
Abstract
Halopiger xanaduensis is the type species of the genus Halopiger and belongs to the euryarchaeal family Halobacteriaceae. H. xanaduensis strain SH-6, which is designated as the type strain, was isolated from the sediment of a salt lake in Inner Mongolia, Lake Shangmatala. Like other members of the family Halobacteriaceae, it is an extreme halophile requiring at least 2.5 M salt for growth. We report here the sequencing and annotation of the 4,355,268 bp genome, which includes one chromosome and three plasmids. This genome is part of a Joint Genome Institute (JGI) Community Sequencing Program (CSP) project to sequence diverse haloarchaeal genomes.
Similar content being viewed by others
Introduction
Halopiger xanaduensis is the type species of the genus Halopiger, and strain SH-6 is the type strain of the species. It was isolated from the sediment of a salt lake, Lake Shangmatala, in Inner Mongolia, China [1]. The name Halopiger refers to its slow growth in the laboratory. There is one other described species in the genus Halopiger, H. aswanensis, which was isolated from a saline soil in Egypt [2]. We report here the first genome sequence from the genus Halopiger.
Classification and features
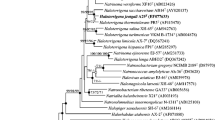
In 16S rRNA trees the Halopiger species are most closely related to Natronolimnobius species [1,2]. Currently there are fifteen complete genomes of haloarchaea in GenBank. Figure 1 shows the relationship of H. xanaduensis to other haloarchaea for which complete genomes have been sequenced. For Halobacterium salinarum and Haloquadratum walsbyi, only one sequence is included in Figure 1, although for both of these species two genomes have been sequenced.

Phylogenetic tree showing the relationships between haloarchaea with sequenced genomes. The sequences were aligned with the Ribosomal Database Project (RDP) aligner [3], which uses the Jukes-Cantor corrected distance model to construct a distance matrix based on alignment model positions without the use of alignment inserts, and uses a minimum comparable position of 200. The tree was generated with the Tree Builder from the RDP which uses Weighbor [4] with an alphabet size of 4 and length size of 1,000. The building of the tree also involves a bootstrapping process repeated 100 times to generate a majority consensus tree. Methanosarcina acetivorans was used as the outgroup.
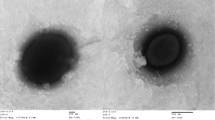
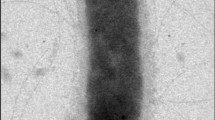
H. xanaduensis was isolated from a sediment sample of Lake Shangmatala in Inner Mongolia, China. The sample was enriched in liquid medium containing salts and yeast extract; the culture was then plated on agar to obtain pure colonies [1]. At the time of sample collection, the salinity of the lake was 16.7%, the temperature was 21.8°C, and the pH was 8.5 [1]. The cells were pleomorphic with the most common shape being rods. Motility was not observed [1]. An electron micrograph is shown in Figure 2. Growth was observed between 28 and 45°C with an optimum at 37°C [1]. The pH range for growth was 6.0–11.0 with an optimal pH of 7.5–8.0 [1]. Growth occurred within a salinity range of 2.5 M to 5.0 M NaCl and was optimal at 4.3M NaCl [1]. The organism is strictly aerobic but was able to reduce nitrate and nitrite with production of gas [1]. Several sugars and amino acids can serve as sole carbon and energy sources, and amino acids are not required in the growth medium [1]. The features of the organism are listed in Table 1.

Electron micrograph of H. xanaduensis SH-6.
Genome sequencing information
Genome project history
H. xanaduensis was selected for sequencing as part of a JGI CSP project to sequence a representative from every genus of haloarchaea. The genome project is listed in the Genomes On Line Database [16], and the complete genome sequence has been deposited in GenBank. Sequencing was carried out at the JGI Production Genomics Facility (PGF). Finishing was done at Los Alamos National Laboratory. Annotation was done at both the PGF and Oak Ridge National Laboratory. Table 2 presents the project information and its association with MIGS version 2.0 compliance [5].
Growth conditions and DNA isolation
Cells were grown in DSMZ medium 372 (Halobacteria medium) [17] at 37°C. DNA was isolated from 1.0–1.5 g cell paste with the MasterPure Gram Positive DNA Purification Kit (Epicentre).
Genome sequencing and assembly
The draft genome of Halopiger xanaduensis SH-6 was generated at the DOE Joint genome Institute (JGI) using a combination of Illumina [18] and 454 technologies [19]. For this genome we constructed and sequenced an Illumina GAII shotgun library which generated 55,857,474 reads totaling 4,245.2 Mb, a 454 Titanium standard library which generated 159,242 reads, and 1 paired end 454 library with an average insert size of 8 kb which generated 341,165 reads totaling 141.8 Mb of 454 data. All general aspects of library construction and sequencing performed at the JGI can be found at the JGI website [20]. The initial draft assembly contained 15 contigs in 2 scaffolds. The 454 Titanium standard data and the 454 paired end data were assembled together with Newbler, version 2.3-PreRelease-6/30/2009. The Newbler consensus sequences were computationally shredded into 2 kb overlapping fake reads (shreds). Illumina sequencing data was assembled with VELVET, version 1.0.13 [21], and the consensus sequences were computationally shredded into 1.5 kb overlapping fake reads (shreds). We integrated the 454 Newbler consensus shreds, the Illumina VELVET consensus shreds and the read pairs in the 454 paired end library using parallel phrap, version 1.080812 (High Performance Software, LLC). The software Consed [22–24] was used in the following finishing process. Illumina data was used to correct potential base errors and increase consensus quality using the software Polisher developed at JGI (Alla Lapidus, unpublished). Possible mis-assemblies were corrected using gapResolution (Cliff Han, unpublished), Dupfinisher [25], or sequencing cloned bridging PCR fragments with subcloning. Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR (Jan-Fang Cheng, unpublished) primer walks. A total of 64 additional reactions were necessary to close gaps and to raise the quality of the finished sequence. The total size of the genome is 4,355,268 bp and the final assembly is based on 117.9 Mb of 454 draft data which provides an average 26.8× coverage of the genome and 4,112.2 Mb of Illumina draft data which provides an average 934.6× coverage of the genome.
Genome annotation
Genes were identified using Prodigal [26], followed by a round of manual curation using GenePRIMP [27]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. The tRNAScan-SE tool [28] was used to find tRNA genes, whereas ribosomal RNAs were found by using BLASTn against the ribosomal RNA databases. The RNA components of the protein secretion complex and the RNase P were identified by searching the genome for the corresponding Rfam profiles using INFERNAL [29]. Additional gene prediction analysis and manual functional annotation was performed within the Integrated Microbial Genomes (IMG) platform [30] developed by the JGI [31].
Genome properties
The genome includes one circular chromosome and three plasmids, for a total size of 4,355,268 bp (Table 3, Table 4). A map of the chromosome is shown in Figure 3 and maps of the plasmids are shown in Figures 4, 5, and 6. A total of 4,370 genes were predicted, 4,310 of which are protein-coding genes and 60 of which are RNA genes. There are three ribosomal RNA operons with one additional copy of 5S rRNA. Putative functions were assigned to 2,560 protein coding genes, with the remaining genes annotated as hypothetical proteins. There are 89 pseudogenes, accounting for 2.06% of protein-coding genes. Table 5 shows the distribution of genes in COG categories.

Graphical circular map of the chromosome. From outside to center: Genes on forward strand (colored by COG categories), genes on reverse strand (colored by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, and GC skew.

Graphical circular map of plasmid pHALXA01. From outside to center: Genes on forward strand (colored by COG categories), genes on reverse strand (colored by COG categories), GC content, and GC skew.

Graphical circular map of plasmid pHALXA02. From outside to center: Genes on forward strand (colored by COG categories), genes on reverse strand (colored by COG categories), GC content, and GC skew.

Graphical circular map of plasmid pHALXA03. From outside to center: Genes on forward strand (colored by COG categories), genes on reverse strand (colored by COG categories), GC content, and GC skew.
Genome analysis
H. xanaduensis grows on only a few of the carbohydrates that were tested (glucose, galactose, and xylose) [1], but surprisingly it has 40 glycosyl hydrolases and 5 polysaccharide lyases [32]. It also has quite a large number of ABC transporters for carbohydrates: 10 full transporters and one additional substrate-binding protein. Among the sequenced haloarchaea, only Haloferax volcanii has a greater number of carbohydrate ABC transporters [33]. Taken together, these findings suggest that H. xanaduensis is capable of growth on other sugars that have not been tested.
While many of the glycosyl hydrolases have no characterized close homologs, for some of them, functions can be predicted. Halxa_0484 has 73% similarity to beta-galactosidase of Haloferax lucentense [34], while Halxa_3778 has 75% similarity to a xylanase from Streptomyces sp. S27 [35]. Two of the polysaccharide lyases from family PL11 have greater than 65% similarity to rhamnogalacturonan lyases YesW and YesX from Bacillus subtilis [36], suggesting that H. xanaduensis may be capable of pectin degradation.
Degradation pathways for the three sugars that H. xanaduensis is known to utilize can be identified in the genome. Glucose is likely degraded by the semiphosphorylated Entner-Doudoroff pathway as in other haloarchaea [37]. Three enzymes of the pathway, glucose dehydrogenase, gluconate dehydratase, and 2-keto-3-deoxyphosphogluconate aldolase, are found in an operon (Halxa_4119-4121). The 2-keto-3-deoxygluconate kinase is found elsewhere in the genome (Halxa_2064). Galactose is probably metabolized via the De Ley-Doudoroff pathway as a galactonate dehydratase is present (Halxa_3608). Adjacent to this gene are a possible alpha-galactosidase (Halxa_3609) and a kinase and aldolase that may take part in this pathway (Halxa_3607, Halxa_3606). Xylose utilization appears to be via the pathway found in H. volcanii [38] which results in formation of 2-oxoglutarate. Again three enzymes of the pathway form an operon - xylonate dehydratase, 2-keto-3-deoxyxylonate dehydratase, and 2,5-dioxopentanoate dehydrogenase (Halxa_3763-3765).
Despite the fact that it was isolated from lake sediment, H. xanaduensis has an operon of gas vesicle proteins (Halxa_0820-0830). It is lacking GvpC, GvpD, GvpE, and GvpH, but mutation studies have shown that these four proteins are not required for gas vesicle formation [39], so H. xanaduensis can probably form functional gas vesicles. This suggests that H. xanaduensis may spend part of its life close to the surface of the lake.
H. xanaduensis has several genes involved in polysaccharide synthesis and transport that are not found in any other sequenced haloarchaea. It has two genes (Halxa_0209, Halxa_2361) belonging to the Capsular Polysaccharide Exporter family (TC 9.A.41). This is unusual as one of the members of this family is thought to transport polysaccharide across the outer membrane of Gram-negative bacteria [40]. Adjacent to these two exporter genes are two genes (Halxa_0208, Halxa_2362) belonging to COG1861, cytidylyl transferases involved in polysaccharide biosynthesis. H. xanaduensis also has one gene (Halxa_2364) related to PseG, a UDP-sugar hydrolase involved in polysaccharide production [41]. The presence of these genes in H. xanaduensis suggests that it may be capable of extracellular polysaccharide synthesis using a process unlike any found in other haloarchaea.
References
Gutiérrez MC, Castillo AM, Kamekura M, Xue Y, Ma Y, Cowan DA, Jones BE, Grant WD, Ventosa A. Halopiger xanaduensis gen. nov., sp. nov., an extremely halophilic archaeon isolated from saline Lake Shangmatala in Inner Mongolia, China. Int J Syst Evol Microbiol 2007; 57:1402–1407. PubMed http://dx.doi.org/10.1099/ijs.0.65001-0
Hezayen FF, Gutiérrez MC, Steinbüchel A, Tindall BJ, Rehm BHA. Halopiger aswanensis sp. nov., a polymer-producing and extremely halophilic archaeon isolated from hypersaline soil. Int J Syst Evol Microbiol 2010; 60:633–637. PubMed http://dx.doi.org/10.1099/ijs.0.013078-0
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 2009; 37(Database issue):D141–D145. PubMed http://dx.doi.org/10.1093/nar/gkn879
Bruno WJ, Socci ND, Halpern AL. Weighted neighbor joining: a likelihood-based approach to distance-based phylogeny reconstruction. Mol Biol Evol 2000; 17:189–197. PubMed
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed http://dx.doi.org/10.1038/nbt1360
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed http://dx.doi.org/10.1073/pnas.87.12.4576
Garrity GM, Holt JG. Phylum AII. Euryarchaeota phy. nov. In: Bergey’s Manual of Systematic Bacteriology, vol. 1, 2nd ed. Edited by: Garrity GM, Boone DR, and Castenholz RW. Springer, New York; 2001:211–355.
List Editor. Validation of publication of new names and new combinations previously effectively published outside the IJSEM. Validation List no. 85. Int J Syst Evol Microbiol 2002; 52:685–690. PubMed http://dx.doi.org/10.1099/ijs.0.02358-0
Grant WD, Kamekura M, McGenity TJ, Ventosa A. Class III. Halobacteria class. nov. In: Bergey’s Manual of Systematic Bacteriology, vol. 1, 2nd ed. Edited by: Garrity GM, Boone DR, and Castenholz RW. Springer, New York; 2001:294.
Grant WD, Larsen H. Extremely halophilic archaebacteria, order Halobacteriales ord. nov. In: Bergey’s Manual of Systematic Bacteriology, vol. 3, 1st ed. Edited by: Staley JT, Bryant MP, Pfennig N, and Holt JG. Williams & Wilkins, Baltimore, MD 1989:2216–2228.
Validation List no. 31. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. Int J Syst Bacteriol 1989; 39:495–497. http://dx.doi.org/10.1099/00207713-39-4-495
Judicial Commission of the International Committee on Systematics of Prokaryotes. The nomenclatural types of the orders Acholeplasmatales, Halanaerobiales, Halobacteriales, Methanobacteriales, Methanococcales, Methanomicrobiales, Planctomycetales, Prochlorales, Sulfolobales, Thermococcales, Thermoproteales and Verrucomicrobiales are the genera Acholeplasma, Halanaerobium, Halobacterium, Methanobacterium, Methanococcus, Methanomicrobium, Planctomyces, Prochloron, Sulfolobus, Thermococcus, Thermoproteus and Verrucomicrobium, respectively. Opinion 79. Int J Syst Evol Microbiol 2005; 55:517–518. PubMed http://dx.doi.org/10.1099/ijs.0.63548-0
Gibbons NE. Family V. Halobacteriaceae fam. nov. In: Bergey’s Manual of Determinative Bacteriology, 8th ed. Edited by: Buchanan RE and Gibbons NE. Williams & Wilkins, Baltimore, MD 1974:269–273.
Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol 1980; 30:225–420. http://dx.doi.org/10.1099/00207713-30-1-225
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25–29. PubMed http://dx.doi.org/10.1038/75556
Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Hugenholtz P, Markowitz VM, Kyrpides NC. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2010; 38(Database Issue):D346–D354. PubMed http://dx.doi.org/10.1093/nar/gkp848
List of growth media used at DSMZ: http://www.dsmz.de/microorganisms/media_list.php
Bennett S. Solexa Ltd. Pharmacogenomics 2004; 5:433–438. PubMed http://dx.doi.org/10.1517/14622416.5.4.433
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005; 437:376–380. PubMed
DOE Joint Genome Institute. http://www.jgi.doe.gov
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821–829. PubMed http://dx.doi.org/10.1101/gr.074492.107
Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 1998; 8:175–185. PubMed
Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 1998; 8:186–194. PubMed
Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res 1998; 8:195–202. PubMed
Han C, Chain P. Finishing repeat regions automatically with Dupfinisher. In Proceedings of the 2006 international conference on bioinformatics and computational biology, ed. Arabnia HR, Valafar H. CSREA Press, 2006:141–146.
Hyatt D, Chen GL, Lacascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119. PubMed http://dx.doi.org/10.1186/1471-2105-11-119
Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455–457. PubMed http://dx.doi.org/10.1038/nmeth.1457
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997; 25:955–964. PubMed http://dx.doi.org/10.1093/nar/25.5.955
INFERNAL. Inference of RNA alignments. http://infernal.janelia.org
Integrated Microbial Genomes (IMG). http://img.jgi.doe.gov
Markowitz VM, Mavromatis K, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271–2278. PubMed http://dx.doi.org/10.1093/bioinformatics/btp393
Carbohydrate-Active enZYmes Database. http://www.cazy.org
Hartman AL, Norais C, Badger JH, Delmas S, Haldenby S, Madupu R, Robinson J, Khouri H, Ren Q, Lowe TM, et al. The complete genome sequence of Haloferax volcanii DS2, a model archaeon. PLoS ONE 2010; 5:e9605. PubMed http://dx.doi.org/10.1371/journal.pone.0009605
Holmes ML, Dyall-Smith ML. Sequence and expression of a halobacterial beta-galactosidase gene. Mol Microbiol 2000; 36:114–122. PubMed http://dx.doi.org/10.1046/j.1365-2958.2000.01832.x
Li N, Shi P, Yang P, Wang Y, Luo H, Bai Y, Zhou Z, Yao B. Cloning, expression, and characterization of a new Streptomyces sp. S27 xylanase for which xylobiose is the main hydrolysis product. Appl Biochem Biotechnol 2009; 159:521–531. PubMed http://dx.doi.org/10.1007/s12010-008-8411-0
Ochiai A, Itoh T, Kawamata A, Hashimoto W, Murata K. Plant cell wall degradation by saprophytic Bacillus subtilis strains: gene clusters responsible for rhamnogalacturonan depolymerization. Appl Environ Microbiol 2007; 73:3803–3813. PubMed http://dx.doi.org/10.1128/AEM.00147-07
Falb M, Müller K, Königsmaier L, Oberwinkler T, Horn P, von Gronau S, Gonzalez O, Pfeiffer F, Bornberg-Bauer E, Oesterhelt D. Metabolism of halophilic archaea. Extremophiles 2008; 12:177–196. PubMed http://dx.doi.org/10.1007/s00792-008-0138-x
Johnsen U, Dambeck M, Zaiss H, Fuhrer T, Soppa J, Sauer U, Schönheit P. D-xylose degradation pathway in the halophilic archaeon Haloferax volcanii. J Biol Chem 2009; 284:27290–27303. PubMed http://dx.doi.org/10.1074/jbc.M109.003814
Offner S, Hofacker A, Wanner G, Pfeifer F. Eight of fourteen gvp genes are sufficient for formation of gas vesicles in halophilic archaea. J Bacteriol 2000; 182:4328–4336. PubMed http://dx.doi.org/10.1128/JB.182.15.4328-4336.2000
Sukupolvi-Petty S, Grass S, St Geme JW, III. The Haemophilus influenzae type b hcsA and hcsB gene products facilitate transport of capsular polysaccharide across the outer membrane and are essential for virulence. J Bacteriol 2006; 188:3870–3877. PubMed http://dx.doi.org/10.1128/JB.01968-05
Liu F, Tanner ME. PseG of pseudaminic acid biosynthesis: a UDP-sugar hydrolase as a masked glycosyltransferase. J Biol Chem 2006; 281:20902–20909. PubMed http://dx.doi.org/10.1074/jbc.M602972200
Acknowledgements
The work conducted by the U.S. Department of Energy Joint Genome Institute is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Anderson, I., Tindall, B.J., Rohde, M. et al. Complete genome sequence of Halopiger xanaduensis type strain (SH-6T). Stand in Genomic Sci 6, 31–42 (2012). https://doi.org/10.4056/sigs.2505605
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.2505605