genome in clockwise and counterclockwise direction, respectively. Red bars indicate the integration positions mapped for randomly selected clones and black bars map those investigated by auxotrophy analysis. Additional circles (from inward to outward) represent relative G+C content and coding regions transcribed in clockwise and counterclockwise direction, respectively. A positive deviation in G+C content from the average (53.8%) is shown by bars pointing outward and a negative deviation by bars pointing inward. Annotated genes are coloured according to the colour scheme of the functional classes system COG (cluster of orthologous groups) [89]. The plot was generated with GenDB version 2.2 [90].
An additional test for target site preferences was carried out by performing sequence pattern searches with the TEIRESIAS algorithm [33]. For this purpose a total of 172 target sites acquired from the random selected clones and from mutants of the auxanographic analyses (see below) were used. No sequence pattern, palindromic sequences or regions of nucleotide symmetry could be detected with this bioinformatic approach (data not shown). Additionally, the occurrence of the bases at a distinct position of the 8-bp target sequence was calculated (data not shown). The values and therefore the appearance of bases appear to be distributed evenly. Neither the pattern searches nor the nucleotide distributions in the TSD indicate a significant target site preference for insertion.
The cointegration of the transposon vector forms long direct repeats in the form of IS6100 copies at the site of insertion. Such direct repeats might cause instability by replication slippage or by homologous recombination, thereby losing the integrated vector and one or both of the IS6100 elements in the absence of antibiotic selection. To assess this, five randomly selected individual clones were subcultured in antibiotic-free liquid complex medium in twelve rounds of 48 h each. The last culture was plated on solid complex medium. From this culture, 900 single colonies from each clone were tested on kanamycin-containing plates. No loss of the resistance phenotype and therefore the integrated transposon could be observed. It has to be concluded that the integrations are stably maintained in C. glutamicum even in the absence of selective pressure. Additionally, isolated DNA from randomly selected clones which had not been grown under selective growth conditions was used to perform PCR experiments. In all cases the presence of the cointegrate was verified (data not shown).
Auxanographic screening and targeted gene identification using PCR techniques
The initial step of performing genome-scale auxanographic analyses was the plating of the entire transposon library on minimal medium plates. After 32 h of incubation this screening approach delivered 342 mutants which exhibit a clear growth-deficient phenotype. The appearance of potential auxotrophic mutants which are unable to form colonies under such growth conditions was used as an initial criterion for efficient mutagenesis. These mutants were further characterized by the rapid method for identification of bacterial biochemical requirements according to Holliday [34]. Therefore, 12 plates of minimal medium were each supplemented with different combinations of six growth factors. Mutants with a single biochemical requirement would grow on two plates. The double auxotrophic mutants could be determined by growth on only a single plate and an alternative requirement by growth on more than two plates. For the latter two cases the exact nutritional requirements have to be determined by further tests combining individual growth factors.
With this method, a total of 36 different growth factors, which are adapted to corynebacterial needs by omitting biotin, choline, and thiosulfate, and adding cobalamine, glutamine, as well as asparagine, could be tested simultaneously. These growth factors comprise 21 amino acids, six nucleotides and nine vitamins, including some of their precursors. Each candidate auxotrophic clone was inoculated on all twelve plates and incubated for 32 h. Forty-seven out of the 342 clones tested formed slow growing colonies on every plate, suggesting that these are generally growth-deficient but not auxotrophic mutants. With the redefined number of 295 clones circa 2.9% of the transposon library are genuine auxotrophic mutants. This roughly correlates to prior studies with different transposons in which auxotrophic mutants were obtained with frequencies of 1.3% in C. glutamicum [24], 2.5% in Rhodococcus spp. [35] and 2% in Rhodococcus fascians [36].
Out of these 295 clones 167 display a requirement for a single growth factor (57%). Among these, 141 exhibit an auxotrophy for one of eleven different amino acids, 24 for one of four different nucleotides and two for different vitamins. Additionally, we have found twelve clones that show a double and six with an alternative requirement. Altogether, the auxotrophic requirements of nearly two thirds of the mutants could be identified.
From all of the auxotrophy categories a number of clones was selected and the transposon integration sites were determined using the plasmid rescue technique described above. Table 2 summarizes the phenotypic and genotypic data obtained from these clones.
In the majority of clones the observed auxotrophic phenotype is well explained by the existing annotation of the disrupted gene. For example amino acid biosynthesis genes like argG (arginine), thrB (threonine), proC (proline), serA (serine), leuD (leucine) or lysA (lysine) were subject to intensive studies in C. glutamicum before, and the transposon insertions in these genes resulted in the expected auxotrophic phenotypes. This was also the case for insertions into genes that show high similarities to genes with a known function in closely related species, e.g. guaA and guaB2, known to be involved in the guanosine monophosphate biosynthesis in Corynebacterium ammoniagenes [37], or to genes in more remote organisms, e.g. purA and purB which are involved in adenosine monophosphate biosynthesis in E. coli [38]. Another example are the carAB genes: even though these genes are not characterized in C. glutamicum they are well known to cause a double requirement for both arginine and uracil in E. coli since they encode for the subunits of a protein that provides the essential carbamoylphosphate for arginine and pyrimidine biosynthesis [39]. The ilvC gene is well studied in C. glutamicum and is known to cause a double auxotrophy for isoleucine and valine when disrupted [40]. The successful supplementation of a metE transposon mutant strain with either methionine or cobalamin (vitamin B12) is explained by the fact that a cobalamin-dependent (MetH) and a cobalamin-independent enzyme (MetE) perform the same step in methionine biosynthesis in C. glutamicum [41]. The loss of the MetE enzyme, the preferred route during aerobic growth, can be compensated by supporting the MetH function with appropriate amounts of cobalamin.
In some cases the correlation of observed phenotype and the gene targeted by transposon integration are not so obvious. An example is the guanine auxotrophy of the nadA insertion. The nadA gene is located together with nadB and nadC and encodes a protein which is apparently involved in quinolinate and nicotinamide adenine dinucleotide biosynthesis. It is currently unclear why guanine supplements growth of this mutant. Also in the case of the integration in gene thiO the connection between gene function and auxotrophy is not clear yet. The gene as part of the thiEOSGF cluster encodes a putative D-amino acid oxidase flavoprotein oxidoreductase that is known to be involved in thiamine biosynthesis [42]. Although a polar effect on the downstream genes thiSGF is not experimentally verified, such an effect would furthermore affect a more extensive part of the thiamine synthesis. The mutant is supplementable by histidine and not, as expected, by thiamine.
Since the de novo purine and pyrimidine nucleotide synthesis pathways are poorly characterized in C. glutamicum especially the functions of genes like purF or pyrB require further studies as well. Mutants with transposon insertions in the C. glutamicum genes purC, purE, and purK, known as purine biosynthesis genes in E. coli [38], were found among those which grew on more than three plates (data not shown). These mutants appear interesting to be investigated in further studies.
The genes of most biosynthetic pathways in C. glutamicum are scattered in different genetic loci. A special case where the distribution of transposon insertions in a limited genomic region could be tested is the trp operon containing six genes which encode all functions of tryptophan biosynthesis in C. glutamicum [43]. For this, the transposon insertion sites of 43 tryptophan-auxotrophic clones were determined. The insertion sites were all different and located within every gene from trpE to trpA (Fig. 2). Although the trp operon contains more insertions than statistically expected, this example gives additional evidence that the transposon library is random and that the transposon system can be used to mutagenise the C. glutamicum chromosome up to a high density.

Physical map of the tryptophan gene cluster in C. glutamicum ATCC 13032. Within the trpPEGDCFBA genes, indicated by blue arrows, the coloured bars denote the mapped integrations of 43 independent tryptophan auxotrophic mutants. The two orientations with respect to the genome are indicated in black (clockwise) and green (counterclockwise direction). The transposon integration identified by PCR screening is also marked (orange bar). The left stem-loop symbol denotes the transcriptional attenuator involved in the regulation of the tryptophan biosynthesis [43] and the right loop a Rho-independent transcriptional terminator structure.
To find a gene of interest that is mutated by a transposon is sometimes not possible by phenotypic screening. We were interested to map additional insertions upstream from the trp operon. For this, a PCR-based screening strategy was chosen and the workflow by Hobom et al. [44] was adapted so that the entire library could be screened for insertion events by iterative rounds of three-step PCR experiments simultaneously. This strategy delivered another transposon insertion which was mapped 35 bp upstream from the predicted trpP translation start. This mutant did not exhibit a tryptophan-auxotrophic phenotype in subsequent growth tests which coincides with the assumption that the trpP gene encodes a permease necessary for tryptophan- and 5-methyltryptophan uptake in C. glutamicum [45].
Identification of a novel C. glutamicum gene closing the last gap in histidine biosynthesis
The whole genome sequence of C. glutamicum ATCC 13032 is known and the genes are annotated based on sequence similarity and experimental data [10]. Due to the fact that a large number of amino acid biosynthesis genes have been characterized before, only few pathways remained incomplete. With the help of sequence similarities and subsequent genetic analyses further genes have been identified [46] leaving only few gaps. These "missing" genes mostly encode transaminases which are notorious for their involvement in more then one pathway and were also partly characterized by bioinformatic and biochemical studies [47, 48]. Beside this, only a single step in histidine biosynthesis remained, where no gene was known or a candidate gene could be identified by sequence similarity.
The metabolic pathway of the histidine biosynthesis, reactions, enzymes, as well as the organization of the corresponding genes display a high degree of conservation in bacteria [49]. According to the KEGG PATHWAY database [50] the histidinol-phosphate phosphatase (HolPase) is an exception which was not characterized at a genetic level in C. glutamicum and closely related Actinobacteria. This enzyme (L-histidinol-phosphate phosphohydrolase; EC 3.1.3.15) catalyses the penultimate step of the biosynthesis, the dephosphorylation of histidinol phosphate to histidinol, the direct precursor of histidine. In the reference organisms E. coli and Salmonella typhimurium the HolPase activity is associated with the N-terminal domain of the HisB bifunctional enzyme. The C-terminal domain exhibit similarity to an imidazoleglycerol-phosphate dehydratase (EC 4.2.1.19), which catalyses the sixth step of the histidine synthesis. However, amino acid sequence homology searches revealed that in C. glutamicum, like in most bacteria, the imidazoleglycerol-phosphate dehydratase is encoded as a monofunctional enzyme. Additional similarity searches for the E. coli homologs of the HolPase in C. glutamicum and other organisms, including the entire class of Actinobacteria, does neither provide any significant sequence homologies to ORFs in the C. glutamicum genome nor to well-conserved domains in other Actinobacteria.
Since the loss of the HolPase activity is supposed to exhibit a histidine-auxotrophic phenotype the corresponding unidentified gene in C. glutamicum was presumed to be among the 16 histidine-auxotrophic transposon mutants. Determination of the transposon integration sites in these mutants revealed insertions in seven of the nine histidine synthesis genes known so far, hisGE, hisHAFI and hisDCB, and another one in the gene cg0910 (Table 2). The integration in this gene, which comprises 783 bp, is located 125 bp downstream from the predicted cg0910 translation start, suggesting that the gene product is non-functional. The cg0910 gene is preceded by the gene cg0911 and both might be transcribed together (Fig. 3). The cg0911cg0910 locus is located far from known histidine biosynthesis genes. BLASTP searches with the deduced protein sequence of both genes revealed significant similarities to each other and to inositol-1(or4)-monophosphatases (IMP; EC 3.1.3.25) which hydrolyse the ester bond of inositol phosphate to generate inositol.

Schematic representation of the 3-kb chromosomal region around cg0910 (blue arrow) in C. glutamicum. His- and his+ denote a histidine-auxotrophic or -prototrophic phenotype of the corresponding mutant or strain, respectively. The transposon integration (A) is marked with a green flag. Red arrows indicate the binding positions of the primers used to amplify the deletion fragments (B; yellow boxes). The features of the deletion- (B) and complementation (C) constructs are also designated. The ruler indicates the absolute position in the genome.
Validation of cg0910 gene function by deletion and homologous complementation
In order to confirm the phenotype of the transposon mutation, a cg0910 deletion mutant was constructed using the "gene splicing by overlap extension" (gene SOEing) technique [51]. Therefore, a fusion product of chromosomal DNA regions of 622 bp and 541 bp directly up- and downstream, respectively, of the cg0910 target locus was created, which was subsequently cloned into the pK18mobsacB vector system [5]. The resulting vector pK18mobsacB-Δcg0910 was finally used for targeted gene deletion. The deleted internal cg0910 fragment was 702 bp of size (Fig. 3).
The initially observed phenotype of the transposon mutant with the integration in cg0910 (clone 99G01) could be reproduced with the resulting deletion mutant, termed CG281, which was tested negative for its ability to grow on minimal medium without and positive with histidine supplementation. This indicated that possible secondary effects in the transposon mutant could be ruled out and that the loss of the cg0910 gene product is responsible for histidine auxotrophy. Both the mutants 99G01 and CG281 also grew on minimal medium supplemented with histidinol, the end product of the histidinol phosphatase reaction, comparable to the wild-type strain. These results suggest that cg0910, assigned with the gene name hisN, encodes the hitherto unknown but essential HolPase activity in C. glutamicum, and is the functional homolog of the E. coli N-terminal HisB protein domain (Table 2).
To provide further evidence that the cg0910 gene encodes the HisN function, homologous complementation was carried out in both mutants 99G01 and CG281. For this, the complete cg0910 coding sequence was amplified by PCR with primers providing an optimized ribosome binding site and additional restriction enzyme recognition sites. The amplificate was subsequently cloned into the IPTG-inducible shuttle expression vector pEC-XT99A [52]. The resulting plasmid pEC-XT99A-cg0910 ex (Fig. 3) was subsequently transferred to the corresponding mutant strains which were then tested for histidine prototrophy. In both cases the extrachromosomally located cg0910 gene product complemented the loss of the corresponding chromosomal region in trans (data not shown).
Phylogenetic analysis of Cg0910 and related inositol monophosphatases (IMP)
BLAST searches revealed four IMP paralogs in the C. glutamicum ATCC 13032 genome with significant similarity to cg0910, by name cg0911, cg0967 (cysQ), cg2090 (suhB), and cg2298 (impA). For a functional classification of the inositol monophosphatase family-like proteins, phylogenetic analysis in comparison to sequence-related proteins from other Actinobacteria and the model organism E. coli was conducted. For this, similarity searches with the deduced amino acid sequences of the C. glutamicum IMPs in non-redundant databases were carried out, comprising, beside E. coli K-12, the genomes of Actinobacteria including completed as well as draft status genomes. Proteins with reliable similarity scores were used to perform a multiple alignment. Based on this alignment a phylogenetic tree was created by the neighbour-joining method and, subsequently, the tree was evaluated by bootstrap analysis (Fig. 4).

Dendrogram showing the relationship of inositol monophosphatase family proteins (IMP) in Actinobacteria and E. coli K-12. A multiple alignment with amino acid sequences of proteins with high similarity to the C. glutamicum IMPs was generated with the use of the DIALIGN2 software. Based on this alignment an unrooted phylogenetic tree was constructed using the neighbour-joining algorithm integrated in the CLUSTALX package and visualized as a radial tree by the TreeTool software. The branches were combined to classes that delivered within the bootstrapping analyses in at least two thirds of the cases the same subtree. These classes, marked by different colours, were named according to the designations in the boxed leaves. The locus tags (leaves) were obtained from the GenBank genome entries. C. glutamicum proteins are printed in bold letters. Locus tag prefixes denote following organisms (c, complete genome sequence; da, draft assembly): Arth (Arthrobacter sp. FB24; da), BL (Bifidobacterium longum NCC2705; c), BLinB01 (Brevibacterium linens BL2; da), DIP (Corynebacterium diphtheriae NCTC13129; c), CE (C. efficiens YS-314; c), cg (C. glutamicum ATCC 13032; c), jk (C. jeikeium K411; c), Ecoli (Escherichia coli K-12; c), Francci3 (Frankia sp. CcI3; c), Franean1 (Frankia sp. EAN1pec; da), JNB (Janibacter sp. HTCC2649; da), Krad (Kineococcus radiotolerans SRS30216; da), Lxx (Leifsonia xyli subsp. xyli str. CTCB07; c), Micol (Micromonospora olivasterospora; da), MAP (Mycobacterium avium subsp. paratuberculosis K-10; c), ML (M. leprae TN; c), Rv (M. tuberculosis H37Rv; c), Mycsm (Mycobacterium smegmatis str. MC2 155; da), nfa (Nocardia farcinica IFM 10152; c), Noca (Nocardioides sp. JS614; da), PPA (Propionibacterium acnes KPA171202; c), RhoDS7 (Rhodococcus sp. DS7; da), Rxyl (Rubrobacter xylanophilus DSM 9941; da), SAV (Streptomyces avermitilis MA-4680; c), SCO (S. coelicolor A3(2); c) and Tfu (Thermobifida fusca YX; c).
The phylogenetic tree separates the five C. glutamicum IMP paralogs (Fig. 4, bold) into distinct classes which all include apparent orthologs from other Actinobacteria. The denotation of the respective class was derived from members for which a physical function has been described (Fig. 4, box): CysQ in E. coli [53], ImpA in Mycobacterium smegmatis [54], SuhB in M. tuberculosis [55] and HisN (Cg0910) in C. glutamicum.
The putative orthologs of Cg0911 form a separate class. None of its members, comprising only corynebacterial and one Nocardioides species, was functionally characterized up to now.
Discussion
In this study we describe a transposon system applicable for efficient random mutagenesis in C. glutamicum ATCC 13032. The use of an IS6100-based transposon vector gave rise to a transposon library of statistically representative size comprising independent mutant clones.
It was shown earlier that IS6100 is capable of transposing in vivo in C. glutamicum with unique transposition events by forming a cointegrate with the chromosome [31]. The excision frequency and therefore the stability of a replicative transposon integration is usually controlled by a resolvase protein, which is not encoded by the pAT6100 transposon vector. Nevertheless, cointegrate resolution involving either the two identical IS6100 copies or the 8-bp direct repeats might be caused by homologous recombination or by replication slippage resulting in the loss of the vector part and one or both copies of the IS6100 element. In this study an antibiotic sensitivity could not be observed after prolonged growth in the absence of antibiotic pressure during cultivation, indicating that IS6100 generates cointegrations that are stably maintained in C. glutamicum. This finding conforms with the study of Weaden and Dyson [56] in which the stable cointegration of IS6100 was observed in Streptomyces avermitilis.
The already developed transposon systems are not well applicable for random mutagenesis in the sequenced C. glutamicum type strain ATCC 13032 because of similar endogenous insertion sequences, pronounced target site preferences, or low transposition frequencies. For instance, seven copies of ISL3 family-like sequences in the ATCC 13032 genome prevent the usage of IS31831 [18] and related elements (e.g. IS1207) [24] as well as their derived transposons (e.g. Tn5531) [24]. Very recently a mutagenesis system was described that used IS31831- and Tn5-based minitransposons to generate a comprehensive library of the C. glutamicum strain R which covers nearly 80% of the presently unpublished genome [57]. The C. glutamicum strain R has the advantage of not possessing insertion elements of the IS31831 family. In contrast to this, IS6100 is absent from the strain ATCC 13032 chromosome, ruling out integration by homologous recombination and thus it is suitable to perform transposon mutagenesis in this strain. Beside IS6100 quite a few active mobile elements have been published for C. glutamicum which are of restricted usability of generating a comprehensive random transposon library since they prefer specific sequences, e.g. a triple A or T (IS1249) [20] or palindromic sequences (e.g. IS14999) [23], as a target for integration. On the other hand, prior studies with IS6100-based transposons suggested independent transposition and absence of a target site preference. This was first shown in Streptomyces lividans and S. coelicolor by nucleotide sequence comparison for a small number of mutants [58] and, on the basis of Southern hybridization studies in S. lividans [59] and C. glutamicum [31].
In this study, the usability of IS6100 for mutagenesis in the type strain ATCC 13032 was shown by analysis of a larger number of clones. The sequence data determined from 172 insertions delivered definite position informations and, consequently, allowed comprehensive analyses on the transposon target sites. Each clone investigated carried the transposon in a different chromosomal location. The absence of regional preferences together with the absence of sequence preferences (sequence pattern, nucleotide-usage for the TSD positions or G+C content) as a target demonstrated the randomness of the library constructed and applied in auxanography analyses. Furthermore, this system might be a practicable genetic tool in other organisms as well because of the broad host-spectrum of the IS6100 element.
This study integrated genomic sequence data with genome-scale auxotrophy analyses. By means of auxanographic assays, for 63% of the 295 isolated auxotrophic mutants with distinct phenotypes various nutritional requirements could be identified. Additionally, out of this contingent the different integration loci of 101 clones were determined. The value of 2.9% auxotrophic clones only slightly differs from those in other transposon mutagenesis studies (1.3% [24], 2.5% [35] and 2% [36]) but is remarkably higher compared to studies in which auxotrophic mutants were obtained with frequencies of 0.2% in "Brevibacterium flavum" with IS31831 [18], 0.2% in C. glutamicum with IS1249 (Tn5432) [20] and 0.5% in Streptomyces avermitilis with IS6100 [56]. This might be explained by the fact that latter systems use mobile elements for which an insertion sequence specificity was identified (IS31831 and IS1249).
Transposon integrations were found in a variety of known amino acid, nucleotide and vitamin pathway genes as well in genes encoding hypothetical proteins or such of presently unknown function. The vast majority of the observed auxotrophic phenotypes could be correlated and explained with the knowledge of the mutated genomic region since most genes of amino acid, nucleotide and vitamin biosyntheses are annotated in C. glutamicum. In contrast, for some observed phenotypes the connection between gene and auxotrophy is not yet clear, delivering interesting targets for further studies. The loss of gene products associated with de novo synthesis or recycling pathways may be compensated by influx of necessary metabolite entries from other pathways that share common intermediates or precursors to a given intermediate. For instance, the PurF protein, an amidophosphoribosyltransferase, is known to be involved in more than one pathway. In Salmonella typhimurium PurF is the first of five enzymes shared by the de novo purine and HMP (hydroxymethylpyrimidine) synthesis, an essential compound of thiamine biosynthesis. Thus, PurF is expected to be required for both purine and thiamine biosynthesis [60]. It is not obvious why a purF transposon mutant is supplementable solely by hypoxanthine. Alternative pathways were discovered which could bypass the requirement for all pur genes in thiamine synthesis [61, 62]. Therefore, hypoxanthine, a purine derivative, might be sufficient to compensate the growth-deficiency of this mutant.
It is known that transposon insertions near the beginning or within an operon can attenuate or interrupt the expression of downstream genes preventing RNA polymerase readthrough [63, 64]. A polar effect on downstream genes was experimentally shown for an IS6100 integration in the gene cysI (cg3118) with transcriptional analysis using Real Time RT-PCR [65]. This example points out that the transposon system is not only applicable for high density mutagenesis of chromosomal regions (e.g. tryptophan operon, Fig. 2) but might be utilised for the determination of operons.
The screening approach for characterization of auxotrophic mutants used in this study shows a limitation in resolving complex growth phenotypes, exhibited by one thirds of these mutants. However, determination of transposon positions in randomly selected members of this auxotrophy category revealed for example mutations within purC, purE, and purK, genes known to be involved in purine biosynthesis. Such genes, as mentioned above, might be of interest to be investigated in further experimental studies. These findings indicate that also the complex phenotypes are not a result of additional spontaneously occurring mutations, but caused by disruption of the corresponding gene or by the accompanying polar effect.
For detailed studies on gene functions, transposon mutants have to be confirmed experimentally in order to rule out secondary mutations, polar effects or leaky phenotypes. In this study, this was carried out by a concrete example in which, by the means of supplementation, genetic deletion and complementation assays, the last gap in the histidine biosynthesis pathway of C. glutamicum could be closed.
Initial similarity searches revealed that the Cg0910 protein (HisN), together with another four paralogs in the C. glutamicum genome, apparently belongs to the monophosphatase-family proteins that usually hydrolyse the ester bond of myo-inositol-1(or4) phosphate. Inositol monophosphatases (IMP) play a crucial role in the biosynthesis of inositol and inositol phospholipids [66]. The role of IMP in bacteria is not completely clear yet. Bacteria of the genus Mycobacterium contain a number of inositol-derived cell wall constituents, like phosphatidylinositol (PI), phosphatidylinositol mannosides (PIM), lipoarabinomannan (LAM) and lipomannan (LM) [67–69]. LAM-like molecules and other inositol-containing phospholipids are not only present in mycobacteria but also in other Actinobacteria, including the genus Corynebacterium [70–72]. Actually, little is known about inositol synthesis in bacteria. The known de novo pathway in mycobacteria basically comprises the cyclization of glucose-6-phosphate to inositol-1-phosphate (I-1-P) and, subsequently, the dephosphorylation of I-1-P by IMP producing inositol [73].
Although the IMP-like proteins analysed in this study appear to be sequence homologs phylogenetic analysis revealed that they are significantly different as they branch into distinct classes. Despite their amino acid conservation, they seem to form a protein family of diverse functions and/or with diverse substrates. The assumption of different substrate specificities corresponds to previously published studies performed either in closely related mycobacteria or in E. coli.
The E. coli CysQ homolog is required for cysteine synthesis during aerobic growth. The sulfate assimilation branch of the cysteine pathway comprises sulfate uptake, its activation by formation of adenosine 5'-phosphosulfate (APS) and conversion to 3'-phosphoadenosine 5'-phosphosulfate (PAPS) by APS kinase, and its reduction to sulfite. It has been suggested that CysQ acts on PAPS as a target. It is proposed to help controlling the levels of PAPS, which may be toxic to the cell in high concentrations, or the generation of sulfite [53]. The actinobacterial CysQ IMP homologs in the phylogenetic tree branch later as compared to CysQ of E. coli, thus functional differences between the E. coli and the actinobacterial CysQ proteins might be possible. In C. glutamicum an APS kinase homolog is missing and thus PAPS as an intermediate is not formed. Sulfite is released from APS by direct reduction through APS reductase [65]. Therefore, the target substrate for a PAPS CysQ protein would be missing as well. It might be considered that CysQ in C. glutamicum and the other actinobacterial members of this phylogenetic class indeed does not possess a PAPS phosphatase activity, but most likely an inositol-phosphate phosphatase activity.
Initially, impA has been proposed to encode the missing HolPase, as it is clustered with the histidine biosynthesis genes in the Actinobacteridae [74]. This theory has been falsified for a M. smegmatis impA mutant. This mutant is not auxotrophic for histidine, but exhibits altered cell envelope permeability properties, with a notable reduction in the synthesis of phosphatidylinositol dimannoside, the precursor of LAM [54].
The SuhB inositol monophosphatase activity has been characterized biochemically in M. tuberculosis. Inositol-1-phosphate was shown to be the preferred target of SuhB for dephosphorylation in order to provide the PI synthase with inositol [75].
The specificity of Cg0910 (HisN) for histidinol phosphate was experimentally proven for C. glutamicum. For all other members of the phylogenetic IMP class labelled Cg0910 the intrinsic HolPase function could be proposed because of the close relationship and the high sequence conservation. Furthermore, in the respective Actinobacteria a protein having the enzymatic function of histidinol phosphate dephosphorylation was not identified.
The Cg0911 class comprises only corynebacterial members and one Nocardioides subspecies. The fact that in all these organisms cg0911 and cg0910 orthologs are located next to each other leads to the assumption of a recent gene duplication that exclusively occurred in corynebacteria or in a common ancestor. However, the finding that the respective proteins of cg0910 and cg0911 are members of different IMP classes indicate that they accomplish different functions and thus solely the cg0910 gene encodes the HolPase activity in C. glutamicum. Additional evidence that the unknown Cg0911 function, in contrast to Cg0910, does not play an important role in the cell was obtained by the inactivation of the respective gene (data not shown). The mutant showed no obvious phenotype under the applied growth conditions.
Conclusion
In conclusion, the random transposon system described in this study together with the availability of the complete genome sequence of C. glutamicum ATCC 13032 [10] represents a powerful tool for genome-scale functional analyses in this type strain. Moreover, the transposon library is a resource of specific mutants which contribute to the genetic understanding of this organism in the future.
The closure of the last gap in the histidine biosynthesis pathway by identification of the hisN gene encoding a histidinol-phosphate phosphatase not only complements the knowledge of the pathway in C. glutamicum but in the entire class of Actinobacteria as well as in several Proteobacteria and Chlorobia [76]. Furthermore, with the classification of the inositol monophosphate proteins based on sequence similarities, it could be proposed that the IMP homologs in Actinobacteria, although generally acting on phosphorylated metabolites, appear to have diverse substrate specificities.
Methods
Bacterial strains, plasmids, oligonucleotides and culture conditions
Relevant bacterial strains, plasmids, the transposon vector and oligonucleotides constructed and used in this study are listed in Table 3. E. coli and C. glutamicum strains were routinely grown on Tryptic Soy Broth (TSB) complex media (Merck, Darmstadt, Germany) supplemented with 1.5% (w/v) agar (Invitrogen, Karlsruhe, Germany) at 37°C and 30°C, respectively. Antibiotics used for selection were kanamycin (50 μg ml-1 for E. coli and 25 μg ml-1 for C. glutamicum), tetracycline (5 μg ml-1) and nalidixic acid (50 μg ml-1). The oligonucleotides used as primers were designed with the Clone Manager software (Scientific & Educational Software, Cary, USA) and purchased from Operon (Cologne, Germany). The optical density of a culture in liquid medium was determined at a wavelength of 600 nm with a BioPhotometer (Eppendorf, Hamburg, Germany) spectralphotometer.
DNA isolation, manipulation and transfer
Plasmid DNA was extracted from E. coli and C. glutamicum by an alkaline lysis technique using the QIAprep Spin Miniprep Kit (Qiagen, Hilden, Germany) with a preliminary incubation for 2 h at 37°C in resuspension buffer P1 containing 50 mg ml-1 lysozyme (185,000 U mg-1; Serva, Heidelberg, Germany) for C. glutamicum. DNA modification, analyses by gel-electrophoresis and ligation were carried out by standard procedures [77]. Restriction endonucleases and T4 DNA ligase, together with enzyme buffers, were purchased from Fermentas (St. Leon-Rot, Germany) and used according to the manufacturer's instructions. DNA restriction fragments required for cloning were recovered from agarose gels by using QIAEX II gel extraction kit (Qiagen). E. coli and corynebacterial cells were transformed by electroporation [78] using the Bio-Rad Gene Pulser system (Bio-Rad, Munich, Germany).
Transformation of C. glutamicum
The artificial transposon vector pAT6100 was electrotransferred into the restriction-deficient strain C. glutamicum RES167 by means of a method previously described [79]. To force transposition, the cells were additionally incubated by shaking at 200 rpm for 50 min at 37°C directly after electroporation procedure before pelleting and spreading them on solid complex media plates containing 25 μg ml-1 kanamycin to select transformants. Kanamycin-resistant colonies were picked with sterile sticks, transferred to separate wells of 96-well microtiter plates (Greiner, Solingen, Germany) containing 200 μl selective complex medium and inoculated for 48 h at 30°C. Long-term storage was carried out in 96-well microtiter plates containing 10% Hogness modified freezing medium (HMFM), composed of 87% (v/v) glycerol, 1.7 mM tri-sodium citrate, 6.8 mM (NH4)2SO4, 0.4 mM MgSO4 · 4 H2O, 36 mM Na2HPO4 · 2 H2O, 13.2 mM KH2PO4, and 90% TSB at -80°C.
Determination of transposon integration sites
Genomic DNA from C. glutamicum transposon mutants was extracted using the GenElute Bacterial Genomic DNA Kit (Sigma-Aldrich, Taufkirchen, Germany). Cloning and sequencing of insertion sites were carried out by plasmid rescue techniques as described before [20] with the exception that the chromosomal DNA was digested with EcoRI and XbaI to clone either side of the insertion. Sequencing of the resulting plasmids was performed with the synthetic oligonucleotide primers s6100e and s6100x for the EcoRI and the XbaI sites, respectively, by IIT Biotech (Bielefeld, Germany).
Sequence data/accession numbers
Nucleotide homology searches were applied against the annotated C. glutamicum ATCC 13032 genome sequence [GenBank:BX927147] [10, 80] obtained from GenBank [81]. Amino acid sequences of predicted inositol monophosphatase proteins were obtained from the Swissprot/TrEMBL databases [82]: Cg0910 [Swiss-Prot:Q8NS80], Cg0911 [Swiss-Prot:Q6M6Y2], CysQ [Swiss-Prot:Q8NS37], SuhB [Swiss-Prot:Q6M4B2] and ImpA [Swiss-Prot:Q8NNT8]. Further protein sequence information used for comparative analyses were retrieved from the following GenBank genome entries: [GenBank:AE014295] Bifidobacterium longum NCC2705, [GenBank:BA000035] C. efficiens YS-314, [GenBank:BX248353] C. diphtheriae NCTC13129, [GenBank:CR931997] C. jeikeium K411, [GenBank:U00096] Escherichia coli K-12, [GenBank:CP000249] Frankia sp. CcI3, [GenBank:AE016822] Leifsonia xyli subsp. xyli str. CTCB07, [GenBank:AE016958] Mycobacterium avium subsp. paratuberculosis K-10, [GenBank:AL450380] M. leprae TN, [GenBank:AL123456] M. tuberculosis H37Rv, [GenBank:AP006618] Nocardia farcinica IFM 10152, [GenBank:AE017283] Propionibacterium acnes KPA171202, [GenBank:BA000030] Streptomyces avermitilis MA-4680, [GenBank:AL645882] S. coelicolor A3(2), [GenBank:CP000088] Thermobifida fusca YX, [GenBank:AAHG00000000] Arthrobacter sp. FB24, [GenBank:AAGP00000000] Brevibacterium linens BL2, [GenBank:AAII00000000] Frankia sp. EAN1pec, [GenBank:AAMN00000000] Janibacter sp. HTCC2649, [GenBank:AAEF00000000] Kineococcus radiotolerans SRS30216, [GenBank:AAJB00000000] Nocardioides sp. JS614, and [GenBank:AAEB00000000] Rubrobacter xylanophilus DSM 9941.
Bioinformatic analyses and tools for interpretation of C. glutamicum sequences
Database searches and sequence similarity-based searches with nucleotide and amino acid sequences were performed with the BLASTN- and BLASTP- algorithms [83], respectively. The multiple amino acid sequence alignment of the inositol monophosphatases-family proteins was performed with the DIALIGN2 software [84]. The phylogenic tree was calculated using the neighbour-joining method [85] integrated in the CLUSTALX software package [86] and visualized as a radial tree with the interactive phylogenetic tree plotting program TreeTool [87]. Sequence pattern searches were performed with the TEIRESIAS algorithm [33] provided by the IBM Bioinformatics Group.
Auxotrophy screening and supplementation of auxotrophic phenotypes
The identification of auxotrophic mutants was performed by parallel plating on minimal media MM1 (MMYE without yeast extract [88]) and TSB complex media. The auxanographic characterizations were carried out by picking the colonies on MM1 plates supplemented with growth factors which were added as sterile filtered stock solutions by means of the purchaser's instructions, following a plating pattern described by Holliday [34], and 25 μg ml-1 kanamycin. The list of growth factors comprises 20 biogenic L-amino acids (alanine, arginine, asparagine, aspartic acid, cysteine, glutamine, glutamic acid, glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine, valine; final concentrations 1 mM each), nucleic acid bases (adenine, cytosine, guanine, thymine, uracil; 1 mM final cc. each), vitamins and precursors (thiamine (vitamin B1; 500 μg l-1), riboflavin (B2; 200 μg l-1), nicotinic acid (B3; 400 μg l-1), pantothenic acid (B5; 400 μg l-1), pyridoxine (B6; 400 μg l-1), cobalamin (B12; 10 μg l-1), folic acid (20 μg l-1), p-aminobenzoic acid (200 μg l-1), inositol (20 μg l-1), ornithine (1 mM), and hypoxanthine (1 mM)). The chemicals were purchased from VWR (Darmstadt, Germany) and Merck.
PCR techniques and conditions
DNA fragments for deletion and complementation experiments were amplified from chromosomal DNA of C. glutamicum ATCC 13032 using Pwo DNA polymerase (Invitrogen), and Taq DNA polymerase (Peqlab, Erlangen, Germany) for the verification of chromosomal deletions. PCR reaction conditions were as follows: initial denaturation at 94°C for 2 min followed by 35 cycles of denaturation for 30 s, annealing for 1 min at a primer-dependent temperature, extension at 72°C for 2 min and a final extension for 4 min at 72°C. PCR products were purified using the QIAquick PCR purification kit (Qiagen).
PCR mutant screening was carried out with Eppendorf Taq DNA polymerase (Eppendorf) with pooled transposon mutants as templates, IS6100-specific primers IS6100x and IS6100y for both possible orientations of the transposon and a position-specific search primer gs_trpP (Table 3). The basic procedure was adapted from a method published by Hobom et al. [44]. Iterative rounds of PCR experiments were performed in order to identify the clone of interest in the library. In the first round clone-pools were examined containing all mutants in one microtiter plate. Sequential PCR rounds were carried out with the mutant-pools representing the clones of those individual plates that were tested positively in prior rounds until the exact position of the clone in the library could be determined. The bacterial cells were directly set in the PCR reaction and lysed with an initial PCR step of 94°C for 10 min to release the chromosomal DNA. PCR reaction conditions were as follows: initial denaturation at 94°C for 10 min followed by 40 cycles of denaturation for 30 s, annealing for 45 s at a primer-dependent temperature, extension at 72°C for 2 min and a final extension for 4 min at 72°C. PCR experiments were performed with the delivered buffers according to the manufacturer's instructions and with the use of a DNA Engine DYAD thermocycler from MJ Research (Watertown, MA, USA).
Deletion and genetic complementation of cg0910
Defined chromosomal deletions within the cg0910 gene were constructed with the pK18mobsacB vector system which allows to identify an allelic exchange by homologous recombination [5]. The deletion was introduced into the target gene by "gene SOEing" [51]. Therefore, upstream and downstream regions of the gene to be deleted were amplified by two different PCR reactions using primer pairs cg0910_del1+2 and cg0910_del3+4 (Table 3), respectively. After purification the resulting amplificates were used as templates for the second round of PCR. The final products were digested with the restriction enzymes BamHI and XhoI, corresponding to the cleavage sites introduced via the PCR primers, and subsequently cloned into appropriately digested pK18mobsacB vector. The ligation mixture was used to transform E. coli DH5αMCR and the transformants were selected on TSB plates containing 50 μg ml-1 kanamycin and 40 mg l-1 X-Gal (5-bromo-4-chloro-indolyl-β-D-galactopyranoside). The resulting deletion plasmid pK18mobsacB-Δcg0910 was extracted and transformed into C. glutamicum ATCC 13032. Integration of the introduced plasmid into the chromosome by single-crossover was selected on TSB plates supplemented with 25 μg ml-1 kanamycin. The kanamycin-resistant clones were grown overnight in liquid media and spread on TSB plates containing 10% (w/v) sucrose. Colonies from this plates were tested for the desired kanamycin-sensitive and sucrose-resistant phenotype by parallel picking. PCR experiments were used to verify the deletion in the C. glutamicum chromosome. Construction of plasmid pEC-XT99A-cg0910 ex for in trans- expression of the Cg0910 protein was initiated with the amplification of the corresponding gene by PCR using the primer pair cg0910_ex1 and cg0910_ex2 (Table 3). Primer cg0910_ex1 was provided with a sequence of an optimal ribosome binding site upstream from the cg0910 start codon. The resulting 821-bp DNA fragment was purified and cleaved using MunI and BamHI. Appropriate restriction sites were added with the 5'-extension of the PCR primers. The fragment was subsequently ligated into the pEC-XT99A vector, which was prior cut with the corresponding enzymes. The ligation mixture was used for transformation of E. coli DH5αMCR, the transformants were selected on TSB plates containing 5 μg ml-1 tetracycline.
References
Leuchtenberger W: Amino acids-technichal production and use. Products of primary metabolism. Edited by: Rehm HJ, Reed G, Pühler A, Stadler P. 1996, Weinheim, Germany , Biotechnology, VCH, 6: 465–502-
Ikeda M: Amino acid production processes. Microbial production of l-amino acids. Edited by: Faurie R, Thommel J. 2003, Berlin, Heidelberg, New York , Adv Biochem Eng Biotechnol, Springer, 79: 1-35.
Rowlands RT: Industrial strain improvement: mutagenesis and random screening procedures. Enzyme Microb Technol. 1984, 6: 3-10. 10.1016/0141-0229(84)90070-X.
Schwarzer A, Pühler A: Manipulation of Corynebacterium glutamicum by gene disruption and replacement. Biotechnology (N Y). 1991, 9 (1): 84-87. 10.1038/nbt0191-84.
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A: Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994, 145 (1): 69-73. 10.1016/0378-1119(94)90324-7.
Dusch N, Pühler A, Kalinowski J: Expression of the Corynebacterium glutamicum panD gene encoding L-aspartate-alpha-decarboxylase leads to pantothenate overproduction in Escherichia coli. Appl Environ Microbiol. 1999, 65 (4): 1530-1539.
Sahm H, Eggeling L, de Graaf AA: Pathway analysis and metabolic engineering in Corynebacterium glutamicum. Biol Chem. 2000, 381 (9-10): 899-910. 10.1515/BC.2000.111.
Peters-Wendisch PG, Schiel B, Wendisch VF, Katsoulidis E, Möckel B, Sahm H, Eikmanns BJ: Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J Mol Microbiol Biotechnol. 2001, 3 (2): 295-300.
Ohnishi J, Hayashi M, Mitsuhashi S, Ikeda M: Efficient 40 degrees C fermentation of L-lysine by a new Corynebacterium glutamicum mutant developed by genome breeding. Appl Microbiol Biotechnol. 2003, 62 (1): 69-75. 10.1007/s00253-003-1254-2.
Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns BJ, Gaigalat L, Goesmann A, Hartmann M, Huthmacher K, Krämer R, Linke B, McHardy AC, Meyer F, Möckel B, Pfefferle W, Pühler A, Rey DA, Rückert C, Rupp O, Sahm H, Wendisch VF, Wiegräbe I, Tauch A: The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J Biotechnol. 2003, 104 (1-3): 5-25. 10.1016/S0168-1656(03)00154-8.
Berg CM, Berg DE: Transposable Element Tools for Microbial Genetics. Escherichia coli and Salmonella cellular and molecular biology. Edited by: Neidhardt FC, Curtis IR, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE. 1996, Washington, DC, USA , ASM Press, 2588−2612-
Hamer L, DeZwaan TM, Montenegro-Chamorro MV, Frank SA, Hamer JE: Recent advances in large-scale transposon mutagenesis. Curr Opin Chem Biol. 2001, 5 (1): 67-73. 10.1016/S1367-5931(00)00162-9.
Craig NL: Target site selection in transposition. Annu Rev Biochem. 1997, 66: 437-474. 10.1146/annurev.biochem.66.1.437.
Berg CM, Berg DE, Groisman E: Transposable elements and the genetic engineering of bacteria. Mobile DNA. Edited by: Berg DW, Howe MM. 1989, Washington, D.C., USA , American Society for Microbiology, 879–925-
Berg DE, Howe MM: Mobile DNA. 1989, Washington, D.C., USA , American Society for Microbiology
Hayes F: Transposon-based strategies for microbial functional genomics and proteomics. Annu Rev Genet. 2003, 37: 3-29. 10.1146/annurev.genet.37.110801.142807.
Mahillon J, Chandler M: Insertion sequences. Microbiol Mol Biol Rev. 1998, 62 (3): 725-774.
Vertes AA, Asai Y, Inui M, Kobayashi M, Kurusu Y, Yukawa H: Transposon mutagenesis of coryneform bacteria. Mol Gen Genet. 1994, 245 (4): 397-405. 10.1007/BF00302251.
Fomukong NG, Dale JW: Transpositional activity of IS986 in Mycobacterium smegmatis. Gene. 1993, 130 (1): 99-105. 10.1016/0378-1119(93)90351-3.
Tauch A, Kassing F, Kalinowski J, Pühler A: The Corynebacterium xerosis composite transposon Tn5432 consists of two identical insertion sequences, designated IS1249, flanking the erythromycin resistance gene ermCX. Plasmid. 1995, 34 (2): 119-131. 10.1006/plas.1995.9995.
Tauch A, Zheng Z, Pühler A, Kalinowski J: Corynebacterium striatum chloramphenicol resistance transposon Tn5564: genetic organization and transposition in Corynebacterium glutamicum. Plasmid. 1998, 40 (2): 126-139. 10.1006/plas.1998.1362.
Inui M, Tsuge Y, Suzuki N, Vertes AA, Yukawa H: Isolation and characterization of a native composite transposon, Tn14751, carrying 17.4 kilobases of Corynebacterium glutamicum chromosomal DNA. Appl Environ Microbiol. 2005, 71 (1): 407-416. 10.1128/AEM.71.1.407-416.2005.
Tsuge Y, Ninomiya K, Suzuki N, Inui M, Yukawa H: A new insertion sequence, IS14999, from Corynebacterium glutamicum. Microbiology. 2005, 151 (Pt 2): 501-508. 10.1099/mic.0.27567-0.
Bonamy C, Labarre J, Cazaubon L, Jacob C, Le Bohec F, Reyes O, Leblon G: The mobile element IS1207 of Brevibacterium lactofermentum ATCC21086: isolation and use in the construction of Tn5531, a versatile transposon for insertional mutagenesis of Corynebacterium glutamicum. J Biotechnol. 2003, 104 (1-3): 301-309. 10.1016/S0168-1656(03)00150-0.
Martin C, Timm J, Rauzier J, Gomez-Lus R, Davies J, Gicquel B: Transposition of an antibiotic resistance element in mycobacteria. Nature. 1990, 345: 739–743-
Kato K, Ohtsuki K, Mitsuda H, Yomo T, Negoro S, Urabe I: Insertion sequence IS6100 on plasmid pOAD2, which degrades nylon oligomers. J Bacteriol. 1994, 176 (4): 1197-1200.
Hall RM, Brown HJ, Brookes DE, Stokes HW: Integrons found in different locations have identical 5' ends but variable 3' ends. J Bacteriol. 1994, 176 (20): 6286-6294.
Sundin GW, Bender CL: Expression of the strA-strB streptomycin resistance genes in Pseudomonas syringae and Xanthomonas campestris and characterization of IS6100 in X. campestris. Appl Environ Microbiol. 1995, 61 (8): 2891-2897.
Sorum H, L'Abee-Lund TM, Solberg A, Wold A: Integron-containing IncU R plasmids pRAS1 and pAr-32 from the fish pathogen Aeromonas salmonicida. Antimicrob Agents Chemother. 2003, 47 (4): 1285-1290. 10.1128/AAC.47.4.1285-1290.2003.
Galas DJ, Chandler M: Bacterial insertion sequences. Mobile DNA. Edited by: Berg DE, Howe MM. 1989, Washington, D.C., USA , American Society for Microbiology, 109-162.
Tauch A, Götker S, Pühler A, Kalinowski J, Thierbach G: The 27.8-kb R-plasmid pTET3 from Corynebacterium glutamicum encodes the aminoglycoside adenyltransferase gene cassette aadA9 and the regulated tetracycline efflux system Tet 33 flanked by active copies of the widespread insertion sequence IS6100. Plasmid. 2002, 48 (2): 117-129. 10.1016/S0147-619X(02)00120-8.
Sassetti CM, Boyd DH, Rubin EJ: Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003, Cold Spring Harbor, N.Y., USA , Cold Spring Harbor Laboratory Press, 48 (1): 77-84. 10.1046/j.1365-2958.2003.03425.x. 2nd
Rigoutsos I, Floratos A: Combinatorial pattern discovery in biological sequences: The TEIRESIAS algorithm. Bioinformatics. 1998, 14 (1): 55-67. 10.1093/bioinformatics/14.1.55. [http://cbcsrv.watson.ibm.com/Tspd.html]
Holliday R: A new method for the identification of biochemical mutants of microorganisms. Nature. 1956, 178: 987-10.1038/178987a0.
Fernandes PJ, Powell JA, Archer JA: Construction of Rhodococcus random mutagenesis libraries using Tn5 transposition complexes. Microbiology. 2001, 147 (Pt 9): 2529-2536.
Desomer J, Crespi M, Van Montagu M: Illegitimate integration of non-replicative vectors in the genome of Rhodococcus fascians upon electrotransformation as an insertional mutagenesis system. Mol Microbiol. 1991, 5 (9): 2115-2124.
Fujio T, Nishi T, Ito S, Maruyama A: High level expression of XMP aminase in Escherichia coli and its application for the industrial production of 5'-guanylic acid. Biosci Biotechnol Biochem. 1997, 61 (5): 840-845.
Bachmann BJ, Low KB: Linkage map of Escherichia coli K-12, edition 6. Microbiol Rev. 1980, 44 (1): 1-56.
Gigot D, Crabeel M, Feller A, Charlier D, Lissens W, Glansdorff N, Pierard A: Patterns of polarity in the Escherichia coli car AB gene cluster. J Bacteriol. 1980, 143 (2): 914-920.
Cordes C, Möckel B, Eggeling L, Sahm H: Cloning, organization and functional analysis of ilvA, ilvB and ilvC genes from Corynebacterium glutamicum. Gene. 1992, 112 (1): 113-116. 10.1016/0378-1119(92)90311-C.
Rückert C, Pühler A, Kalinowski J: Genome-wide analysis of the L-methionine biosynthetic pathway in Corynebacterium glutamicum by targeted gene deletion and homologous complementation. J Biotechnol. 2003, 104 (1-3): 213-228. 10.1016/S0168-1656(03)00158-5.
Miranda-Rios J, Morera C, Taboada H, Davalos A, Encarnacion S, Mora J, Soberon M: Expression of thiamin biosynthetic genes (thiCOGE) and production of symbiotic terminal oxidase cbb3 in Rhizobium etli. J Bacteriol. 1997, 179 (22): 6887-6893.
Heery DM, Dunican LK: Cloning of the trp gene cluster from a tryptophan-hyperproducing strain of Corynebacterium glutamicum: identification of a mutation in the trp leader sequence. Appl Environ Microbiol. 1993, 59 (3): 791-799.
Hobom U, Brune W, Messerle M, Hahn G, Koszinowski UH: Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J Virol. 2000, 74 (17): 7720-7729. 10.1128/JVI.74.17.7720-7729.2000.
Heery DM, Fitzpatrick R, Dunican LK: A sequence from a tryptophan-hyperproducing strain of Corynebacterium glutamicum encoding resistance to 5-methyltryptophan. Biochem Biophys Res Commun. 1994, 201 (3): 1255-1262. 10.1006/bbrc.1994.1840.
Hartmann M, Tauch A, Eggeling L, Bathe B, Möckel B, Pühler A, Kalinowski J: Identification and characterization of the last two unknown genes, dapC and dapF, in the succinylase branch of the L-lysine biosynthesis of Corynebacterium glutamicum. J Biotechnol. 2003, 104 (1-3): 199-211. 10.1016/S0168-1656(03)00156-1.
McHardy AC, Tauch A, Rückert C, Pühler A, Kalinowski J: Genome-based analysis of biosynthetic aminotransferase genes of Corynebacterium glutamicum. J Biotechnol. 2003, 104 (1-3): 229-240. 10.1016/S0168-1656(03)00161-5.
Marienhagen J, Kennerknecht N, Sahm H, Eggeling L: Functional analysis of all aminotransferase proteins inferred from the genome sequence of Corynebacterium glutamicum. J Bacteriol. 2005, 187 (22): 7639-7646. 10.1128/JB.187.22.7639-7646.2005.
Alifano P, Fani R, Lio P, Lazcano A, Bazzicalupo M, Carlomagno MS, Bruni CB: Histidine biosynthetic pathway and genes: structure, regulation, and evolution. Microbiol Rev. 1996, 60 (1): 44-69.
Kanehisa M: The KEGG database. Novartis Found Symp. 2002, 247: 91-101; discussion 101-3, 119-28, 244-52.
Horton RM: PCR-mediated recombination and mutagenesis. SOEing together tailor-made genes. Mol Biotechnol. 1995, 3 (2): 93-99.
Kirchner O, Tauch A: Tools for genetic engineering in the amino acid-producing bacterium Corynebacterium glutamicum. J Biotechnol. 2003, 104 (1-3): 287-299. 10.1016/S0168-1656(03)00148-2.
Neuwald AF, Krishnan BR, Brikun I, Kulakauskas S, Suziedelis K, Tomcsanyi T, Leyh TS, Berg DE: cysQ, a gene needed for cysteine synthesis in Escherichia coli K-12 only during aerobic growth. J Bacteriol. 1992, 174 (2): 415-425.
Parish T, Liu J, Nikaido H, Stoker NG: A Mycobacterium smegmatis mutant with a defective inositol monophosphate phosphatase gene homolog has altered cell envelope permeability. J Bacteriol. 1997, 179 (24): 7827-7833.
Nigou J, Besra GS: Characterization and regulation of inositol monophosphatase activity in Mycobacterium smegmatis. Biochem J. 2002, 361 (Pt 2): 385-390.
Weaden J, Dyson P: Transposon mutagenesis with IS6100 in the avermectin-producer Streptomyces avermitilis. Microbiology. 1998, 144 ( Pt 7): 1963-1970.
Suzuki N, Okai N, Nonaka H, Tsuge Y, Inui M, Yukawa H: High-throughput transposon mutagenesis of Corynebacterium glutamicum and construction of a single-gene disruptant mutant library. Appl Environ Microbiol. 2006, 72 (5): 3750-3755. 10.1128/AEM.72.5.3750-3755.2006.
Herron PR, Evans MC, Dyson PJ: Low target site specificity of an IS6100-based mini-transposon, Tn1792, developed for transposon mutagenesis of antibiotic-producing Streptomyces. FEMS Microbiol Lett. 1999, 171 (2): 215-221. 10.1111/j.1574-6968.1999.tb13435.x.
Smith B, Dyson P: Inducible transposition in Streptomyces lividans of insertion sequence IS6100 from Mycobacterium fortuitum. Mol Microbiol. 1995, 18 (5): 933-941. 10.1111/j.1365-2958.1995.18050933.x.
Newell PC, Tucker RG: New pyrimidine pathway involved in the biosynthesis of the pyrimidine of thiamine. Nature. 1967, 215 (108): 1384-1385. 10.1038/2151384a0.
Downs DM, Petersen L: apbA, a new genetic locus involved in thiamine biosynthesis in Salmonella typhimurium. J Bacteriol. 1994, 176 (16): 4858-4864.
Enos-Berlage JL, Downs DM: Involvement of the oxidative pentose phosphate pathway in thiamine biosynthesis in Salmonella typhimurium. J Bacteriol. 1996, 178 (5): 1476-1479.
Berg DE, Weiss A, Crossland L: Polarity of Tn5 insertion mutations in Escherichia coli. J Bacteriol. 1980, 142 (2): 439-446.
Grindley ND, Reed RR: Transpositional recombination in prokaryotes. Annu Rev Biochem. 1985, 54: 863-896. 10.1146/annurev.bi.54.070185.004243.
Rückert C, Koch DJ, Rey DA, Albersmeier A, Mormann S, Pühler A, Kalinowski J: Functional genomics and expression analysis of the Corynebacterium glutamicum fpr2-cysIXHDNYZ gene cluster involved in assimilatory sulphate reduction. BMC Genomics. 2005, 6: 121-10.1186/1471-2164-6-121.
Majerus PW: Inositol phosphate biochemistry. Annu Rev Biochem. 1992, 61: 225-250. 10.1146/annurev.bi.61.070192.001301.
Brennan PJ, Nikaido H: The envelope of mycobacteria. Annu Rev Biochem. 1995, 64: 29-63. 10.1146/annurev.bi.64.070195.000333.
Chatterjee D, Khoo KH: Mycobacterial lipoarabinomannan: an extraordinary lipoheteroglycan with profound physiological effects. Glycobiology. 1998, 8 (2): 113-120. 10.1093/glycob/8.2.113.
Vercellone A, Nigou J, Puzo G: Relationships between the structure and the roles of lipoarabinomannans and related glycoconjugates in tuberculosis pathogenesis. Front Biosci. 1998, 3: e149-63.
Nigou J, Gilleron M, Puzo G: Lipoarabinomannans: from structure to biosynthesis. Biochimie. 2003, 85 (1-2): 153-166. 10.1016/S0300-9084(03)00048-8.
Nampoothiri KM, Hoischen C, Bathe B, Möckel B, Pfefferle W, Krumbach K, Sahm H, Eggeling L: Expression of genes of lipid synthesis and altered lipid composition modulates L-glutamate efflux of Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2002, 58 (1): 89-96. 10.1007/s00253-001-0861-z.
Yague G, Segovia M, Valero-Guillen PL: Phospholipid composition of several clinically relevant Corynebacterium species as determined by mass spectrometry: an unusual fatty acyl moiety is present in inositol-containing phospholipids of Corynebacterium urealyticum. Microbiology. 2003, 149 (Pt 7): 1675-1685. 10.1099/mic.0.26206-0.
Chen IW, Charalampous CF: Biochemical studies on inositol. IX. D-Inositol 1-phosphate as intermediate in the biosynthesis of inositol from glucose 6-phosphate, and characteristics of two reactions in this biosynthesis. J Biol Chem. 1966, 241 (10): 2194-2199.
Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, de Crecy-Lagard V, Diaz N, Disz T, Edwards R, Fonstein M, Frank ED, Gerdes S, Glass EM, Goesmann A, Hanson A, Iwata-Reuyl D, Jensen R, Jamshidi N, Krause L, Kubal M, Larsen N, Linke B, McHardy AC, Meyer F, Neuweger H, Olsen G, Olson R, Osterman A, Portnoy V, Pusch GD, Rodionov DA, Rückert C, Steiner J, Stevens R, Thiele I, Vassieva O, Ye Y, Zagnitko O, Vonstein V: The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33 (17): 5691-5702. 10.1093/nar/gki866.
Nigou J, Dover LG, Besra GS: Purification and biochemical characterization of Mycobacterium tuberculosis SuhB, an inositol monophosphatase involved in inositol biosynthesis. Biochemistry. 2002, 41 (13): 4392-4398. 10.1021/bi0160056.
Haft DH, Selengut JD, Brinkac LM, Zafar N, White O: Genome Properties: a system for the investigation of prokaryotic genetic content for microbiology, genome annotation and comparative genomics. Bioinformatics. 2005, 21 (3): 293-306. 10.1093/bioinformatics/bti015.
Sambrook J, Fritsch EF, Maniatis T: Molecular Cloning: A Laboratory Manual. 1989, Cold Spring Harbor, NY, USA , Cold Spring Harbor Laboratory Press, 2nd
Tauch A, Kirchner O, Wehmeier L, Kalinowski J, Pühler A: Corynebacterium glutamicum DNA is subjected to methylation-restriction in Escherichia coli. FEMS Microbiol Lett. 1994, 123 (3): 343-347. 10.1111/j.1574-6968.1994.tb07246.x.
Tauch A, Kirchner O, Löffler B, Götker S, Pühler A, Kalinowski J: Efficient electrotransformation of Corynebacterium diphtheriae with a mini-replicon derived from the Corynebacterium glutamicum plasmid pGA1. Curr Microbiol. 2002, 45 (5): 362-367. 10.1007/s00284-002-3728-3.
Tauch A, Homann I, Mormann S, Rüberg S, Billault A, Bathe B, Brand S, Brockmann-Gretza O, Rückert C, Schischka N, Wrenger C, Hoheisel J, Möckel B, Huthmacher K, Pfefferle W, Pühler A, Kalinowski J: Strategy to sequence the genome of Corynebacterium glutamicum ATCC 13032: use of a cosmid and a bacterial artificial chromosome library. J Biotechnol. 2002, 95 (1): 25-38. 10.1016/S0168-1656(01)00443-6.
Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Rapp BA, Wheeler DL: GenBank. Nucleic Acids Res. 2000, 28 (1): 15-8. 10.1093/nar/28.1.15. [http://www.ncbi.nlm.nih.gov/Genbank/index.html]
Bairoch A, Apweiler R: The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28 (1): 45-8. [http://www.expasy.ch/sprot]. 10.1093/nar/28.1.45.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25 (17): 3389-3402. 10.1093/nar/25.17.3389.
Morgenstern B: DIALIGN 2: improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics. 1999, 15 (3): 211-218. 10.1093/bioinformatics/15.3.211.
Saitou N, Nei M: The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987, 4 (4): 406-425.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25 (24): 4876-4882. 10.1093/nar/25.24.4876.
TreeTool - Interactive phylogenetic tree plotting program. [http://rdp8.cme.msu.edu/download/programs/TreeTool/]
Katsumata R, Ozaki A, Oka T, Furuya A: Protoplast transformation of glutamate-producing bacteria with plasmid DNA. J Bacteriol. 1984, 159 (1): 306-311.
Tatusov RL, Galperin MY, Natale DA, Koonin EV: The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28 (1): 33-36. 10.1093/nar/28.1.33.
Meyer F, Goesmann A, McHardy AC, Bartels D, Bekel T, Clausen J, Kalinowski J, Linke B, Rupp O, Giegerich R, Pühler A: GenDB--an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003, 31 (8): 2187-2195. 10.1093/nar/gkg312.
Flachmann R, Kunz N, Seifert J, Gütlich M, Wientjes FJ, Laufer A, Gassen HG: Molecular biology of pyridine nucleotide biosynthesis in Escherichia coli. Cloning and characterization of quinolinate synthesis genes nadA and nadB. Eur J Biochem. 1988, 175 (2): 221-228. 10.1111/j.1432-1033.1988.tb14187.x.
Taylor AL, Trotter CD: Linkage map of Escherichia coli strain K-12. Bacteriol Rev. 1972, 36 (4): 504-524.
Keer J, Smeulders MJ, Williams HD: A purF mutant of Mycobacterium smegmatis has impaired survival during oxygen-starved stationary phase. Microbiology. 2001, 147 (Pt 2): 473-481.
Chun JY, Lee MS: Cloning of the argF gene encoding the ornithine carbamoyltransferase from Corynebacterium glutamicum. Mol Cells. 1999, 9 (3): 333-337.
Liu DX, Fan CS, Tao JH, Liang GX, Gao SE, Wang HJ, Li X, Song DX: Integration of E. coli aroG-pheA tandem genes into Corynebacterium glutamicum tyrA locus and its effect on L-phenylalanine biosynthesis. World J Gastroenterol. 2004, 10 (24): 3683-3687.
Follettie MT, Sinskey AJ: Molecular cloning and nucleotide sequence of the Corynebacterium glutamicum pheA gene. J Bacteriol. 1986, 167 (2): 695-702.
Jung SI, Han MS, Kwon JH, Cheon CI, Min KH, Lee MS: Cloning of the histidine biosynthetic genes of Corynebacterium glutamicum: organization and sequencing analysis of the hisA, impA, and hisF gene cluster. Biochem Biophys Res Commun. 1998, 247 (3): 741-745. 10.1006/bbrc.1998.8850.
Ankri S, Serebrijski I, Reyes O, Leblon G: Mutations in the Corynebacterium glutamicum proline biosynthetic pathway: a natural bypass of th proA step. J Bacteriol. 1996, 178 (15): 4412-4419.
Peters-Wendisch P, Netzer R, Eggeling L, Sahm H: 3-Phosphoglycerate dehydrogenase from Corynebacterium glutamicum: the C-terminal domain is not essential for activity but is required for inhibition by L-serine. Appl Microbiol Biotechnol. 2002, 60 (4): 437-441. 10.1007/s00253-002-1161-y.
Patek M, Krumbach K, Eggeling L, Sahm H: Leucine synthesis in Corynebacterium glutamicum: enzyme activities, structure of leuA, and effect of leuA inactivation on lysine synthesis. Appl Environ Microbiol. 1994, 60 (1): 133-140.
Peoples OP, Liebl W, Bodis M, Maeng PJ, Follettie MT, Archer JA, Sinskey AJ: Nucleotide sequence and fine structural analysis of the Corynebacterium glutamicum hom-thrB operon. Mol Microbiol. 1988, 2 (1): 63-72.
Yeh P, Sicard AM, Sinskey AJ: Nucleotide sequence of the lysA gene of Corynebacterium glutamicum and possible mechanisms for modulation of its expression. Mol Gen Genet. 1988, 212 (1): 112-119. 10.1007/BF00322452.
Pierard A, Glansdorff N, Gigot D, Crabeel M, Halleux P, Thiry L: Repression of Escherichia coli carbamoylphosphate synthase: relationships with enzyme synthesis in the arginine and pyrimidine pathways. J Bacteriol. 1976, 127 (1): 291-301.
Grant SGN, Jessee J, Bloom FR, Hanahan D: Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc Natl Acad Sci USA. 1990, 87: 4645–4649-10.1073/pnas.87.12.4645.
Acknowledgements
We like to thank Prof. Göran Kauermann (StatBeCE, Universität Bielefeld) for help with statistical analyses and the Degussa AG as well as the Bundesministerium für Bildung und Forschung for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
SM carried out the experimental work and drafted the manuscript. AL participated during plasmid rescue analyses. CR constructed the cg0910 deletion mutant and performed the phylogenetic analyses. LG performed the cointegrate stability assays. AT provided the transposon vector and participated in supervision. AP aided in coordination and participated in supervision. JK conceived of the study and participated in coordination and supervision. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Mormann, S., Lömker, A., Rückert, C. et al. Random mutagenesis in Corynebacterium glutamicum ATCC 13032 using an IS6100-based transposon vector identified the last unknown gene in the histidine biosynthesis pathway. BMC Genomics 7, 205 (2006). https://doi.org/10.1186/1471-2164-7-205
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-7-205