
Abstract
Objective
The objective of this study was to investigate the safety and pharmacokinetics of edaravone administered by single or successive intravenous infusions in healthy Chinese volunteers.
Methods
A total of 30 subjects (15 males and 15 females) were recruited and randomly assigned to three groups receiving edaravone doses of 20, 30, and 60 mg. All subjects received a single dose of edaravone during a 30-minute period, and only the 30 mg dose group continued to receive the same dose successively by intravenous infusion twice daily for the next 5 days. Plasma concentrations of edaravone were monitored by high-performance liquid chromatography at the following times: 15, 30, 45, 60, 75, 105, 165, 225, 300, 390, 480, 600, and 720 minutes after edaravone administration.
Results
The area under the plasma concentration-time curve during a dosage interval (AUCτ) values of the single dose in the 20, 30, and 60 mg dose groups were 3.64±1.37, 5.17 ± 0.93, and 11.25 ± 3.42 mg · h/L, respectively, while in the group receiving repeated dosing of 30 mg, the mean AUCτ value was 5.06 ± 0.89mg · h/L. The corresponding maximum plasma drug concentration (Cmax) values were 1599.0 ± 382.6, 2378.7 ± 316.7, and 4540.1 ± 901.1 ng/mL, respectively, in the single-dose groups, and 2479.1 ± 477.9 ng/mL in the 30 mg repeated-dose group. The mean AUCτ and Cmax ratios between the repeated-dose group and the single-dose groups were 0.98 and 1.04. All laboratory test abnormalities (including increased alanine transaminase and triacylglycerol levels, and decreased white blood cell counts and creatinine levels) were mild and tolerable. All abnormal blood biochemical indices returned to normal levels after 7 days.
Conclusion
Edaravone was safe and well tolerated in the volunteers and displayed linear increases in the Cmax and AUCτ values.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Free radicals have been considered one of the most harmful factors that contribute to the development of cardiovascular disease, cancer, neurodegenerative disease, etc.[1–5] The term ‘free-radical scavengers’ refers to chemicals (such as vitamins, minerals, or enzymes) that are able to destroy free radicals. Although many free-radical scavengers are utilized clinically, only a few of them (such as NXY-059, 21-aminosteroid tirilazad, and edaravone [3-methyl-1-phenyl-2-pyrazolin-5-one; see figure 1]) have been used in the conduct of clinical trials in ischemic stroke.[6] Among those, edaravone is widely utilized in clinical trials to protect tissues from ischemia reperfusion injury after acute ischemic stroke.[7–9]

Chemical structure of edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one; MCI-185).
Edaravone can scavenge both hydroxyl radicals and peroxynitrite radicals, but it has no significant effect on superoxide anion radicals.[10,11] According to recent reports, edaravone has various functions, such as relieving neuropathic pain induced by spinal nerves,[10] attenuating nerve injury induced by ischemia,[12] elevating the metabolism rate,[13] reducing the inflammatory response,[14] and ameliorating experimental autoimmune encephalomyelitis.[15] Edaravone can be useful for the treatment of diseases and clinical conditions in which oxidative stress plays a key role in the pathogenesis.[16] Some studies have also indicated that edaravone shows beneficial effects in treatment of idiopathic sudden sensorineural hearing loss with profound hearing-loss encephalomyelitis.[17] The major side effects of edaravone are hepatic impairment or renal function disorder.[18] For that reason, it is prescribed with care to patients who have a clinical history of hepatic or renal disorder.
A few analytical methods of measuring edaravone plasma concentrations have been reported, such as liquid chromatography with tandem mass spectrometry (LC-MS/MS) and gas chromatography with mass spectrometry (GC-MS).[19,20] These two methods are feasible but have their limitations. The objective of this study was to investigate the safety and pharmacokinetics of edaravone administered by single or successive intravenous infusions in healthy Chinese volunteers.
Materials and Methods
Design and Demographic Characteristics
The protocol was approved in advance by the hospital ethics committee and conducted in accordance with Good Clinical Practice and the Helsinki Declaration. After receiving oral and written explanations of the study, the subjects gave written informed consent prior to starting the study. All subjects (15 males and 15 females) were recruited into three edaravone dose groups (20, 30, and 60 mg). The volunteers who had been given a 30 mg single dose continued to receive multiple administrations from the second day, twice daily for 5 days. None of the subjects consumed excessive amounts of alcohol or smoked, and none took any drugs during or at least 1 week prior to the study. Subjects were excluded on the basis of a clinically significantly abnormal electrocardiogram, the results of blood chemistry or urinalysis tests, or a positive result on the pregnancy test. The demographic characteristics of the study cohort are presented in table I. The subjects fasted from 10 hours before to 2 hours after edaravone administration. The volunteers were observed for 24 hours post-drug administration, during which time both safety laboratory data and the results of physical examinations were assessed. The edaravone parenteral solution was provided by Yangtze River Pharmaceutical Group–Nanjing Hailing Pharmaceutical Co. Ltd. (Nanjing, China).

Demographic characteristics of the subjects
Sample Collection and Assays of Edaravone
Peripheral blood samples were drawn from an intravenous cannula (inserted into a forearm vein) into 5 mL heparinized tubes prior to and after intravenous administration of edaravone at the following times: 5, 10, 15, 30, 45, 60, 120, 180, 240, 360, 480, 600, and 720 minutes. After collection, the blood samples were immediately centrifuged at 3500 rpm for 6 minutes, and the plasma was separated and stored at -80°C until analysis. The plasma concentrations were measured by HPLC with an ultraviolet (UV) detector (LC-2010-CTH; Shimadzu, Kyoto, Japan). The assay was performed in accordance with the following procedure. An aliquot of 0.2 mL of plasma was vortex mixed with 40 μL of HClO4 (30%) to acidify the plasma and precipitate plasma protein for 40 seconds, then centrifuged at 4°C at 16 000 rpm for 6 minutes. The supernate fluid was then prepared for analysis. An aliquot of 20 μL of the supernate fluid was analyzed using a Syncronis C18 column (250 mm × 4.6 mm; 5 μm) [Thermo Scientific, Waltham, MA, USA]. The mobile phase consisted of ammonium acetate buffer (pH 6.6; 0.05 mol/L) and methanol [Merck, Darmstadt, Germany] (55 : 45, v/v). The flow rate was 1.0 mL/min; the detector wave was set at 240 nm. The limit of quantification was 30 ng/mL, and the intra- and inter-batch relative standard deviations were less than 9% and 13%, respectively.
Data and Statistical Analyses
The results are expressed as means ± standard deviations. The area under the plasma concentration-time curve (AUC), elimination half-life (t1/2), volume of distribution (Vd), and total plasma drug clearance (CL) were obtained by noncompartmental analysis, utilizing the pharmacokinetic analysis package DAS 2.0 (Drug And Statistics, Shanghai University of Traditional Chinese Medicine, Shanghai, China). Statistically significant differences in the mean values between the different dosage groups were determined by one-way analysis of variance (ANOVA) with an unpaired two-tailed heteroscedastic t-test. The paired t-test was utilized to compare the AUC during a dosage interval (AUCτ), AUC from time zero to infinity (AUC∞), maximum plasma drug concentration (Cmax), t1/2, CL, and Vd values for single and multiple dosing. Statistical significance was set at p < 0.05.
Results
Area under the Plasma Concentration-Time Curve Values and Maximum Plasma Drug Concentration Values of Edaravone in Plasma
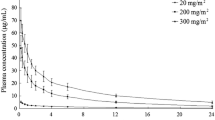
The mean AUCτ, AUC∞, Cmax, t1/2, CL, and Vd values for the three groups after single and repeated doses are shown in table II. There were no significant differences between the three groups, regardless of the number of doses received. The mean AUC∞ ratio and mean Cmax ratio for multiple-dose/single-dose administration of 30 mg were 0.99 and 1.04, respectively. These values indicate that there was no accumulation after repeated doses. The mean plasma concentration-time curves after single and repeated doses are shown in figure 2. As shown in the linear equation and the scatter diagrams (table III and figure 3, respectively), the Cmax and AUCτ values in the three single-dose groups appeared linear in accordance with the doses.

2 Plots of the mean plasma concentration-time curves of intravenous edaravone for the three dose groups (20, 30, and 60 mg) on the first day after a single dose, and on the fifth day after repeated twice-daily doses of 30 mg. Values are given as means ± standard deviations.

Scatter diagrams of the relationship between the dose and (a) the log-normal maximum plasma drug concentration (ln Cmax); and (b) the log-normal area under the plasma concentration-time curve during a dosage interval (ln AUCτ).

Pharmacokinetic parameters on the first day after a single 30-minute intravenous infusion of edaravone in the three dose groups, and on the fifth day after repeated twice daily doses in the 30 mg dose group (n = 10)

Relationships of edaravone doses to log-normal maximum plasma concentration (ln Cmax) and log-normal area under the plasma concentration-time curve (ln AUCT) values during a dosage interval at steady state
Safety Results
Edaravone, given by intravenous infusion, was well tolerated at doses of up to 60 mg administered once or 30 mg administered twice daily for 5 days. No symptomatic adverse effects were observed.
Although some laboratory test abnormalities were observed, the symptoms were mild and tolerable, and were considered not to diminish the value of the study. All serum biochemistry indices returned to normal levels after 7 days, without any treatment. All adverse events were possibly related to the drug. The changes in serum biochemistry in subjects who experienced adverse events and the numbers of adverse events that occurred after single or multiple doses are shown in table IV.

Changes of serum biochemistry in subjects with adverse events after single or multiple doses of edaravone parenteral solution
Discussion and Conclusion
Edaravone has been widely used clinically in Japan. It has been reported that the binding rate of 14C-MCI-186 to human serum protein is 91.0–91.9%.[21] After precipitation of plasma protein by perchloric acid, edaravone shows good linearity in the sample, thus it is unnecessary to add an internal standard. In our study, we also carried out relevant research on edaravone metabolism in the human body, but we could not detect the accurate concentration of edaravone in urine, because of impurity interference. An isotope-labeling method was used to determine the concentration of edaravone in urine, but it could only be used to measure the urinary concentrations during the first 2 hours.[20] This is consistent with the results of our study. Edaravone is excreted as the unmetabolized drug (∼1%) or is metabolized by sulfation (5–13%) or glucuronidation (68–83%) and excreted in urine within 24 hours of administration.[22] There were no significant differences between the multiple-dose group and the single-dose groups, indicating that there was no accumulation after multiple administrations.
In our study, we observed that edaravone displayed a linear increase in the Cmax and AUCτ values over a dose range of 20–60 mg administered by intravenous infusion. The Cmax values were measured 30 minutes after the intravenous infusion of edaravone. The Cmax values (table II) were significantly higher than the values reported in a previous study (Cmax 222.53 ± 16.77 ng/mL, dosage 0.2 mg/kg; Cmax 658.89 ± 96.88 ng/mL, dosage 0.5 mg/kg; Cmax 1727.19 ± 210.88 ng/mL, dosage 1.0 mg/kg; and Cmax 3060.73 ± 236.88 ng/mL, dosage 1.5 mg/kg).[20] The related explanations are as follows:
-
1.
The intravenous infusion time in our study was 30 minutes, while in the previous study it was 40 minutes.
-
2.
We developed a simple, rapid, sensitive method for determination of the edaravone plasma concentration with HPLC, which took less than 10 minutes to obtain the supernatant, making it more convenient and stable. Edaravone is unstable in human plasma in air,[23] and the extraction method always takes more than 30 minutes, meaning that edaravone is exposed to air for a long time.[20]
-
3.
In a preliminary experiment, we found that edaravone in human plasma was unstable when stored at room temperature for more than 45 minutes.[24] This was consistent with the dramatic decrease in the edaravone plasma concentration. Thus we tested all plasma samples within 24 hours after administration of the drug.
The LC-MS/MS method, as another analytical method for measuring the plasma edaravone concentration, has also been used by another group. The calibration curve is linear in the range of 10–500 ng/mL but is not linear above 500 ng/mL.[19]
In conclusion, edaravone parenteral solution is both well tolerated and safe when administered as a single dose or as multiple doses.
References
Berliner JA, Heinecke JW. Review: the role of oxidized lipoproteins in atherogenesis. Free Radic Biol Med 1996; 20: 707–27.
Breen AP, Murphy JA. Review: reactions of oxyl radicals with DNA. Free Radic Biol Med 1995; 18: 1033–77.
Burdon RH. Review: superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic Biol Med 1995; 18: 775–94.
Markesbery WR. Review: oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med 1997; 23: 134–47.
Adams JD, Odunze IN. Review: oxygen free radicals and Parkinson’s disease. Free Radic Biol Med 1991; 10: 161–9.
Doeppner TR, Hermann DM. Free radical scavengers and spin traps — therapeutic implications for ischemic stroke. Best Pract Res Clin Anaesthesiol 2010; 24: 511–20.
The Edaravone Acute Brain Infarction Study Group. Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction: randomized, placebo-controlled, double-blind study at multicenters. Cerebrovasc Dis 2003; 15: 222–9.
Feng S, Yang Q, Liu M, et al. Edaravone for acute ischaemic stroke (review). Cochrane Database Syst Rev 2011; (12): CD007230.
Yang J, Liu M, Zhou J, et al. Edaravone for acute intracerebral haemorrhage (review). Cochrane Database Syst Rev 2011;(2):CD007755.
Mao YF, Yan N, Xu H, et al. Edaravone, a free radical scavenger, is effective on neuropathic pain in rats. Brain Res 2009; 1248: 68–75.
Yoshida H, Yanai H, Namiki Y, et al. Neuroprotective effects of edaravone: a novel free radical scavenger in cerebrovascular injury. CNS Drug Rev 2006; 12: 9–20.
Takeda T, Takeda S, Takumida M, et al. Protective effects of edaravone against ischemia-induced facial palsy. Auris Nasus Larynx 2007; 35: 321–7.
Ishizawa M, Mizushige K, Noma T, et al. An antioxidant treatment potentially protects myocardial energy metabolism by regulating uncoupling protein 2 expression in a chronic beta-adrenergic stimulation rat model. Life Sci 2006; 78: 2974–82.
Zhang N, Komine-Kobayashi M, Tanaka R, et al. Edaravone reduces early accumulation of oxidative products and sequential inflammatory responses after transient focal ischemia in mice brain. Stroke 2005; 36: 2220–5.
Moriya M, Nakatsuji Y, Miyamoto K, et al. Edaravone, a free radical scavenger, ameliorates experimental autoimmune encephalomyelitis. Neurosci Lett 2008; 440: 323–6.
Kikucki K, Uchikado H, Miyagi N, et al. Beyond neurological disease: new targets for edaravone (review). Int J Mol Med 2011; 28: 899–906.
Sano H, Kamijo T, Ino T, et al. Edaravone, a free radical scavenger, in the treatment of idiopathic sudden sensorineural hearing loss with profound hearing loss. Auris Nasus Larynx 2010; 37: 42–6.
Higashi Y, Jitsuiki D, Chayama K, et al. Edaravone (3-me-thyl-1-phenyl-2-pyrazolin-5-one), a novel free radical scavenger, for treatment of cardiovascular diseases. Recent Pat Cardiovasc Drug Dis 2006; 1: 85–93.
Gu LQ, Xin YF, Zhang S, et al. Determination of edaravone in plasma of beagle dog by LC-MS. Zhejiang Provincial Academy of Medical Sciences 2010; 21: 24–7.
Shibata H, Arai S, Izawa M, et al. Phase I clinical study of MCI-186 (edaravone, 3-methyl-1-phenyl-2-pyrazolin-5-one) in healthy volunteers: safety and pharmacokinetics of single and multiple administrations. Japanese Journal of Clinical Pharmacology and Therapeutics 1998; 29: 863–76.
Yamamoto M, Takamatus Y. Pharmacokinetic studies of 3-methyl-1-phenyl-2-pyrazolin-5-one (MCI-186): protein binding and distribution to red blood cells. Japanese Pharmacology and Therapeutics 1997; 25: 245–53.
Lapchak P. A critical assessment of edaravone acute ischemic stroke efficacy trials: is edaravone an effective neuroprotective therapy? Expert Opin Pharmacother 2010 July; 11 (10): 1753–63.
Rolando B, Filieri A, Chegaev K, et al. Synthesis physicochemical profile and PAMPA study of new NO-donor edaravone co-drugs. Bioorganic & Med Chem 2012; 20: 841–50.
Data on file, Yongqing Wang, 2011.
Acknowledgments
This study was supported by Nanjing Yudao Pharmaceutical Science & Technology Co. (Nanjing, China), Nanjing Hailing Pharmaceutical Co. Ltd. (Nanjing, China), the National Science and Technology Major Projects for “Major New Drugs Innovation and Development” (grant no. 2011ZX09302-003-02), Jiangsu Province Science and Technology Major Projects (grant no. BM2011017), the Foundation of the Health Bureau of Jiangsu Province (Nanjing, China; grant no. H201108), and the Foundation of the Nanjing Pharmaceutical Association (Nanjing, China; grant no. H2011YX001).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Li, H., Xu, K., Wang, Y. et al. Phase I Clinical Study of Edaravone in Healthy Chinese Volunteers. Drugs R D 12, 65–70 (2012). https://doi.org/10.2165/11634290-000000000-00000
Published:
Issue Date:
DOI: https://doi.org/10.2165/11634290-000000000-00000