
Abstract
Parvibaculum lavamentivorans DS-1T is the type species of the novel genus Parvibaculum in the novel family Rhodobiaceae (formerly Phyllobacteriaceae) of the order Rhizobiales of Alphaproteobacteria. Strain DS-1T is a non-pigmented, aerobic, heterotrophic bacterium and represents the first tier member of environmentally important bacterial communities that catalyze the complete degradation of synthetic laundry surfactants. Here we describe the features of this organism, together with the complete genome sequence and annotation. The 3,914,745 bp long genome with its predicted 3,654 protein coding genes is the first completed genome sequence of the genus Parvibaculum, and the first genome sequence of a representative of the family Rhodobiaceae.
Similar content being viewed by others

Introduction
Parvibaculum lavamentivorans strain DS-1T (DSM13023 = NCIMB13966) was isolated for its ability to degrade linear alkylbenzenesulfonate (LAS), a major laundry surfactant with a world-wide use of 2.5 million tons per annum [1]. Strain DS-1T was difficult to isolate, is difficult to cultivate, and represents a novel genus in the Alphaproteobacteria [2,3]. Strain DS-1 catalyzes not only the degradation of LAS, but also of 16 other commercially important anionic and non-ionic surfactants (hence the species name lavamentivorans = consuming [chemicals] used for washing [3]). The initial degradation as catalyzed by strain DS-1T involves the activation and shortening of the alkyl-chain of the surfactant molecules, and the excretion of short-chain degradation intermediates. These intermediates are then completely utilized by other bacteria in the community [4,5]. P. lavamentivorans DS-1T is therefore an example of a first tier member of a two-step process that mineralizes environmentally important surfactants.
Other representatives of the novel genus Parvibaculum have been recently isolated. Parvibaculum sp. strain JP-57 was isolated from seawater [6] and is also difficult to cultivate [3]. Parvibaculum indicum sp. nov. was also isolated from seawater, via an enrichment culture that degraded polycyclic aromatic hydrocarbons (PAH) and crude oil [7]. Another Parvibaculum sp. strain was isolated from a PAH-degrading enrichment culture, using river sediment as inoculum [8]. Parvibaculum species were also reported in a study on marine alkane-degrading bacteria [9]. Parvibaculum species are frequently detected by cultivation-independent methods, predominantly in habitats or settings with hydrocarbon degradation. These include a bacterial community on marine rocks polluted with diesel oil [10], a bacterial community from diesel-contaminated soil [11], a petroleum-degrading bacterial community from seawater [12], an oil-degrading cyanobacterial community [13] and biofilm communities in pipes of a district heating system [14]. Parvibaculum species have also been detected in denitrifying, linear-nonylphenol (NP) degrading enrichment cultures from NP-polluted river sediment [15] and in groundwater that had been contaminated by linear alkyl benzenes (LABs; non-sulfonated LAS] [16]. Additionally, Parvibaculum species were detected in biofilms that degraded polychlorinated biphenyls (PCBs) using pristine soil as inoculum [17], and in a PAH-degrading bacterial community from deep-sea sediment of the West Pacific [18]. Finally, Parvibaculum species were detected in an autotrophic Fe(II)-oxidizing, nitrate-reducing enrichment culture [19], as well as in Tunisian geothermal springs [20]. The widespread occurrence of Parvibaculum species in habitats or settings related to hydrocarbon degradation implies an important function and role of these organisms in environmental biodegradation, despite their attribute as being difficult to cultivate in a laboratory.
Here we present a summary classification and a set of features for P. lavamentivorans DS-1T, together with the description of a complete genome sequence and annotation. The genome sequencing and analysis was part of the Microbial Genome Program of the DOE Joint Genome Institute.
Classification and features
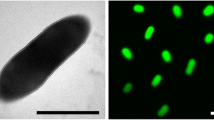
P. lavamentivorans DS-1T is a Gram-negative, non-pigmented, very small (approx. 1.0 × 0.2 µm), slightly curved rod-shaped bacterium that can be motile by means of a polar flagellum (Figure 1, Table 1). Strain DS-1T grows very slowly on complex medium (e.g. on LB- or peptone-agar plates) and forms pinpoint colonies only after more than two weeks of incubation. The organism can be quickly overgrown by other organisms. Larger colonies are obtained when the complex medium is supplemented with a surfactant, e.g. Tween 20 (see DSM-medium 884 [29]) or LAS [3]. When cultivated in liquid culture with mineral-salts medium, strain DS-1T grows within one week with the single carbon sources acetate, ethanol, or succinate, or alkanes, alkanols and alkanoates (C8–C16); no sugars tested were utilized [3].

Scanning electron micrograph of P. lavamentivorans DS-1T. Cells derived from a liquid culture that grew in acetate/mineral salts medium.
To allow for growth in liquid culture with most of the 16 different surfactants at high concentrations (e.g. for LAS, >1 mM; see [3].), the culture fluid needs to be supplemented with a solid surface, e.g. polyester fleece or glass fibers [2,3]. The additional solid surface is believed to support biofilm formation, especially in the early growth phase when the surfactant concentration is high, and the organism grows as single, suspended cells (non-motile) during the later growth phase. Growth with a non-membrane toxic substrate (e.g. acetate) is independent of a solid surface, and constitutes suspended, single cells (motile). We presume that the biofilm formation by strain DS-1T is a protective response to the exposure to membrane-solubilizing agents (cf. [30]).
Based on the 16S rRNA gene sequence, strain DS-1T was described as the novel genus Parvibaculum, which was originally placed in the family Phyllobacteriaceae within the order Rhizobiales of Alphaproteobacteria [3,31]. The nearest well-described organism to strain DS-1T is Afifella marina (formerly Rhodobium marinum) (92% 16S rRNA gene sequence identity), a photosynthetic purple, non-sulfur bacterium. The genus Rhodobium was later re-classified as a member of the novel family Rhodobiaceae [26,32], together with two novel genera of other photosynthetic purple non-sulfur bacteria (Afifella and Roseospirillum), as well as with two novel genera of heterotrophic aerobic bacteria, represented by the red-pigmented Anderseniella baltica (gen. nov., sp. nov.) [33,34] and non-pigmented Tepidamorphus gemmatus (gen. nov., sp. nov.) [35,36]. A phylogenetic tree (Figure 2) was constructed with the 16S rRNA gene sequence of P. lavamentivorans DS-1T and that of (i) other isolated Parvibaculum strains, (ii) representatives of other genera within the family Rhodobiaceae, (iii) representatives of the genera in the family Phyllobacteriaceae, as well as, (iv) representatives of other families within the order Rhizobiales. The phylogenetic tree shows now the placement of Parvibaculum species within the family Rhodobiaceae, and that the Parvibaculum sequences clustered as a distinct evolutionary lineage within this family (Figure 2). This classification of Parvibaculum has been adopted in the Ribosomal Database Project (RDP) and SILVA rRNA Database Project, but not in the GreenGenes database. The family Rhodobiaceae has also not been included in the NCBI-taxonomy, IMG-taxonomy, and GOLD databases.

Phylogenetic tree of 16S rRNA gene sequences showing the position of P. lavamentivorans DS-1T relative to other type strains within the families Rhodobiaceae, Phyllobacteriaceae and other families in the order Rhizobiales (see the text). Strains within the Rhodobiaceae and Phyllobacteriaceae shown in bold have genome projects underway or completed. The corresponding 16S rRNA gene accession numbers (or draft genome sequence identifiers) are indicated. The sequences were aligned using the GreenGenes NAST alignment tool [37]; neighbor-joining tree building and visualization involved the CLUSTAL and DENDROSCOPE software [38]. Caulobacterales sequences were used as outgroup. Bootstrap values >30 % are indicated; bar, 0.01 substitutions per nucleotide position.
Currently, 360 genome sequences of members of the order Rhizobiales of Alphaproteobacteria have been made available (GOLD database; August 2011), and within the family Phyllobacteriaceae there are 21 genome sequences available (Chelativorans sp. BNC1, Hoeflea phototrophica DFL-43, and 18 Mesorhizobium strains). No genome sequences currently exist for a representative of the novel family Rhodobiaceae, except of the genome of P. lavamentivorans DS-1T.
Chemotaxonomy
Examination of the respiratory lipoquinone composition of strain DS-1T showed that ubiquinones are the sole respiratory quinones present, and the major lipoquinone is ubiquinone 11 (Q11) [3]. The fatty acids of P. lavamentivorans are straight chain saturated and unsaturated, as well as ester- and amide-linked hydroxy-fatty acids, in membrane fractions [3]. The major polar lipids are phosphatidyl glycerol, diphosphatidyl glycerol, phosphatidyl ethanolamine, phosphatidyl choline, and two, unidentified aminolipids; the presence of the two additional aminolipids appears to be distinctive of the organism [3]. The G+C content of the DNA was determined to be 64% [3], which corresponds well to the G+C content observed for the complete genome sequence (see below).
Genome sequencing information
Genome project history
The genome was selected for sequencing as part of the U.S. Department of Energy - Microbial Genome Program 2006. The DNA sample was submitted in April 2006 and the initial sequencing phase was completed in October 2006. The genome finishing and assembly phase were completed in June 2007, and presented for public access on December 2007; a modified version was presented in February 2011. Table 2 presents the project information and its association with MIGS version 2.0 compliance [39].
Growth conditions and DNA isolation
P. lavamentivorans DS-1T was grown on LB agar plates (2 weeks) and pinpoint colonies were transferred into selective medium (1 mM LAS/minimal salts medium; with glass-fiber supplement, 5-ml scale [3]). This culture was sub-cultivated to larger scale (100-ml and 1-liter scale) in 30 mM acetate/minimal salts medium; cell pellets were stored frozen until DNA preparation. DNA was prepared following the JGI DNA Isolation Bacterial CTAB Protocol [40].
Genome sequencing and assembly
The genome of P. lavamentivorans DS-1T was sequenced at the Joint Genome Institute (JGI) using a combination of 3.5 kb, 9 kb and 37 kb DNA libraries. All general aspects of library construction and sequencing performed at the JGI can be found at the JGI website [41]. Draft assemblies were based on 76,870 reads. Combined, the reads from all three libraries provided 16-fold coverage of the genome. The Phred/Phrap/Consed software package [42] was used for sequence assembly and quality assessment [43–45]. After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with Dupfinisher [46], PCR amplification, or transposon bombing of bridging clones (Epicentre Biotechnologies, Madison, WI, USA). Gaps between contigs were closed by editing in Consed, custom primer walk or PCR amplification (Roche Applied Science, Indianapolis, IN, USA). A total of 24 primer walk reactions were necessary to close gaps and to raise the quality of the finished sequence. The completed genome assembly contains 76,885 reads, achieving an average of 16-fold sequence coverage per base with an error rate less than 5 in 100,000.
Genome annotation
Genes were identified using a combination of Critica [47] and Glimmer [48] as part of the genome annotation pipeline at Oak Ridge National Laboratory (ORNL), Oak Ridge, TN, USA, followed by a round of manual curation. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) non-redundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases; miscellaneous features were predicted using TMHMM [49] and signalP [50]. These data sources were combined to assert a product description for each predicted protein. The tRNAScanSE tool [51] was used to find tRNA genes, whereas ribosomal RNAs were found by using BLASTn against the ribosomal RNA databases. The RNA components of the protein secretion complex and the RNaseP were identified by searching the genome for the corresponding Rfam profiles using INFERNAL [52]. Additional gene prediction analysis and manual functional annotation was performed within the Integrated Microbial Genomes (IMG) platform [41] developed by the Joint Genome Institute, Walnut Creek, CA, USA [53].
Genome properties
The genome of P. lavamentivorans DS-1T comprises one circular chromosome of 3,914,745 bp (62.33% GC content) (Figure 3), for which a total number of 3,714 genes were predicted. Of these predicted genes, 3,654 are protein-coding genes, and 2,723 of the protein-coding genes were assigned to a putative function and the remaining annotated as hypothetical proteins; 18 pseudogenes were also identified. A total of 60 RNA genes and one rRNA operon are predicted; the latter is reflective of the slow growth of P. lavamentivorans DS-1T [54,55]. Furthermore, one Clustered Regularly Interspaced Short Palindromic Repeats element (CRISPR) including associated protein genes were predicted. The properties and the statistics of the genome are summarized in Table 3, and the distribution of genes into COGs functional categories is presented in Table 4.

Graphical circular map of the genome of P. lavamentivorans DS-1T. From outside to center: Genes on forward strand (color by COG categories), genes on reverse strand (color by COG categories), RNA genes (tRNA, green; rRNA, red; other RNAs, black), GC content, GC skew.
Metabolic features
The genome of P. lavamentivorans encodes complete pathways for synthesis of all proteinogenic amino acids and essential co-factors, and the central metabolism is represented by a complete pathway for the citrate cycle, glycolysis/gluconeogenesis, and the non-oxidative branch of the pentose-phosphate pathway; no candidate genes for the oxidative branch of the pentose-phosphate pathway or for the Entner-Doudoroff pathway are predicted.
P. lavamentivorans DS-1T does not grow on D-glucose, D-fructose, maltose, D-mannitol, D-mannose, and N-acetylglucosamine [3,7], and there are no valid candidate genes predicted in the genome for ATP-dependent sugar uptake systems or for D-glucose uptake via a phosphotransferase system. Similarly, no valid candidate genes were predicted for ATP-dependent amino-acid and di/oligo-peptide transport systems or for other amino-acid/peptide transporters, which reflects the poor growth of strain DS-1T in complex medium (LB-medium).
For the assimilation of acetyl-CoA from the degradation of alkanes and surfactants [2,3,5], or during growth with acetate, the genome of P. lavamentivorans encodes the glyoxylate cycle (isocitrate lyase, Plav_0592; malate synthase, Plav_0593) to generate succinate for the synthesis of carbohydrates. The genome also encodes the complete ethyl-malonyl-CoA pathway to assimilate acetate [56]. This observation, i.e. glyoxylate cycle and ethyl-malonyl-CoA pathway in the same organism, has been made before [57], and these two pathways in P. lavamentivorans DS-1T might be differentially expressed under varying environmental conditions.
For the degradation of alkanes and surfactants through abstraction of acetyl-CoA [54], the genome contains a wealth of candidate genes for the entry into alkyl-chain degradation (omega-oxygenation to activate the chain) supplemented by a variety of genes predicted for omega-oxidations (to generate the corresponding fatty-acids) and fatty-acid beta-oxidations (to excise acetyl-CoA units). We are currently exploring this high abundance of genes for alkane/alkyl-utilization in strain DS-1T by transcriptional and translational analysis [unpublished]. For example, at least nine cytochrome-P450 (CYP) alkane monooxygenase (COG2124), 44 alcohol dehydrogenase (COG1028), 11 aldehyde dehydrogenase (COG1012), 20 acyl-CoA synthetase (COG0318), 40 acyl-CoA dehydrogenase (COG1960), 31 enoyl-CoA hydratase (COG1024), 14 acyl-CoA acetyl-transferase (COG0183), six thioesterase (COG0824), and 17 putative long-chain acyl-CoA thioester hydrolase (PF03061) candidate genes are predicted in the genome.
Other predicted oxygenase genes comprise three putative Baeyer-Villiger-type FAD-binding monooxygenase genes (COG2072). Cyclohexanone and hydroxyacetophenone, which are putative substrates for such oxygenases (e.g. [58,59]) were tested as carbon source for growth of strain DS-1T, as well as cycloalkanes (C6, C8, C12), however, none supported growth. The terpenoids camphor (for the involvement of a cytochrome-P450 oxygenase in the degradation pathway [60]) and geraniol, citronellol, linalool, menthol and eucalyptol (for the involvement of acyl-CoA interconversion enzymes in the degradation pathways) as substrates for growth were also tested negative.
In contrast to the high abundance of genes for aliphatic-hydrocarbon degradation, the genome contains few genes for aromatic-hydrocarbon degradation. One gene set for an aromatic-ring dioxygenase component (Plav_1761 and 1762; BenAB-type), three aromatic-ring monooxygenase component genes (Plav_1541 and 0131, MhpA-type; Plav_1785, HpaB-type), and three valid candidate genes for extradiol ring-cleavage dioxygenase (Plav_1539 [61] and 1787, BphC-type; Plav_0983, LigB-type) were predicted in the genome. Strain DS-1T did not grow with benzoate, protocatechuate, phenylacetate, phenylpropionate, or phenylalanine and tyrosine as carbon source when tested.
Finally, P. lavamentivorans DS-1T is predicted to store carbon in form of intracellular polyhydroxyalkanoate/butyrate (PHB) as its genome encodes a PHB-synthase (PhbC) gene (Plav_1129), PHB-depolymerase (PhaZ) gene (Plav_0012), and PHB-synthesis repressor (PhaR) gene (Plav_1572).
References
Website of linear alkylbenzene sulphonate. http://www.lasinfo.org
Schleheck D, Dong W, Denger K, Heinzle E, Cook AM. An alpha-proteobacterium converts linear alkylbenzenesulfonate surfactants into sulfophenylcarboxylates and linear alkyldiphenyletherdisulfonate surfactants into sulfodiphenyl-ethercarboxylates. Appl Environ Microbiol 2000; 66:1911–1916. PubMed doi:10.1128/AEM.66.5.1911-1916.2000
Schleheck D, Tindall BJ, Rossello-Mora R, Cook AM. Parvibaculum lavamentivorans gen. nov., sp. nov., a novel heterotroph that initiates catabolism of linear alkylbenzenesulfonate. Int J Syst Evol Microbiol 2004; 54:1489–1497. PubMed doi:10.1099/ijs.0.03020-0
Schleheck D, Knepper TP, Fischer K, Cook AM. Mineralization of individual congeners of linear alkylbenzenesulfonate by defined pairs of heterotrophic Bacteria. Appl Environ Microbiol 2004; 70:4053–4063. PubMed doi:10.1128/AEM.70.7.4053-4063.2004
Schleheck D, Lechner M, Schonenberger R, Suter MJ, Cook AM. Desulfonation and degradation of the disulfodiphenylethercarboxylates from linear alkyldiphenyletherdisulfonate surfactants. Appl Environ Microbiol 2003; 69:938–944. PubMed doi:10.1128/AEM.69.2.938-944.2003
Eilers H, Pernthaler J, Peplies J, Glöckner FO, Gerdts G, Amann R. Isolation of novel pelagic bacteria from the German Bight and their seasonal contributions to surface picoplankton. Appl Environ Microbiol 2001; 67:5134–5142. PubMed doi:10.1128/AEM.67.11.5134-5142.2001
Lai Q, Wang L, Liu Y, Yuan J, Sun F, Shao Z. Parvibaculum indicum sp. nov., isolated from deep sea water of Indian Ocean. Int J Syst Evol Microbiol 2010.
Hilyard EJ, Jones-Meehan JM, Spargo BJ, Hill RT. Enrichment, isolation, and phylogenetic identification of polycyclic aromatic hydrocarbon-degrading bacteria from Elizabeth River sediments. Appl Environ Microbiol 2008; 74:1176–1182. PubMed doi:10.1128/AEM.01518-07
Wang L, Wang W, Lai Q, Shao Z. Gene diversity of CYP153A and AlkB alkane hydroxylases in oil-degrading bacteria isolated from the Atlantic Ocean. Environ Microbiol 2010; 12:1230–1242. PubMed doi:10.1111/j.1462-2920.2010.02165.x
Alonso-Gutiérrez J, Figueras A, Albaigés J, Jiménez N, Viñas M, Solanas AM, Novoa B. Bacterial communities from shoreline environments (Costa da Morte, Northwestern Spain) affected by the Prestige oil spill. Appl Environ Microbiol 2009; 75:3407–3418. PubMed doi:10.1128/AEM.01776-08
Paixão DA, Dimitrov MR, Pereira RM, Accorsini FR, Vidotti MB, Lemos EG. Molecular analysis of the bacterial diversity in a specialized consortium for diesel oil degradation. Rev Bras Cienc Solo 2010; 34:773–781. doi:10.1590/S0100-06832010000300019
Li B, Shen L, Zhang D-M. Dynamics of petroleum-degrading bacterial community with biodegradation of petroleum contamination in seawater. J Ningbo University (Natural Science & Engineering Edition) 2009–04.
Sánchez O, Diestra E, Esteve I, Mas J. Molecular characterization of an oil-degrading cyanobacterial consortium. Microb Ecol 2005; 50:580–588. PubMed doi:10.1007/s00248-005-5061-4
Kjeldsen KU, Kjellerup BV, Egli K, Frolund B, Nielsen PH, Ingvorsen K. Phylogenetic and functional diversityof bacteria in biofilms from metal surfaces of an alkaline district heating system. FEMS Microbiol Ecol 2007; 61:384–397. PubMed doi:10.1111/j.1574-6941.2006.00255.x
De Weert JPA, Grotenhuis MVT, Rijnaarts HHM, Langenhoff AAM. Degradation of 4-n-nonylphenol under nitrate reducing conditions. Biodegradation 2011; 22:175–187. PubMed doi:10.1007/s10532-010-9386-4
Martínez-Pascual E, Jimenez N, Vidal-Gavilan G, Vinas M, Solanas AM. Chemical and microbial community analysis during aerobic biostimulation assays of non-sulfonated alkyl-benzene-contaminated groundwater. Appl Microbiol Biotechnol 2010; 88:985–995. PubMed doi:10.1007/s00253-010-2816-8
Macedo AJ, Timmis KN, Abraham WR. Widespread capacity to metabolize polychlorinated biphenyls by diverse microbial communities in soils with no significant exposure to PCB contamination. Environ Microbiol 2007; 9:1890–1897. PubMed doi:10.1111/j.1462-2920.2007.01305.x
Wang B, Lai Q, Cui Z, Tan T, Shao Z. A pyrene-degrading consortium from deep-sea sediment of the West Pacific and its key member Cycloclasticus sp. P1. Environ Microbiol 2008; 10:1948–1963. PubMed doi:10.1111/j.1462-2920.2008.01611.x
Blöthe M, Roden EE. Composition and activity of an autotrophic Fe(II)-oxidizing, nitrate-reducing enrichment culture. Appl Environ Microbiol 2009; 75:6937–6940. PubMed doi:10.1128/AEM.01742-09
Sayeh R, Birrien JL, Alain K, Barbier G, Hamdi M, Prieur D. Microbial diversity in Tunisian geothermal springs as detected by molecular and culture-based approaches. Extremophiles 2010; 14:501–514. PubMed doi:10.1007/s00792-010-0327-2
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Bacteria, Archaea and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed doi:10.1073/pnas.87.12.4576
Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part B, Springer, New York, 2005, p. 1.
Validation List No. 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 2006; 56:1–6. PubMed doi:10.1099/ijs.0.64188-0
Garrity GM, Bell JA, Lilburn T. Class I. Alphaproteobacteria class. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 1.
Kuykendall LD. Order VI. Rhizobiales ord. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 324.
Garrity GM, Bell JA, Lilburn T. Family X. Rhodobiaceae fam. nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s Manual of Systematic Bacteriology, second edition (The Proteobacteria), part C (The Alpha-, Beta-, Delta-, and Epsilonproteobacteria). Volume 2. New York: Springer; 2005. p 571.
Dong W, Eichhorn P, Radajewski S, Schleheck D, Denger K, Knepper TP, Murrell JC, Cook AM. Parvibaculum lavamentivorans converts linear alkylbenzenesulphonate surfactant to sulphophenylcarboxylates, alpha, beta-unsaturated sulphophenylcarboxylates and sulphophenyldicarboxylates, which are degraded in communities. J Appl Microbiol 2004; 96:630–640. PubMed doi:10.1111/j.1365-2672.2004.02200.x
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25–29. PubMed doi:10.1038/75556
German Collection of Microorganisms and Cell Cultures. http://www.dsmz.de
Klebensberger J, Rui O, Fritz E, Schink B, Philipp B. Cell aggregation of Pseudomonas aeruginosa strain PAO1 as an energy-dependent stress response during growth with sodium dodecyl sulfate. Arch Microbiol 2006; 185:417–427. PubMed doi:10.1007/s00203-006-0111-y
Notification that new names and new combinations have appeared in volume 54, part 5, of the IJSEM. Int J Syst Evol Microbiol 2005; 55:3–5. PubMed doi:10.1099/ijs.0.63563-0
Validation list No. 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 2006; 56:1–6. PubMed doi:10.1099/ijs.0.64188-0
Brettar I, Christen R, Bötel J, Lünsdorf H, Höfle MG. Anderseniella baltica en. nov., sp. nov., a novel marine bacterium of the isolated from sediment in the central Baltic Sea. Int J Syst Evol Microbiol 2007; 57:2399–2405. PubMed doi:10.1099/ijs.0.65007-0
Notification that new names and new combinations have appeared in volume 57, part 10, of the IJSEM. Int J Syst Evol Microbiol 2008; 58:3–4. PubMed doi:10.1099/ijs.0.65722-0
Albuquerque L, Rainey FA, Pena A, Tiago I, Veri’ssimo A, Nobre MF, da Costa MS. Tepidamorphus gemmatus gen. nov., sp. nov., a slightly thermophilic member of the Alphaproteobacteria. Syst Appl Microbiol 2010; 33:60–66. PubMed doi:10.1016/j.syapm.2010.01.002
Euzéby J. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 2010; 60:1477–1479. doi:10.1099/ijs.0.026252-0
DeSantis TZ, Hugenholtz P, Keller K, Brodie EL, Larsen N, Piceno YM, Phan R, Andersen GL. NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res 2006; 34:W394–W399. PubMed doi:10.1093/nar/gkl244
Huson DH, Richter DC, Rausch C, Dezulian T, Franz M, Rupp R. Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinformatics 2007; 8:460. PubMed doi:10.1186/1471-2105-8-460
Field D, Garrity GM, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed doi:10.1038/nbt1360
JGI protocols. http://my.jgi.doe.gov/general/index.html
DOE Joint Genome Institute. http://www.jgi.doe.gov
The Phred/Phrap/Consed software package. http://www.phrap.com
Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 1998; 8:175–185. PubMed
Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 1998; 8:186–194. PubMed
Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res 1998; 8:195–202. PubMed
Han CS, Chain P. Finishing repeat regions automatically with Dupfinisher. In: Arabnia HR, Valafar H, editors. Proceeding of the 2006 international conference on bioinformatics & computational biology: CSREA Press; 2006. p 141–146.
Badger JH, Olsen GJ. CRITICA: Coding region identification tool invoking comparative analysis. Mol Biol Evol 1999; 16:512–524. PubMed
Delcher AL, Bratke K, Powers E, Salzberg S. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007; 23:673–679. PubMed doi:10.1093/bioinformatics/btm009
Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol 2001; 305:567–580. PubMed doi:10.1006/jmbi.2000.4315
Dyrløv Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 2004; 340:783–795. PubMed doi:10.1016/j.jmb.2004.05.028
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997; 25:955–964. PubMed doi:10.1093/nar/25.5.955
Infernal: inference of RNA alignments. http://infernal.janelia.org
Markowitz VM, Szeto E, Palaniappan K, Grechkin Y, Chu K, Chen IMA, Dubchak I, Anderson I, Lykidis A, Mavromatis K, et al. The Integrated Microbial Genomes (IMG) system in 2007: data content and analysis tool extensions. Nucleic Acids Res 2007; 36:D528–D533. PubMed doi:10.1093/nar/gkm846
Schleheck D, Cook AM. Omega-oxygenation of the alkyl sidechain of linear alkylbenzenesulfonate (LAS) surfactant in Parvibaculum lavamentivorans (T). Arch Microbiol 2005; 183:369–377. PubMed doi:10.1007/s00203-005-0002-7
Schleheck D, Knepper TP, Eichhorn P, Cook AM. Parvibaculum lavamentivorans DS-1T degrades centrally substituted congeners of commercial linear alkylbenzenesulfonate to sulfophenyl carboxylates and sulfophenyl dicarboxylates. Appl Environ Microbiol 2007; 73:4725–4732. PubMed doi:10.1128/AEM.00632-07
Erb TJ, Berg IA, Brecht V, Muller M, Fuchs G, Alber BE. Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: the ethylmalonyl-CoA pathway. Proc Natl Acad Sci USA 2007; 104:10631–10636. PubMed doi:10.1073/pnas.0702791104
Erb TJ, Fuchs G, Alber BE. (2S)-Methylsuccinyl-CoA dehydrogenase closes the ethylmalonyl-CoA pathway for acetyl-CoA assimilation. Mol Microbiol 2009; 73:992–1008. PubMed doi:10.1111/j.1365-2958.2009.06837.x
Chen YC, Peoples OP, Walsh CT. Acinebacter cyclohexanone monooxygenase: gene cloning and sequence determination. J Bacteriol 1988; 170:781–789. PubMed
Rehdorf J, Zimmer CL, Bornscheuer UT. Cloning, expression, characterization, and biocatalytic investigation of the 4-hydroxyacetophenone monooxygenase from Pseudomonas putida JD1. Appl Environ Microbiol 2009; 75:3106–3114. PubMed doi:10.1128/AEM.02707-08
Hedegaard J, Gunsalus IC. Mixed function oxidation. IV. An induced methylene hydroxylase in camphor oxidation. J Biol Chem 1965; 240:4038–4043. PubMed
Sipilä TP, Keskinen AK, Akerman ML, Fortelius C, Haahtela K, Yrjala K. High aromatic ring-cleavage diversity in birch rhizosphere: PAH treatment-specific changes of I.E.3 group extradiol dioxygenases and 16S rRNA bacterial communities in soil. ISME J 2008; 2:968–981. PubMed doi:10.1038/ismej.2008.50
Acknowledgements
We thank Joachim Hentschel for SEM operation. The work was supported by the University of Konstanz and the Konstanz Research School Chemical Biology, the University of New South Wales and the Centre for Marine Bio-Innovation, and the Deutsche Forschungsgemeinschaft (DFG grant SCHL 1936/1-1 to D.S.). The work conducted by the U.S. Department of Energy Joint Genome Institute was supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231, and that of the University of California, Lawrence Livermore National Laboratory under Contract No. W-7405-Eng-48, Lawrence Berkeley National Laboratory under contract No. DE-AC03-76SF00098, and Los Alamos National Laboratory under contract No. W-7405-ENG-36.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Schleheck, D., Weiss, M., Pitluck, S. et al. Complete genome sequence of Parvibaculum lavamentivorans type strain (DS-1T). Stand in Genomic Sci 5, 298–310 (2011). https://doi.org/10.4056/sigs.2215005
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.2215005