
Abstract
The promise afforded by attenuated sporozoite vaccines in the 1970s led many researchers to believe that an efficacious malaria vaccine was an attainable medium-term goal. Over 30 years later, no licensed vaccine is currently available for public health intervention. This is despite global expenditure on research and development for malaria vaccines that is estimated to have increased from $US42 million in 1999 to $US84 million in 2004. Serious questions must therefore be asked: is this a good investment of research and public health funds, and are we really any nearer to producing a viable product for global use?
Proponents of a malaria vaccine promote this technology as a viable way to combat both the current economic and humanitarian burden of malaria and the decreasing efficacy of many front-line antimalaria drug therapies. The recent successful phase IIb trial of the RTS,S/AS02A vaccine showed that the production of a subunit vaccine with significant efficacy is technically possible. The combined efforts and financial commitment of researchers, pharmaceutical companies, and not-for-profit organizations, including the Malaria Vaccines Initiative, have resulted in a significant scaling up in the number of products suitable for testing in humans. In addition, new technologies, such as genetically attenuated vaccines and the exploitation of malaria genomes, offer exciting possibilities for vaccine development. There is now a real possibility of producing a malaria vaccine licensed for public health. However, this positive outlook must be tempered with the challenges facing vaccine development and distribution. The efficacy levels seen with RTS,S/AS02A are well below those of all vaccines currently in use for public health. Furthermore, poor preclinical and clinical predictors of efficacy, allele-specific immunity, and an imperfect understanding of natural and induced immunity to malaria may yet delay (or even prevent) the development of a vaccine suitable for global use.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1. The Need for a Vaccine
With the exception of Plasmodium knowlesi,[1,2] there are four species of Plasmodium that infect humans: P. falciparum, P. vivax, P. ovale, and P. malariae. The majority of research and development into malaria vaccines is currently directed towards those products that would protect against Plasmodium falciparum, the most virulent of the human malarias.[3] This species is responsible for an estimated 300–500 million cases of clinical malaria and well in excess of 1 million deaths per year.[4] This in turn causes a major impact on the gross domestic product (GDP) of countries where malaria is endemic and the cost, in terms of lost productivity and medical care, exceeds $US1.7 billion each year. Sachs and Melaney[5] estimated that the gross national product per capita in malaria endemic countries has been reduced by >50% compared with non-malarious countries.
Globally, there is a marked increase in resistance to front-line antimalarial drugs, such as chloroquine[6,7] and sulfadoxine/pyrimethamine.[8] Even the newly licensed artemisinin derivatives run the risk of being rendered ineffective by the occurrence and spread of mutations within the P. falciparum genome.[9] Thus, there is a real need for novel intervention programs. However, were a successful vaccine to be licensed, it is uncertain whether a global market could effectively sustain such a product. Major concerns include the problem of a limited global vaccine production capacity and global production and distribution costs, together with an inability to deliver the vaccine to the target populations within existing infrastructures such as the Expanded Program on Immunization.[10,11] Nonetheless, the acceptance that these problems are insurmountable is not universal,[12,13] and many strategies have been proposed to overcome them. If the economics and logistics of vaccine manufacture, distribution, and delivery can be dealt with, then the advantages of an effective vaccine are clear.
2. What Stage Should a Malaria Vaccine Work Against?
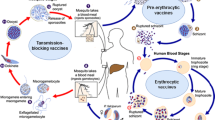
A major factor when considering the component(s) to include in a malaria vaccine is the different outcomes that a vaccine can be devised to produce. Vaccines can be directed to prevent infection, reduce disease/parasite burden, or prevent the spread of infection from existing hosts depending on the stage of the P. falciparum life cycle to be targeted. As shown in figure 1, this life cycle can be divided into four main parts, three of which lie in the human host (pre-erythrocytic, erythrocytic, and sexual stages) while the fourth occurs within the mosquito vector.

Schematic of the Plasmodium falciparum life cycle: different stages to which a malaria vaccine can be targeted.
Pre-erythrocytic-stage vaccines are designed to target either the sporozoites before they can invade hepatocytes, or the infected hepatocytes before they can produce infective merozoites.[14] It is at the pre-erythrocytic stage that the irradiated sporozoite vaccine works.[15–17] Such a vaccine would be expected to produce sterile immunity and would, theoretically, have little effect on the progression of disease once liver stage merozoites have successfully established an erythrocytic infection. Candidate antigens for such a pre-erythrocytic vaccine include those expressed in either sporo-zoites or within the hepatocyte, such as cirscumsporozoite protein (CSP) and liver stage antigen (LSA)-1, respectively.
By comparison, vaccines containing antigens expressed within the erythrocytic stage of the disease would be expected to reduce the overall parasite burden and associated morbidity.[18] Candidate parasite-derived antigens for erythrocytic-stage vaccines include merozoite surface protein (MSP)-1 (the major surface component of the merozoite), apical membrane antigen (AMA)-1 (released from the apical organelles of the merozoite prior to invasion and also expressed in the sporozoite stage), and P. falciparum erythrocyte membrane protein 1 (exported to the surface of infected erythrocytes and associated with parasite virulence). Individuals would most likely be rendered semi-immune, capable of maintaining infections without succumbing to clinical episodes of the disease. An alternative strategy is to induce immune responses to detoxify by-products of infection, such as glycosylphosphatidylinositol, which may play a role in the severity of clinical morbidity.[19]
Finally, transmission-blocking vaccines have been promoted as a way to interrupt the sexual stage of the life cycle. The vaccine would not directly protect an inoculated individual, but rather act to reduce the rate at which new infections are established via the mosquito vector, thereby reducing overall disease rates in endemic regions. Such actions would undoubtedly complement the effects of insecticide-treated bed nets and residual spraying. Transmission-blocking vaccines would induce antibodies against antigens expressed during the sexual stages of the malaria parasite, either on the gametocytes resident within the human blood stream or, in the case of the Pfs25 protein, in the stages found within the mosquito mid gut.
One possible way forward is to include multiple antigens within a vaccine in order to replicate the activity of all three vaccine types. However, the inclusion of multiple antigens within a single vaccine may be hampered by the higher development and production costs to produce a homogeneous product and ensure that no interference occurs between the different antigens.
3. Irradiated Sporozoites and Subunit Vaccines
In the early 1970s, sterile immunity was achieved in naive volunteers using the bites of irradiated mosquitoes infected with P. falciparum. Using this approach, ≤90% sterile protection was achieved against experimental infection.[15–17,20] Unfortunately, the logistics required to produce infected irradiated mosquitoes suitable for a global vaccination program has so far precluded this approach as a viable public health intervention strategy. Even if it were possible, the requirement for thousands of infected bites makes it unfeasible to advocate such a vector-based approach in the field.
Given the problems associated with an irradiated sporozoite vaccine, subunit vaccines have been explored as possible alternatives. Human and animal subjects presenting with sterile immunity due to irradiated sporozoite inoculation appear to show elevated T cell-mediated responses to a limited number of P. falciparum antigens.[17,20–23] Thus, there is a real possibility that subunit vaccines using a defined panel of polypeptides could induce protective sterile immunity.
Such immunity would be different from that presented by people living in areas where malaria is hyperendemic, such as The Gambia. People in these locations develop the capacity to maintain low-level infections without associated clinical disease over the course of repeated infections, with the greatest burden of malaria being carried by children <5 years of age.[24] The requirement for repeated infection is such that in areas of low endemicity no effective immune response is generated, resulting in an equal burden of disease in both adults and children. The humoral component of this immunity has been demonstrated by the passive transfer of immunoglobulin G (IgG) from healthy semi-immune African volunteers to non-immune children infected with P. falciparum, resulting in an accelerated clearance of clinical morbidity.[25,26] It has been shown that people living in locations endemic for malaria possess cellular reactivity to specific malaria antigens[27] and some of these are associated with protection.[28] However, with a lack of antigen processing in erythrocytes the majority of immunity to blood-stage malaria parasites will most likely be conferred by humoral immune responses. Any malaria vaccine formulated to mimic natural immunity against blood-stage parasites would be expected to elicit high titers of antibodies against specific antigenic targets that are capable of controlling an infection. However, the specific mechanisms of protection and how to measure them are poorly understood, limiting the quality of information that can be used to guide vaccine design.[24]
The major problem with subunit vaccines containing recombinantly produced polypeptides is the limited number that can currently be accommodated within a single vaccine. The first licensed subunit vaccine, developed for hepatitis B, contained a single hepatitis B surface antigen (Recombivax HB®,Footnote 1 Merck & Co Inc.).[29] Although vaccination with DNA-based constructs offer the potential to overcome these limitations by the incorporation of epitopes from multiple antigens, success with these systems has been very limited. Recently, Dunachie et al.[30] showed that a prime boost regimen using TRAP (thrombospondin-related anonymous protein) but not CSP produced partial protection in healthy, malaria-naive adults. However, recent vaccine trials have failed to show protective efficacy in either Gambian adults[31] or Kenyan children.[32] This is despite strong immunogenicity data with relation to induction of effector T-cell responses in volunteers.[31]
Subunit vaccines that are produced by fragmenting whole parasites may contain many more components but require the capacity for large-scale culture of the infectious organisms. For example, the Pneumovax® II (Sanofi Pasteur MSD) pneumococcal vaccine contains capsule polysaccharides from 23 common types of Streptococcus pneumoniae. The major problem for subunit vaccines remains which (if any) out of the predicted 5000 plus components of the P. falciparum proteome in isolation are likely to induce an effective immune response capable of combating infection. It is sobering to note that this is a parasite that has been shaped through evolution to evade an immune system that is exposed to its full antigenic repertoire during the course of a natural infection.[33] The quest to answer this question has seen the development of numerous preclinical assays to determine the suitability of a candidate antigen as a vaccine component.
4. Preclinical Analysis of Candidate Antigens for a Vaccine
Table I provides a summary of candidate antigens currently under investigation for use in subunit vaccines. It can be seen that there are numerous lines of evidence that can be used to support development of a particular candidate to a good manufacturing practice (GMP) product suitable for use in human trials. These include in vitro invasion inhibition (with both hepatocyte and erythrocyte models), cytoadherance inhibition, antibody-dependant cellular inhibition (ADCI), immuno-epidemiology studies, opsonization studies, and vaccination of animal models. For example, the addition of anti-AMA-1 antibodies to in vitro cultured malaria parasites causes a significant decrease in the asexual growth rate, interfering with the erythrocyte invasion process.[34] In addition, immuno-epidemiology studies with recombinant AMA-1 have shown a significant association of naturally occurring anti-AMA-1 antibodies with reduced occurrence of clinical disease in the subsequent transmission season in Kenyan children.[35] Finally, immunizations in animal model systems with AMA-1 (alone or as a hybrid) preclincial vaccines have elicited immune responses capable of either preventing or limiting experimental infections,[36,37] supporting their development to GMP products.

Summary of potential malarial vaccine candidates currently under investigation
While positive results with preclincial assays suggest that creating an efficacious malaria vaccine is possible, there are problems inherent in the interpretation of such data that have prevented any one assay being universally accepted as a ‘gold-standard’ appropriate for all candidates. Furthermore, there is no standardized criterion that would allow a like-for-like comparison of candidates analyzed by different methodologies (table I). It is difficult therefore to prioritize candidacy of different antigens, especially when assays may give conflicting results; purified human IgG that is capable of reducing clinical morbidity in passive transfer experiments has no effect on invasion rates of parasites in vitro but is associated with ADCI.[120,141] Even within a single assay, such as in vitro invasion inhibition, there is often no standardized cut-off constituting a positive result. Additionally, different immunoepidemiologic studies will often yield conflicting results for a given antigen, perhaps as a result of differences in the ethnicity of the cohorts, malaria transmission, or misclassification of an individual’s immune responsiveness.[24]
For animal immunization models there are also drawbacks, making it impossible to guarantee how a vaccine candidate will function within an endemic location. In the mouse model system, the rodent malaria parasites P. yoelii and P. berghei are often used to analyze the homologs of P. falciparum vaccine candidates. However, there is a predicted evolutionary distance of >60 million years between these species of malaria.[142] The effect of this divergence is that direct homologs are not always identifiable (as seen with many of the P. falciparum erythrocyte binding antigen genes).[143] Even when the homolog is readily identifiable there is no guarantee the identified genes would have an identical function in the two species, as shown by gene knockout and transcriptional studies (S. Polley, unpublished). It is also of interest to note that mice are not the natural host of these parasites and may not be the best model organisms to use if a natural infection is to be studied. Mice have a strong innate immune response against P. berghei not seen in the natural host Grammomys surdaster (thicket rat).[144] A severe combined immunodeficiency disease (SCID) mouse model with humanized erythrocytes offers the ability to use P. falciparum;[145] however, even this system is hampered as a result of the differences that exist between the human and murine immune system.
The use of monkey and primate models offers a closer approximation to the human immune system for preclinical safety and efficacy studies. The common model organisms are Aotus and rhesus monkeys together with splenectomized chimpanzees. The Aotus monkey model system offers the flexibility to use erythrocytic challenge with strains of P. falciparum that have been adapted to this host;[146] in addition, certain Aotus species have the ability to be infected with sporozoites from infectious mosquito bites.[147] However, the progression of disease in Aotus species deviates from that seen in humans in a number of ways. These include a tendency to life-threatening anemia and a rapid acquisition of effective blood-stage immunity.[148] In addition to this, there are substantial differences between human and Aotus major histocompatibility complex gene sequences. Although genetically closer to humans than Aotus monkeys, the rhesus monkey system is refractory to P. falciparum, necessitating the use of homologous malaria parasites (P. knowlesi and P. cynomolgi) with the associated problems due to evolutionary divergence.[148] Even the chimpanzee (our nearest genetic relative) shows substantial differences in its susceptibility to P. falciparum, most likely due to alterations on the erythrocyte surface.[149] These potential draw backs, along with substantial financial, ethical, and time consideration associated with monkey trials, have prompted some to question the utility of this methodology.
Given the heterogeneous outcomes achieved with the current preclinical models, it is hardly surprising that 2 of the 11 goals of the Malaria Vaccine Technology Roadmap[150] are to improve the understanding of correlates of protection and to establish a systematic approach for prioritizing subunit vaccine candidates using accepted preclincial criteria.
5. The Pipeline for Development
Table II shows the stages a promising vaccine candidate must go through to arrive at a finished product licensed for use in humans. The current total development cost of a single pharmaceutical product is around $US200–800 million over a 7- to 14-year period, and it is unlikely that the costs for a malaria vaccine will be significantly lower. This requirement is a serious limitation to the number of vaccine candidates that can be taken forward, even in the current environment of increased funding. The Bill and Melinda Gates Foundation, a significant financial contributor to malaria vaccine development, admit that the process is complicated, “and more expensive than we anticipated.”[151] To proceed to any clinical trials (phase I onwards), the product must first be manufactured to a strict set of guidelines, set down as GMP. The limited global capacity for GMP production is another cap on the number of products that can advance to clinical trials. Addressing this is also a major goal of the Malaria Vaccine Technology Roadmap. Companies in locations such as India and China offer a viable opportunity to expand GMP production and fast track a number of promising candidates through to clinical trials.

Steps in the vaccine development pipeline[152]
A major problem when considering erythrocytic vaccines is the inability to perform experimental human challenge with blood-stage parasites when assessing the protective efficacy of blood-stage vaccines in phase IIa trials. Although the 3D7 strain is licensed for human experimental infection, there is no standardized measure of correlates of protection when a delay in clinical presentation of disease is the endpoint. In contrast, presence or absence of blood-stage infection as a marker of sterile immunity is a well established endpoint for pre-erythrocytic vaccines.[15] However, even with pre-erythrocytic vaccines, experimental infection can be a poor approximation for natural infection in an endemic location, which is illustrated by the conflicting results obtained with the multiple epitope (ME)-TRAP vaccine when analyzed with experimental (phase Ia) and natural (phase Ib) infections.[32,153] Could this be due in part to the nature of the 3D7 strain licensed for experimental infections? This parasite clone has undergone numerous passages since it was first adapted for in vitro culture. It has been shown that the parasite’s genome has undergone rearrangement and loss of material with unknown consequences for the infectivity or virulence of the parasite.[154,155]
A third limitation on the numbers of vaccine candidates that can be pushed through this pipeline is the number of people needed at each stage of the development process (see table II). This is affected by whether the required endpoint of the trial is protection against infection or protection against clinical manifestation of disease (cohort sizes are significantly larger for the latter).[12] The logistics involved in recruiting volunteers, administering the vaccination programs, and collecting endpoint measurement means that there are only a limited number of locations where the phase IIb and III studies can be implemented. This is because the required infrastructure is often lacking in locations endemic for malaria. A recent venture to establish an HIV vaccine trial in Haiti highlights some of the logistical problems associated with vaccine research in developing countries.[156] These include a weak health infrastructure, a shattered economy, high unemployment, residual political instability, and a high rate of illiteracy, as well as daily logistical hurdles like bad roads, erratic telephone networks, and energy blackouts as some of the problems facing would be trial administrators.
Another goal of the Malaria Vaccine Technology Roadmap is the scaling up of such vaccine trial facilities, although it is also important to remember the ethical as well as logistical considerations of such trials when planning them. Ethical considerations include the vulnerability of child participants, likely benefits versus risks to the research subjects, ability to ensure informed consent, standard of care for research subjects should the intervention fail, and access for the subjects and community to the product given a successful intervention. One ethical consideration particularly relevant to malaria vaccines is the issue of safety versus efficacy of the product. Could more lives be saved with a product that was more efficacious but caused more severe adverse effects? In addition to these concerns, communities involved in such interventions can easily develop research fatigue if they come to view themselves as mere guinea pigs for clinical research and experience a lack of positive outcomes with the trials.[157]
A further complication with trials involving humans is the choice of adjuvant incorporated with the vaccine antigen(s). These immunomodulators include a variety of compounds, which are designed to enhance the immunogenicity of antigens, thereby magnifying, accelerating, and prolonging the immune response. Adjuvants licensed for use in humans include mineral salts such as the commonly used alum, microbial derivatives such as monophosphoryl lipid A, oil-in-water emulsions such as Monta-nide ISA-51, and the proprietary adjuvant AS02A. The choice of adjuvant can have a major effect on the performance of a vaccine, as shown by a recent trial of malaria vaccine in treatment-naive volunteers.[158] In this study the same recombinant protein was conjugated with three different adjuvants as follows:
-
vaccine 1 — alum and monophosphoryl lipid A
-
vaccine 2 — an oil-in-water emulsion
-
vaccine 3 — an oil-in-water emulsion plus monophosphoryl lipidAandQS21.
Vaccines 2 and 3 elicited significantly higher titers of antibodies against the recombinant protein than vaccine 1, while only those volunteers given vaccine 3 demonstrated a high (>80%) level of protection against subsequent experimental infection. However, with many human licensed adjuvants being poor immunomodulators in animal models,[159] it is only at the stage of human testing that antigen/adjuvant combinations can be assessed.
6. The Success of the RTS,S/AS02A Vaccine
Table III provides a summary of malaria vaccines that are currently under development for clinical trials. This list includes subunit vaccines based on recombinant polypeptide products conjugated with a suitable adjuvant and DNA-based vaccine constructs (such as ME-TRAP).


Summary of malarial vaccine candidates in production[160]
As can be seen from table III, only a very limited number of products have made it through to any kind of efficacy testing (phase IIb or higher) in an endemic location. SPf66 (omitted from table III) was the first malaria vaccine to be taken to phase III trials. However, despite promising results from four South American trials with SPf66, these trials produced disappointing results when conducted in Africa. For instance, an efficacy of only 2% was seen in a Tanzanian trial of children in their first year of age, resulting in SPf66 being abandoned by many research agencies as a viable vaccine[195] and causing a considerable degree of controversy.
The disappointment of SPf66 has been superseded by a successful phase IIb trial of the RTS,S/AS02A vaccine. This vaccine has been developed by GlaxoSmithKline (GSK) Biologicals in combination with the Walter Reed Army Institute of Research (WRAIR). It is comprised of the CSP antigen repeat region (R) and T cell epitopes (T) coupled to the core of the licensed surface antigen (S) subunit hepatitis B vaccine conjugated with proprietary adjuvant AS02A. An RTS,S/AS02A trial was carried out in Mozambique on 1142 children, aged 1–4 years old, who received a 3-dose regimen of the vaccine with no booster in the follow-up period. The immunized children showed a reduction in clinical malarial episodes of 35% and severe malaria episodes by 49% in the 18-month period of follow-up, compared with control subjects.[161] An even greater (58%) reduction in severe malaria was seen in the initial 6-month follow-up period.[196] This trial has raised hopes that an efficacious malaria vaccine that will induce long lasting immunity is technically achievable.
7. Allele-Specific Immunity
A major barrier to malaria vaccine development is the polymorphic nature of many of the vaccine candidates. Even the CSP antigen is polymorphic, with an over-representation of polymorphic residues within sites shown to stimulate a T-cell response.[197] If the regions of antigens to be used in a vaccine are polymorphic in nature, then a suitable diversity of allelic forms must be included to protect against the total diversity encountered through natural infection (strain-transcending immunity). Failure to do this will most probably result in the selection of divergent variants, rapidly reducing any efficacy that a vaccine may have initially had. The problem posed by polymorphic candidate antigens is highlighted by the outcome of a phase IIb trial with the Combination B vaccine in Papua New Guinea.[190] While no significant protection was seen, there was an apparent skew in the Msp2 gene frequencies within immunized volunteers, such that the frequency of heterologous allele type increased significantly in the subsequent transmission season. The Combination B vaccine is based around three P. falciparum merozoite- and ring-stage derived recombinant protein antigens (MSP-1/MSP-2/RESA) formulated with Montanide ISA 720 as adjuvant. MSP-2 is an antigen with a highly polymorphic structure, with Msp2 alleles grouping into two major allelic classes (IC-1 and Fc27). The failure of Combination B would appear to the result of induction of allele-specific rather than strain-transcending immunity. Such a phenomenon has been observed in experimental challenges in animal model systems and in vitro growth inhibition assays, whereas allele-specific antibodies are commonly displayed by people in areas where malaria is endemic.[35,56,93,198]
Including multiple allelic forms of a given antigen will undoubtedly increase the complexity, development time, and overall cost of a vaccine. Tetteh et al.[199] demonstrated the effectiveness of using an epitope-mapping strategy in the rational design of a multi-allelic construct comprising highly polymorphic structures, such as those found in the N-terminal MSP-1 block 2 region,[56,200] to induce immunity against a broad spectrum of genotypes. Franks et al.[201] showed that despite the presence of significant polymorphic differences between different members of the same MSP-2 allelic class, conserved epitopes within the IC-1 type (and to a lesser degree the Fc27 type) can elicit antibodies that bind to all antigens of a given allelic type. A rational approach to vaccine design may therefore be capable of limiting the number of allelic forms needed to induce strain-transcending immunity. The ability to genetically engineer different allelic sequences into long chimeric proteins offers the ability to overcome gene diversity. This approach has allowed the development of a recombinant polypeptide vaccine against the diverse group A streptococcus, a causative agent of life-threatening necrotizing fasciitis, toxic shock, and rheumatic fever.[202] Encouragingly, initial trials showed no selective change in allele frequencies induced by the RTS,S/AS02A vaccine in Mozambique, suggesting that allele-specific immunity was not a problem when using this vaccine,[203] despite the polymorphic nature of CSP-1.
An alternative approach to using divergent allele sequences is to concentrate on those antigens/antigenic regions that are conserved amongst isolates. Anti-MSP-3 antibodies were first identified as correlates of immunity through passive transfer studies. The N-terminal region is highly polymorphic (with sequences clustering into two main allelic types) and allele-specific immune responses to this region have been shown to be associated with protection.[93] It could be argued, therefore, that an MSP-3-based vaccine should contain both sequence types to induce strain-transcending immunity. The Pasteur Institute (Paris, France) has instead looked to construct a vaccine containing only the relatively conserved C-terminal region of MSP-3.[204] In human volunteers, this vaccine has been shown to induce antibodies capable of clearing infection in the humanized SCID mouse model,[204] raising hopes for its efficacy and proving the utility of such an approach. It is important to consider why certain antigen regions, such as the C-terminus of MSP-3, show little or no diversity in the wild. It is possible that these sequences may not be under sufficiently strong immune-selection pressure to maintain polymorphisms in natural populations. Alternatively, they may be under such strong purifying selection that mutations can not accumulate without causing a serious deleterious effect on the parasites fitness. It is important to note that under novel positive-selection pressures, even sequences subject to purifying selection can accumulate polymorphisms that would normally be deleterious. This has been seen in HIV infections from patients undergoing antiretroviral therapy, and poses a serious limit to the lifespan of front-line treatments.[205]
8. Genetically Attenuated Vaccines
A number of groups have recently developed attenuated whole-organism vaccine candidates in an attempt to replicate the immunity induced by irradiated sporozoites. Van Dijk et al.[206] described the genetic attenuation of P. berghei sporozoites by disruption of the p36 gene, which encodes a member of the P48/45 family of sporozoite surface proteins. When infected into mice, the attenuated parasites invaded hepatocytes but failed to develop beyond this stage. These arrested infections induced immune responses capable of protecting against subsequent experimental challenge with non-attenuated parasites. More recently, Mueller et al.[207] showed that disruption of the UIS3 gene gave rise to genetically attenuated parasites that were incapable of developing into liver stage merozoites and, therefore, did not lead to disease. Immunization of mice with the uis3(–) parasites led to a sustained, stage-specific protection. Although questions of safety and potency need to be addressed,[208] it is worth noting that neither of the early phase studies for RTS,S or the DNA-based vaccines demonstrated the level of protection observed following immunization with irradiated sporozoites.[207,208] As with irradiated sporozoites, the attenuated vaccine approach can only be viable for global vaccination if sufficient material for large-scale immunization can be produced in culture and a suitable delivery system (preferably not vector-based) can be developed for human use.[209]
9. Current and Future Developments in Malaria Vaccines
Many of the vaccine candidates in table I were initially identified based on the reactivity of human sera against them (due to either natural infection or experimental sporozoite immunization) or an abundance of the antigen in fractionated parasite extracts. The analyses of these candidates through classical techniques (such as cloning from bacterial expression libraries or purification and protein sequencing) were time consuming, resulting in a slow (but steady) progress towards defining candidates antigens. Novel approaches in identifying new vaccine candidates and speeding up their molecular characterization will greatly increase the number of candidates brought to preclincial analysis. If this can be done in a framework of standardized assays that identify correlates for protection, then the number of antigens to go through to GMP products will be greatly increased. A milestone towards achieving this was the sequencing of several malarial genomes. The completion of the P. falciparum genome in 2002[210] provided researchers with the opportunity to develop these approaches through a better understanding of malarial parasite biology and its interaction with its hosts. Approximately 5300 P. falciparum genes were revealed upon completion of the genome and nearly 65% of these were hypothetical genes of unknown function. There is a real need for novel methods of high throughput characterization in order to rapidly interrogate these hypothetical genes to discover their usefulness as novel putative vaccine candidates. Hall et al.[211] recently completed ‘a comprehensive survey of the Plasmodium life cycle’ (reviewed in Fraunholz[212]). This study defined 4391 genes in P. falciparum with homologs in rodent malaria parasites allowing these species (which are more amenable to genetic manipulation)[213] to be used in determining gene function. This integration of genomic, transcriptomic, and proteomic datasets offers the prospect of a rationale approach to identifying key vaccine and drug targets using high throughput methodologies. Such an approach will be more powerful when used in conjunction with classical biochemical and cell biology techniques.[212]
The development of transfection technologies for P. falciparum (including the recent advances in double crossover integration into the genome)[214] provides a powerful molecular tool to define the role of individual genes within the cellular biology of this organism. However, the application of this technique is limited by the process being relatively inefficient, labor intensive, and time consuming. Genetic manipulations of essential asexually expressed genes are often not possible due to deleterious effects. Despite these limitations, gene knockout has been effectively used to characterize genes involved in processes such as the invasion of red blood cells[215] and cellular adhesion,[216–218] many of which represent either current or putative vaccine candidates.
The ability to identify homologs in rodent malaria systems means that the more efficient and rapid knockout technologies of P. Berghei can be used to characterize gene function in P. falciparum.[213] Alternatively, more rapid gene knockout technologies for P. falciparum using transposable elements[219] will speed up gene characterization; Balu and colleagues[219] employed the lepidoptearan transposable element piggyBac to efficiently transform P. falciparum. Such a technique offers the potential to perform large scale mutagenesis of the P. falciparum genome in order to screen for interesting phenotypes and identify the loci which have been disrupted. Other advances in transfection techniques include the use of the mycobacterium Bxb1 integrase to produce genetically and phenotypically homogeneous transgenic parasite populations.[220] This integration occurred within 1 month in the absence of drug pressure.[221] With regards to the knockout of essential genes, the development of a successful inducible promoter (as has been achieved in the apicomplexan Toxoplasma gondii and currently being developed in P. falciparum)[222] remains a key goal. Such a system would allow essential genes to be knocked out and their function studied by regulating the expression level of episomal copies as well as looking at the effects of inappropriate timing of expression. In addition to gene knockout, green fluorescent protein (GFP) tagging has provided the ability to look at the cellular location of putative vaccine candidates in live cells, allowing scientists to investigate processes such as proteolytic cleavage and cellular export.
10. Conclusion
In the last 5 years, malaria vaccine development has seen advances on many fronts. A long-lasting immunity with a significant protective effect generated by RTS,S/AS02A has proved that a vaccine is technically feasible. This is coupled with a dramatic increase in the number of candidates being taken forward into GMP development for human trials and development on vaccine delivery platforms. The development of RTS,S/AS02A has undoubtedly been advanced due to an agreement between GSK Biologicals and the MVI. The MVI is a nonprofit organization dedicated to overcoming the barriers to malaria vaccine development. CSP has been under development as a vaccine for 18 years by GSK Biologicals, with a current cumulative expenditure of between $US75 and $US100 million. The financial and political power behind MVI, coupled with the definition of strategic goals to overcome the barriers to vaccine development and licensing is key to the scale of undertaking necessary to move an increasing number of vaccine candidates through the pipeline. In addition to MVI, the European Malaria Vaccine Initiative, the US Army Medical Research and Materiel Command (via the WRAIR), as well as private pharmaceutical companies such as GSK Biologicals are providing key resources and technical knowledge to push forward vaccine development.
Although there is a real philanthropic nature to the development of malaria vaccines for developing countries, there is realization that an increase in GDP resulting from the distribution of an efficacious vaccine[223] would produce a substantial increase in market potential for European and American countries. Therefore, there is real financial incentive for private companies to become involved in the race for a malaria vaccine. Technological progress in the field of vaccine development to protect against organisms such as group A Streptococcus can also be applied to the development of malaria vaccines. The ability of chimeric constructs to deliver strain-transcending immunity make it reasonable to predict that multiple allelic forms and/or multiple life-cycle stages of P. falciparum could be targeted by a malaria vaccine to increase efficacy. Promising results have also been achieved in genetically attenuating P. falciparum for the production of live attenuated vaccines, an approach which is receiving serious financial investment.
These positives must be tempered with some concerns. So far, the best efficacy seen with a malaria vaccine has been around 30% against clinical malaria, yet currently there is no vaccine widely in use as a public health tool that does not provide at least 75% sustained protection against infection and/or disease. It is also worth noting that all of the vaccines widely used as a public health tool in infants and young children in the developing world were first licensed, manufactured, and sold for use in children and/or adults in the developed world. While there are attempts to define better preclincial and clinical models to predict the efficacy of vaccines, there are no current models to accurately predict the usefulness of an asexual protein as a vaccine candidate. In addition, there are still no reliable immunologic correlates of protection for natural infections or immunizations. The requirement for multiple allelic forms to overcome allelic diversity (although technically possible) will no doubt increase the complexity of a vaccine and its developmental and production costs. This would also be the likely outcome of combining multiple antigens to target different life-cycle stages of the parasite. It can be seen that the field of malaria vaccine research has several challenges to overcome before a viable product for global usage can be produced.
Whether the increase in GDP of developing countries and the philanthropy of developed countries could sustain annual costs of around $US1 billion to buy and deploy an efficacious malaria vaccine is unknown. Were a licensed malaria vaccine to become a reality, and a successful vaccination program begin to reduce malaria-associated morbidity/mortality, then there is a real prospect that the lifespan of an individual malaria vaccine would be limited. The ability of the malaria parasite (and its vectors) to overcome successful intervention programs via a process of mutation and gene flow has been a recurrent theme in public health programs. Even if a vaccine is successfully developed and deployed, perhaps the biggest challenge for researchers and public health specialists will be predicting and responding to future evolutionary changes in this parasite that could render such a vaccine ineffective.
Notes
The use of trade names is for identification purposes only, and does not imply endorsement.
References
Singh B, Kim Sung L, Matusop A, et al. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet 2004 Mar 27; 363 (9414): 1017–24
Stoute JA, Heppner Jr DG, Mason CJ, et al. Phase 1 safety and immunogenicity trial of malaria vaccine RTS,S/AS02A in adults in a hyperendemic region of western Kenya. Am J Trop Med Hyg 2006 Jul; 75 (1): 166–70
Herrera S, Corradin G, Arévalo-Herrera M. An update on the search for a Plasmodium vivax vaccine. Trends Parasitol 2007 Mar; 23 (3): 122–8
Greenwood BM, Bojang K, Whitty CJ, et al. Malaria. Lancet 2005 Apr 23-29; 365 (9469): 1487–98
Sachs J, Malaney P. The economic and social burden of malaria. Nature 2002 Feb 7; 415 (6872): 680–5
Payne D. Spread of chloroquine resistance in Plasmodium falciparum. Parasitol Today 1987 Aug; 3 (8): 241–6
Marsh K. Malaria disaster in Africa. Lancet 1998 Sep 19; 352 (9132): 924
Plowe CV, Cortese JF, Djimde A, et al. Mutations in Plasmodium falciparum dihydrofolate reductase and dihydropteroate synthase and epidemiologic patterns of pyrimethamine-sulfadoxine use and resistance. J Infect Dis 1997 Dec; 176 (6): 1590–6
Uhlemann AC, Cameron A, Eckstein-Ludwig U, et al. A single amino acid residue can determine the sensitivity of SERCAs to artemisinins. Nat Struct Mol Biol 2005 Jul; 12 (7): 628–9
Hemingway J, Bates I. Malaria: past problems and future prospects. After more than a decade of neglect, malaria is finally back on the agenda for both biomedical research and public health politics. EMBO Rep 2003 Jun; 4: S29–31
Smith S. A malaria vaccine: what if? ODI Opinions 2004; 17 [online]. Available from URL: http://www.odi.org.uk/Publications/opinions/17_immunisations_JulyO4.html[Accessed 2007 Aug 27]
Moorthy VS, Good MF, Hill AV. Malaria vaccine developments. Lancet 2004 Jan 10; 363 (9403): 150–6
Berndt ER, Glennerster R, Kremer MR, et al. Advance market commitments for vaccines against neglected diseases: estimating costs and effectiveness. Health Econ 2007 May; 16 (5): 491–511
Hill AV. Pre-erythrocytic malaria vaccines: towards greater efficacy. Nat Rev Immunol 2006 Jan; 6 (1): 21–32
Nussenzweig RS, Vanderberg J, Spitalny GL, et al. Sporozoite-induced immunity in mammalian malaria: a review. Am J Trop Med Hyg 1972 Sep; 21 (5): 722–8
Hoffman SL, Goh LM, Luke TC, et al. Protection of humans against malaria by immunization with radiation-attenuated Plasmodium falciparum sporozoites. J Infect Dis 2002 Apr 15; 185 (8): 1155–64
Egan JE, Hoffman SL, Haynes JD, et al. Humoral immune responses in volunteers immunized with irradiated Plasmodium falciparum sporozoites. Am J Trop Med Hyg 1993 Aug; 49 (2): 166–73
Carvalho LJ, Daniel-Ribeiro CT, Goto H. Malaria vaccine: candidate antigens, mechanisms, constraints and prospects. Scand J Immunol 2002 Oct; 56 (4): 327–43
Schofield L, Hewitt MC, Evans K, et al. Synthetic GPI as a candidate anti-toxic vaccine in a model of malaria. Nature 2002 Aug 15; 418 (6899): 785–9
Herrington D, Davis J, Nardin E, et al. Successful immunization of humans with irradiated malaria sporozoites: humoral and cellular responses of the protected individuals. Am J Trop Med Hyg 1991 Nov; 45 (5): 539–47
Kumar KA, Sano G, Boscardin S, et al. The circumsporozoite protein is an immunodominant protective antigen in irradiated sporozoites. Nature 2006 Dec 14; 444 (7121): 937–40
Khusmith S, Charoenvit Y, Kumar S, et al. Protection against malaria by vaccination with sporozoite surface protein 2 plus CS protein. Science 1991 May 3; 252 (5006): 715–8
Zavala F, Cochrane AH, Nardin EH, et al. Circumsporozoite proteins of malaria parasites contain a single immunodominant region with two or more identical epitopes. J Exp Med 1983 Jun 1; 157 (6): 1947–57
Marsh K, Kinyanjui S. Immune effector mechanisms in malaria. Parasite Immunol 2006 Jan-Feb; 28 (l-2): 51–60
Cohen S, Mc GI, Carrington S. Gamma-globulin and acquired immunity to human malaria. Nature 1961 Nov 25; 192: 733–7
Sabchareon A, Burnouf T, Ouattara D, et al. Parasitologic and clinical human response to immunoglobulin administration in falciparum malaria. Am J Trop Med Hyg 1991 Sep; 45 (3): 297–308
Flanagan KL, Plebanski M, Odhiambo K, et al. Cellular reactivity to the P. Falciparum protein trap in adult kenyans: novel epitopes, complex cytokine patterns, and the impact of natural antigenic variation. Am J Trop Med Hyg 2006 Mar; 74 (3): 367–75
Migot-Nabias F, Deloron P, Ringwald P, et al. Immune response to Plasmodium falciparum liver stage antigen-1: geographical variations within Central Africa and their relationship with protection from clinical malaria. Trans R Soc Trop Med Hyg 2000 Sep-Oct; 94 (5): 557–62
Hammond GW, Parker J, Mimms L, et al. Comparison of immunogenicity of two yeast-derived recombinant hepatitis B vaccines. Vaccine 1991 Feb; 9 (2): 97–100
Dunachie SJ, Walther M, Epstein JE, et al. A DNA prime-modified vaccinia virus Ankara boost vaccine encoding thrombospondin-related adhesion protein but not circumsporozoite protein partially protects healthy malaria-naive adults against Plasmodium falciparum sporozoite challenge. Infect Immun 2006 Oct; 74 (10): 5933–42
Moorthy VS, Imoukhuede EB, Milligan P, et al. A randomised, double-blind, controlled vaccine efficacy trial of DNA/MVA ME-TRAP against malaria infection in Gambian adults. PLoS Med 2004 Nov; 1 (2): e33
Bejon P, Mwacharo J, Kai O, et al. A Phase 2b randomised trial of the candidate malaria vaccines FP9 ME-TRAP and MVA ME-TRAP among children in Kenya. PLoS Clin Trials 2006 Oct 20; 1 (6): e29
Conway DJ, Polley SD. Measuring immune selection. Parasitology 2002; 125 Suppl.: S3–16
Deans JA, Alderson T, Thomas AW, et al. Rat monoclonal antibodies which inhibit the in vitro multiplication of Plasmodium knowlesi. Clin Exp Immunol 1982 Aug; 49 (2): 297–309
Polley SD, Mwangi T, Kocken CH, et al. Human antibodies to recombinant protein constructs of Plasmodium falciparum Apical Membrane Antigen 1 (AMA1) and their associations with protection from malaria. Vaccine 2004 Dec 16; 23 (5): 718–28
Collins WE, Pye D, Crewther PE, et al. Protective immunity induced in squirrel monkeys with recombinant apical membrane antigen-1 of Plasmodium fragile. Am J Trop Med Hyg 1994 Dec; 51 (6): 711–9
Narum DL, Ogun SA, Thomas AW, et al. Immunization with parasite-derived apical membrane antigen 1 or passive immunization with a specific monoclonal antibody protects BALB/c mice against lethal Plasmodium yoelii yoelii YM blood-stage infection. Infect Immun 2000 May; 68 (5): 2899–906
Calvo-Calle JM, Oliveira GA, Watta CO, et al. A linear peptide containing minimal T- and B-cell epitopes of Plasmodium falciparum circumsporozoite protein elicits protection against transgenic sporozoite challenge. Infect Immun 2006 Dec; 74 (12): 6929–39
Yoshida N, Nussenzweig RS, Potocnjak P, et al. Hybridoma produces protective antibodies directed against the sporozoite stage of malaria parasite. Science 1980 Jan 4; 207 (4426): 71–3
John CC, Moormann AM, Pregibon DC, et al. Correlation of high levels of antibodies to multiple pre-erythrocytic Plasmodium falciparum antigens and protection from infection. Am J Trop Med Hyg 2005 Jul; 73 (1): 222–8
Mlambo G, Mutambu SL, Mduluza T, et al. Antibody responses to Plasmodium falciparum vaccine candidate antigens in three areas distinct with respect to altitude. Acta Trop 2006 Nov; 100 (1-2): 70–8
Aley SB, Bates MD, Tam JP, et al. Synthetic peptides from the circumsporozoite proteins of Plasmodium falciparum and Plasmodium knowlesi recognize the human hepatoma cell line HepG2-A16 in vitro. J Exp Med 1986 Dec 1; 164 (6): 1915–22
Rogers WO, Rogers MD, Hedstrom RC, et al. Characterization of the gene encoding sporozoite surface protein 2, a protective Plasmodium yoelii sporozoite antigen. Mol Biochem Parasitol 1992 Jul; 53 (1–2): 45–51
Khusmith S, Sedegah M, Hoffman SL. Complete protection against Plasmodium yoelii by adoptive transfer of a CD8+ cytotoxic T-cell clone recognizing sporozoite surface protein 2. Infect Immun 1994 Jul; 62 (7): 2979–83
John CC, Zickafoose JS, Sumba PO, et al. Antibodies to the Plasmodium falciparum antigens circumsporozoite protein, thrombospondin-related adhesive protein, and liver-stage antigen 1 vary by ages of subjects and by season in a highland area of Kenya. Infect Immun 2003 Aug; 71 (8): 4320–5
Dolo A, Modiano D, Doumbo O, et al. Thrombospondin related adhesive protein (TRAP), a potential malaria vaccine candidate. Parassitologia 1999 Sep; 41 (1-3): 425–8
Kurtis JD, Lanar DE, Opollo M, et al. Interleukin-10 responses to liver-stage antigen 1 predict human resistance to Plasmodium falciparum. Infect Immun 1999 Jul; 67 (7): 3424–9
John CC, Moormann AM, Sumba PO, et al. Gamma interferon responses to Plasmodium falciparum liver-stage antigen 1 and thrombospondin-related adhesive protein and their relationship to age, transmission intensity, and protection against malaria. Infect Immun 2004 Sep; 72 (9): 5135–42
May J, Lell B, Luty AJ, et al. HLA-DQB 1s501-restricted Th1 type immune responses to Plasmodium falciparum liver stage antigen 1 protect against malaria anemia and reinfections. J Infect Dis 2001 Jan 1; 183 (1): 168–72
Brahimi K, Badell E, Sauzet JP, et al. Human antibodies against Plasmodium falciparum liver-stage antigen 3 cross-react with Plasmodium yoelii preerythrocytic-stage epitopes and inhibit sporozoite invasion in vitro and in vivo. Infect Immun 2001 Jun; 69 (6): 3845–52
Perlaza BL, Zapata C, Valencia AZ, et al. Immunogenicity and protective efficacy of Plasmodium falciparum liver-stage Ag-3 in Aotus lemurinus griseimembra monkeys. Eur J Immunol 2003 May; 33 (5): 1321–7
Daubersies P, Thomas AW, Millet P, et al. Protection against Plasmodium falciparum malaria in chimpanzees by immunization with the conserved preerythrocytic liver-stage antigen 3. Nat Med 2000 Nov; 6 (11): 1258–63
Meraldi V, Nebie I, Tiono AB, et al. Natural antibody response to Plasmodium falciparum Exp-1, MSP-3 and GLURP long synthetic peptides and association with protection. Parasite Immunol 2004 Jun-Jul; 26 (6–7): 265–72
Singh S, Miura K, Zhou H, et al. Immunity to recombinant plasmodium falciparum merozoite surface protein 1 (MSP1): protection in Aotus nancymai monkeys strongly correlates with anti-MSP1 antibody titer and in vitro parasite-inhibitory activity. Infect Immun 2006 Aug; 74 (8): 4573–80
deKoning-Ward TF, O’Donnell RA, Drew DR, et al. A new rodent model to assess blood stage immunity to the Plasmodium falciparum antigen merozoite surface protein 119 reveals a protective role for invasion inhibitory antibodies. J Exp Med 2003 Sep 15; 198 (6): 869–75
Conway DJ, Cavanagh DR, Tanabe K, et al. A principal target of human immunity to malaria identified by molecular population genetic and immunological analyses. Nat Med 2000 Jun; 6 (6): 689–92
Perraut R, Marrama L, Diouf B, et al. Antibodies to the conserved C-terminal domain of the Plasmodium falciparum merozoite surface protein 1 and to the merozoite extract and their relationship with in vitro inhibitory antibodies and protection against clinical malaria in a Senegalese village. J Infect Dis 2005 Jan 15; 191 (2): 264–71
Bergmann-Leitner ES, Duncan EH, Mullen GE, et al. Critical evaluation of different methods for measuring the functional activity of antibodies against malaria blood stage antigens. Am J Trop Med Hyg 2006 Sep; 75 (3): 437–42
Hui G, Hashimoto C. Plasmodium falciparum anti-MSP1-19 antibodies induced by MSP1-42 and MSP1-19 based vaccines differed in specificity and parasite growth inhibition in terms of recognition of conserved versus variant epitopes. Vaccine 2007 Jan 15; 25 (5): 948–56
Pan W, Huang D, Zhang Q, et al. Fusion of two malaria vaccine candidate antigens enhances product yield, immunogenicity, and antibody-mediated inhibition of parasite growth in vitro. J Immunol 2004 May 15; 172 (10): 6167–74
Salazar LM, Alba MP, Torres MH, et al. Protection against experimental malaria associated with AMA-1 peptide analogue structures. FEBS Lett 2002 Sep 11; 527 (1–3): 95–100
Burns Jr JM, Flaherty PR, Nanavati P, et al. Protection against Plasmodium chabaudi malaria induced by immunization with apical membrane antigen 1 and merozoite surface protein 1 in the absence of gamma interferon or interleukin-4. Infect Immun 2004 Oct; 72 (10): 5605–12
Cubillos M, Salazar LM, Torres L, et al. Protection against experimental P. falciparum malaria is associated with short AMA-1 peptide analogue alpha-helical structures. Biochimie 2002 Dec; 84 (12): 1181–8
Kocken CH, Withers-Martinez C, Dubbeld MA, et al. High-level expression of the malaria blood-stage vaccine candidate Plasmodium falciparum apical membrane antigen 1 and induction of antibodies that inhibit erythrocyte invasion. Infect Immun 2002 Aug; 70 (8): 4471–6
Stowers AW, Kennedy MC, Keegan BP, et al. Vaccination of monkeys with recombinant Plasmodium falciparum apical membrane antigen 1 confers protection against blood-stage malaria. Infect Immun 2002 Dec; 70 (12): 6961–7
Dutta S, Haynes JD, Moch JK, et al. Invasion-inhibitory antibodies inhibit proteolytic processing of apical membrane antigen 1 of Plasmodium falciparum merozoites. Proc Natl Acad Sci U S A 2003 Oct 14; 100 (21): 12295–300
Miao J, Li X, Liu Z, et al. Immune responses in mice induced by prime-boost schemes of the Plasmodium falciparum apical membrane antigen 1 (PfAMAl)based DNA, protein and recombinant modified vaccinia Ankara vaccines. Vaccine 2006 Sep 11; 24 (37-39): 6187–98
Coley AM, Parisi K, Masciantonio R, et al. The most polymorphic residue on Plasmodium falciparum apical membrane antigen 1 determines binding of an invasion-inhibitory antibody. Infect Immun 2006 May; 74 (5): 2628–36
Healer J, Murphy V, Hodder AN, et al. Allelic polymorphisms in apical membrane antigen-1 are responsible for evasion of antibody-mediated inhibition in Plasmodium falciparum. Mol Microbiol 2004 Apr; 52 (1): 159–68
Mullen GE, Giersing BK, Ajose-Popoola O, et al. Enhancement of functional antibody responses to AMAl-Cl/Alhydrogel, a Plasmodium falciparum malaria vaccine, with CpG oligodeoxynucleotide. Vaccine 2006 Mar 24; 24 (14): 2497–505
Sim BK, Narum DL, Liang H, et al. Induction of biologically active antibodies in mice, rabbits, and monkeys by Plasmodium falciparum EBA-175 region II DNA vaccine. Mol Med 2001 Apr; 7 (4): 247–54
Bermudez A, Cifuentes G, Guzman F, et al. Immunogenicity and protectivity of Plasmodium falciparum EBA-175 peptide and its analog is associated with alpha-helical region shortening and displacement. Biol Chem 2003 Oct-Nov; 384 (10–11): 1443–50
Jones TR, Narum DL, Gozalo AS, et al. Protection of Aotus monkeys by Plasmodium falciparum EBA-175 region II DNA prime-protein boost immunization regimen. J Infect Dis 2001 Jan 15; 183 (2): 303–12
Touré FS, Deloron P, Migot-Nabias F. Analysis of human antibodies to erythrocyte binding antigen 175 peptide 4 of Plasmodium falciparum. Clin Med Res 2006 Mar; 4 (1): 1–6
Pandey KC, Singh S, Pattnaik P, et al. Bacterially expressed and refolded receptor binding domain of Plasmodium falciparum EBA-175 elicits invasion inhibitory antibodies. Mol Biochem Parasitol 2002 Aug 7; 123 (1): 23–33
Singh AP, Puri SK, Chitnis CE. Antibodies raised against receptor-binding domain of Plasmodium knowlesi Duffy binding protein inhibit erythrocyte invasion. Mol Biochem Parasitol 2002 Apr 30; 121 (1): 21–31
Ridley RG, Takacs B, Etlinger H, et al. A rhoptry antigen of Plasmodium falciparum is protective in Saimiri monkeys. Parasitology 1990 Oct; 101 Pt 2: 187–92
Alifrangis M, Lemnge MM, Moon R, et al. IgG reactivities against recombinant Rhoptry-Associated Protein-1 (rRAP-1) are associated with mixed Plasmodium infections and protection against disease in Tanzanian children. Parasitology 1999 Oct; 119 (Pt 4): 337–42
Fonjungo PN, Elhassan IM, Cavanagh DR, et al. A longitudinal study of human antibody responses to Plasmodium falciparum rhoptry-associated protein 1 in a region of seasonal and unstable malaria transmission. Infect Immun 1999 Jun; 67 (6): 2975–85
Jacobson KC, Thurman J, Schmidt CM, et al. A study of antibody and T cell recognition of rhoptry-associated protein-1 (RAP-1) and RAP-2 recombinant proteins and peptides of Plasmodium falciparum in migrants and residents of the state of Rondonia, Brazil. Am J Trop Med Hyg 1998 Aug; 59 (2): 208–16
Stowers AW, Cooper JA, Ehrhardt T, et al. A peptide derived from a B cell epitope of Plasmodium falciparum rhoptry associated protein 2 specifically raises antibodies to rhoptry associated protein 1. Mol Biochem Parasitol 1996 Nov 25; 82 (2): 167–80
Harnyuttanakorn P, McBride JS, Donachie S, et al. Inhibitory monoclonal antibodies recognise epitopes adjacent to a proteolytic cleavage site on the RAP-1 protein of Plasmodium falciparum. Mol Biochem Parasitol 1992 Oct; 55 (1–2): 177–86
Perrin LH, Ramirez E, Lambert PH, et al. Inhibition of P. falciparum growth in human erythrocytes by monoclonal antibodies. Nature 1981 Jan 22; 289 (5795): 301–3
Howard RF, Jacobson KC, Rickel E, et al. Analysis of inhibitory epitopes in the Plasmodium falciparum rhoptry protein RAP-1 including identification of a second inhibitory epitope. Infect Immun 1998 Jan; 66 (1): 380–6
Okech B, Mujuzi G, Ogwal A, et al. High titers of IgG antibodies against Plasmodium falciparum serine repeat antigen 5 (SERA5) are associated with protection against severe malaria in Ugandan children. Am J Trop Med Hyg 2006 Feb; 74 (2): 191–7
Okech BA, Nalunkuma A, Okello D, et al. Natural human immunoglobulin G subclass responses to Plasmodium falciparum serine repeat antigen in Uganda. Am J Trop Med Hyg 2001 Dec; 65 (6): 912–7
Aoki S, Li J, Itagaki S, et al. Serine repeat antigen (SERA5) is predominantly expressed among the SERA multigene family of Plasmodium falciparum, and the acquired antibody titers correlate with serum inhibition of the parasite growth. J Biol Chem 2002 Dec 6; 277 (49): 47533–40
Fox BA, Horii T, Bzik DJ. Plasmodium falciparum: fine-mapping of an epitope of the serine repeat antigen that is a target of parasite-inhibitory antibodies. Exp Parasitol 2002 May; 101 (1): 69–72
Soe S, Singh S, Camus D, et al. Plasmodium falciparum serine repeat protein, a new target of monocyte-dependent antibody-mediated parasite killing. Infect Immun 2002 Dec; 70 (12): 7182–4
Metzger WG, Okenu DM, Cavanagh DR, et al. Serum IgG3 to the Plasmodium falciparum merozoite surface protein 2 is strongly associated with a reduced prospective risk of malaria. Parasite Immunol 2003 Jun; 25 (6): 307–12
Polley SD, Conway DJ, Cavanagh DR, et al. High levels of serum antibodies to merozoite surface protein 2 of Plasmodium falciparum are associated with reduced risk of clinical malaria in coastal Kenya. Vaccine 2006 May 8; 24 (19): 4233–46
Carvalho LJ, Oliveira SG, Theisen M, et al. Immunization of Saimiri sciureus monkeys with Plasmodium falciparum merozoite surface protein-3 and glutamate-rich protein suggests that protection is related to antibody levels. Scand J Immunol 2004 Apr; 59 (4): 363–72
Polley SD, Tetteh KK, Lloyd JM, et al. Plasmodium falciparum merozoite surface protein 3 is a target of allele-specific immunity and alleles are maintained by natural selection. J Infect Dis 2007 Jan 15; 195 (2): 279–87
Soe S, Theisen M, Roussilhon C, et al. Association between protection against clinical malaria and antibodies to merozoite surface antigens in an area of hyperendemicity in Myanmar: complementarity between responses to merozoite surface protein 3 and the 220-kilodalton glutamate-rich protein. Infect Immun 2004 Jan; 72 (1): 247–52
Lundquist R, Nielsen LK, Jafarshad A, et al. Human recombinant antibodies against Plasmodium falciparum merozoite surface protein 3 cloned from peripheral blood leukocytes of individuals with immunity to malaria demonstrate antiparasitic properties. Infect Immun 2006 Jun; 74 (6): 3222–31
Oeuvray C, Bouharoun-Tayoun H, Gras-Masse H, et al. Merozoite surface protein-3: a malaria protein inducing antibodies that promote Plasmodium falciparum killing by cooperation with blood monocytes. Blood 1994 Sep 1; 84 (5): 1594–602
Goschnick MW, Black CG, Kedzierski L, et al. Merozoite surface protein 4/5 provides protection against lethal challenge with a heterologous malaria parasite strain. Infect Immun 2004 Oct; 72 (10): 5840–9
Kedzierski L, Black CG, Goschnick MW, et al. Immunization with a combination of merozoite surface proteins 4/5 and 1 enhances protection against lethal challenge with Plasmodium yoelii. Infect Immun 2002 Dec; 70 (12): 6606–13
Kedzierski L, Black CG, Stowers AW, et al. Comparison of the protective efficacy of yeast-derived and Escherichia coli-derived recombinant merozoite surface protein 4/5 against lethal challenge by Plasmodium yoelii. Vaccine 2001 Sep 14; 19 (32): 4661–8
Rainczuk A, Smooker PM, Kedzierski L, et al. The protective efficacy of MSP4/5 against lethal Plasmodium chabaudi adami challenge is dependent on the type of DNA vaccine vector and vaccination protocol. Vaccine 2003 Jun 20; 21 (21–22): 3030–42
Wang L, Richie TL, Stowers A, et al. Naturally acquired antibody responses to Plasmodium falciparum merozoite surface protein 4 in a population living in an area of endemicity in Vietnam. Infect Immun 2001 Jul; 69 (7): 4390–7
Makobongo MO, Keegan B, Long CA, et al. Immunization of Aotus monkeys with recombinant cysteine-rich interdomain region 1 alpha protects against severe disease during Plasmodium falciparum reinfection. J Infect Dis 2006 Mar 1; 193 (5): 731–40
Moll K, Pettersson F, Vogt AM, et al. Generation of cross-protective antibodies against Plasmodium falciparum sequestration by immunization with an erythrocyte membrane protein 1-duffy binding-like 1alpha domain. Infect Immun 2007 Jan; 75 (1): 211–9
Ahuja S, Pettersson F, Moll K, et al. Induction of cross-reactive immune responses to NTS-DBL-1alpha/x of PfEMP1 and in vivo protection on challenge with Plasmodium falciparum. Vaccine 2006 Aug 28; 24 (35–36): 6140–54
Chen Q, Pettersson F, Vogt AM, et al. Immunization with PfEMPl-DBL1 alpha generates antibodies that disrupt rosettes and protect against the sequestration of Plasmodium falciparum-infected erythrocytes. Vaccine 2004 Jul 29; 22 (21–22): 2701–12
Baruch DI, Gamain B, Barnwell JW, et al. Immunization of Aotus monkeys with a functional domain of the Plasmodium falciparum variant antigen induces protection against a lethal parasite line. Proc Natl Acad Sci U S A 2002 Mar 19; 99 (6): 3860–5
Joergensen L, Vestergaard LS, Turner L, et al. 3D7-derived plasmodium falciparum erythrocyte membrane protein 1 is a frequent target of naturally acquired antibodies recognizing protein domains in a particular pattern independent of malaria transmission intensity. J Immunol 2007 Jan 1; 178 (1): 428–35
Lusingu JP, Jensen AT, Vestergaard LS, et al. Levels of plasma immunoglobulin G with specificity against the cysteine-rich interdomain regions of a semiconserved Plasmodium falciparum erythrocyte membrane protein 1, VAR4, predict protection against malarial anemia and febrile episodes. Infect Immun 2006 May; 74 (5): 2867–75
Dodoo D, Staalsoe T, Giha H, et al. Antibodies to variant antigens on the surfaces of infected erythrocytes are associated with protection from malaria in Ghanaian children. Infect Immun 2001 Jun; 69 (6): 3713–8
Giha HA, Staalsoe T, Dodoo D, et al. Antibodies to variable Plasmodium falciparum-infected erythrocyte surface antigens are associated with protection from novel malaria infections. Immunol Lett 2000 Feb 1; 71 (2): 117–26
Avril M, Gamain B, Lepolard C, et al. Characterization of anti-var2CSA-PfEMP1 cytoadhesion inhibitory mouse monoclonal antibodies. Microbes Infect 2006 Nov-Dec; 8 (14–15): 2863–71
Yipp BG, Baruch DI, Brady C, et al. Recombinant PfEMP1 peptide inhibits and reverses cytoadherence of clinical Plasmodium falciparum isolates in vivo. Blood 2003 Jan 1; 101 (1): 331–7
Hogh B, Petersen E, Dziegiel M, et al. Antibodies to a recombinant glutamate-rich Plasmodium falciparum protein: evidence for protection of individuals living in a holoendemic area of Liberia. Am J Trop Med Hyg 1992 Mar; 46 (3): 307–13
Dziegiel M, Rowe P, Bennett S, et al. Immunoglobulin M and G antibody responses to Plasmodium falciparum glutamate-rich protein: correlation with clinical immunity in Gambian children. Infect Immun 1993 Jan; 61 (1): 103–8
Dodoo D, Theisen M, Kurtzhals JA, et al. Naturally acquired antibodies to the glutamate-rich protein are associated with protection against Plasmodium falciparum malaria. J Infect Dis 2000 Mar; 181 (3): 1202–5
Theisen M, Dodoo D, Toure-Balde A, et al. Selection of glutamate-rich protein long synthetic peptides for vaccine development: antigenicity and relationship with clinical protection and immunogenicity. Infect Immun 2001 Sep; 69 (9): 5223–9
Oeuvray C, Theisen M, Rogier C, et al. Cytophilic immunoglobulin responses to Plasmodium falciparum glutamate-rich protein are correlated with protection against clinical malaria in Dielmo, Senegal. Infect Immun 2000 May; 68 (5): 2617–20
Lusingu JP, Vestergaard LS, Alifrangis M, et al. Cytophilic antibodies to Plasmodium falciparum glutamate rich protein are associated with malaria protection in an area of holoendemic transmission. Malar J 2005; 4: 48
Hermsen CC, Verhage DF, Telgt DS, et al. Glutamate-rich protein (GLURP) induces antibodies that inhibit in vitro growth of Plasmodium falciparum in a phase 1 malaria vaccine trial. Vaccine 2007 Apr 12; 25 (15): 2930–40
Theisen M, Soe S, Oeuvray C, et al. The glutamate-rich protein (GLURP) of Plasmodium falciparum is a target for antibody-dependent monocyte-mediated inhibition of parasite growth in vitro. Infect Immun 1998 Jan; 66 (1): 11–7
Theisen M, Soe S, Jessing SG, et al. Identification of a major B-cell epitope of the Plasmodium falciparum glutamate-rich protein (GLURP), targeted by human antibodies mediating parasite killing. Vaccine 2000 Sep 15; 19 (2–3): 204–12
Collins WE, Anders RF, Pappaioanou M, et al. Immunization of Aotus monkeys with recombinant proteins of an erythrocyte surface antigen of Plasmodium falciparum. Nature 1986 Sep 18-24; 323 (6085): 259–62
Collins WE, Pappaioanou M, Anders RF, et al. Immunization trials with the ringinfected erythrocyte surface antigen of Plasmodium falciparum in owl monkeys (Aotus vociferans). Am J Trop Med Hyg 1988 Mar; 38 (2): 268–82
Astagneau P, Chougnet C, Lepers JP, et al. Antibodies to the 4-mer repeat of the ring-infected erythrocyte surface antigen (Pfl55/RESA) protect against Plasmodium falciparum malaria. Int J Epidemiol 1994 Feb; 23 (1): 169–75
al-Yaman F, Genton B, Anders R, et al. Assessment of the role of the humoral response to Plasmodium falciparum MSP2 compared to RESA and SPf66 in protecting Papua New Guinean children from clinical malaria. Parasite Immunol 1995 Sep; 17 (9): 493–501
Wahlin Flyg B, Siddique AB, Perlmann P, et al. Inhibition of in vitro growth of Plasmodium falciparum field isolates mediated by human antibodies to Pfl55/ RESA and Pf332. Parasite Immunol 1999 Jun; 21 (6): 331–4
Siddique AB, Ahlborg N, Wahlin Flyg B, et al. Antibodies to sequences in a nonrepeat region of Plasmodium falciparum antigen Pfl55/RESA inhibit either cytoadherence or parasite growth in vitro. Parasitology 1998 Sep; 117 (Pt 3): 209–16
Roeffen W, Lensen T, Mulder B, et al. Transmission blocking immunity as observed in a feeder system and serological reactivity to Pfs 48/45 and Pfs230 in field sera. Mem Inst Oswaldo Cruz 1994; 89 Suppl. 2: 13–5
Roeffen W, Mulder B, Teelen K, et al. Association between anti-Pfs48/45 reactivity and P. falciparum transmission-blocking activity in sera from Cameroon. Parasite Immunol 1996 Feb; 18 (2): 103–9
Drakeley CJ, Eling W, Teelen K, et al. Parasite infectivity and immunity to Plasmodium falciparum gametocytes in Gambian children. Parasite Immunol 2004 Apr; 26 (4): 159–65
Bustamante PJ, Woodruff DC, Oh J, et al. Differential ability of specific regions of Plasmodium falciparum sexual-stage antigen, Pfs230, to induce malaria transmission-blocking immunity. Parasite Immunol 2000 Aug; 22 (8): 373–80
Healer J, McGuinness D, Carter R, et al. Transmission-blocking immunity to Plasmodium falciparum in malaria-immune individuals is associated with antibodies to the gamete surface protein Pfs230. Parasitology 1999 Nov; 119 (Pt 5): 425–33
Healer J, McGuinness D, Hopcroft P, et al. Complement-mediated lysis of Plasmodium falciparum gametes by malaria-immune human sera is associated with antibodies to the gamete surface antigen Pfs230. Infect Immun 1997 Aug; 65 (8): 3017–23
Lobo CA, Dhar R, Kumar N. Immunization of mice with DNA-based Pfs25 elicits potent malaria transmission-blocking antibodies. Infect Immun 1999 Apr; 67 (4): 1688–93
Moelans II, Cohen J, Marchand M, et al. Induction of Plasmodium falciparum sporozoite-neutralizing antibodies upon vaccination with recombinant Pfs16 vaccinia virus and/or recombinant Pfs16 protein produced in yeast. Mol Biochem Parasitol 1995 Jun; 72 (1–2): 179–92
Arakawa T, Komesu A, Otsuki H, et al. Nasal immunization with a malaria transmission-blocking vaccine candidate, Pfs25, induces complete protective immunity in mice against field isolates of Plasmodium falciparum. Infect Immun 2005 Nov; 73 (11): 7375–80
Kubler-Kielb J, Majadly F, Wu Y, et al. Long-lasting and transmission-blocking activity of antibodies to Plasmodium falciparum elicited in mice by protein conjugates of Pfs25. Proc Natl Acad Sci U S A 2007 Jan 2; 104 (1): 293–8
Ramjanee S, Robertson JS, Franke-Fayard B, et al. The use of transgenic Plasmodium berghei expressing the Plasmodium vivax antigen P25 to determine the transmission-blocking activity of sera from malaria vaccine trials. Vaccine 2007 Jan 15; 25 (5): 886–94
Collins WE, Barnwell JW, Sullivan JS, et al. Assessment of transmission-blocking activity of candidate Pvs25 vaccine using gametocytes from chimpanzees. Am J Trop Med Hyg 2006 Feb; 74 (2): 215–21
Duffy PE, Kaslow DC. A novel malaria protein, Pfs28, and Pfs25 are genetically linked and synergistic as falciparum malaria transmission-blocking vaccines. Infect Immun 1997 Mar; 65 (3): 1109–13
McGregor IA. The passive transfer of human malarial immunity. Am J Trop Med Hyg 1964 Jan; 13 Suppl.: 237–9
Polley SD, Weedall GD, Thomas AW, et al. Orthologous gene sequences of merozoite surface protein 1 (MSP1) from Plasmodium reichenowi and P. gallinaceum confirm an ancient divergence of P. falciparum alleles. Mol Biochem Parasitai 2005 Jul; 142 (1): 25–31
Adams JH, Sim BK, Dolan SA, et al. A family of erythrocyte binding proteins of malaria parasites. Proc Natl Acad Sci U S A 1992 Aug 1; 89 (15): 7085–9
Chatterjee S, Perignon JL, Van Marck E, et al. How reliable are models for malaria vaccine development? Lessons from irradiated sporozoite immunizations. J Postgrad Med 2006 Oct-Dec; 52 (4): 321–4
Singh S, Soe S, Mejia JP, et al. Identification of a conserved region of Plasmodium falciparum MSP3 targeted by biologically active antibodies to improve vaccine design. J Infect Dis 2004 Sep 1; 190 (5): 1010–8
Herrera S, Herrera MA, Perlaza BL, et al. Immunization of Aotus monkeys with Plasmodium falciparum blood-stage recombinant proteins. Proc Natl Acad Sci U S A 1990 May; 87 (10): 4017–21
Gramzinski RA, Obaldia 3rd N, Jones TR, et al. Susceptibility of Panamanian Aotus lemurinus lemurinus to sporozoite-induced Plasmodium falciparum (Santa Lucia) infection. Am J Trop Med Hyg 1999 Jul; 61 (1): 19–25
Miller LH, Haynes JD, McAuliffe FM, et al. Evidence for deifferences in erythrocyte surface receptors for the malaria parasites Plasmodium falciparum and Plasmodium knowlesi. J Exp Med 1977; 46: 277–81
Martin MJ, Rayner JC, Gagneux P, et al. Evolution of human-chimpanzee differences in malaria susceptibility: relationship to human genetic loss of Nglycolylneuraminic acid. Proc Natl Acad Sci U S A 2005 Sep 6; 102 (36): 12819–24
Malaria vaccine technology roadmap [online]. Available from URL: http://www.malariavaccineroadmap.net/[Accessed 2007 Aug 27]
Supporting development of a vaccine for malaria, the deadliest disease among Africa’s children [online]. Available from URL: http://www.gatesfoundation.org/nr/downloads/aboutus/learning/mvi.pdf [Accessed 2007 Aug 27]
Malaria Vaccine Initiative. Clinical trials: crucial steps on the road to a malaria vaccine [technical series]. 2004 Aug [online]. Available from URL: http://www.malariavaccineroadmap.net/pdfs/vaccine_steps.pdf [Accessed 2007 Aug 27]
Webster DP, Dunachie S, Vuola JM, et al. Enhanced T cell-mediated protection against malaria in human challenges by using the recombinant poxviruses FP9 and modified vaccinia virus Ankara. Proc Natl Acad Sci U S A 2005 Mar 29; 102 (13): 4836–41
Day KP, Karamalis F, Thompson J, et al. Genes necessary for expression of a virulence dete rminant and for transmission of Plasmodium falciparum are located on a 0.3-megabase region of chromosome 9. Proc Natl Acad Sci U S A 1993 Sep 1; 90 (17): 8292–6
Pologe LG, Ravetch JV. A chromosomal rearrangement in a P. falciparum histidine-rich protein gene is associated with the knobless phenotype. Nature 1986 Jul 31-Aug 6; 322 (6078): 474–7
Haiti: scaling the mountains. AIDS vaccine trial begins in the western hemisphere’s hardest-hit country [online]. Available from URL: http://www.aegis.com/pubs/iavi/2001/IAVI2001-0404.html [Accessed 2007 Aug 27]
Maar M, McGregor-Sutherland M, McGregor L. First nations taking charge of health research: the development of a community-based ethics review process on Manitoulin Island, Ontario, Canada. A report prepared for the National Council for Ethics in Human Research (NCEHR) [online]. Available from URL: http://www.ncehr-cnerh.org/pdf/whats_new/manitoulin_report_march_2005_english.pdf[Accessed 2007 Aug 27]
Stoute JA, Slaoui M, Heppner DG, et al. A preliminary evaluation of a recombinant circumsporozoite protein vaccine against Plasmodium falciparum malaria. RTS,S Malaria Vaccine Evaluation Group. N Engl J Med 1997 Jan 9; 336 (2): 86–91
Gupta RK, Siber GR. Adjuvants for human vaccines: current status, problems and future prospects. Vaccine 1995 Oct; 13 (14): 1263–76
World Health Organization. New vaccines against infectious diseases: research and development status IVR. April 2005, updated February 2006 [online]. Available from URL: http://www.who.int/vaccine_research/documents/en/Status_Table.pdf[Accessed 2007 Aug 27]
Alonso PL, Sacarlal J, Aponte JJ, et al. Duration of protection with RTS,S/AS02A malaria vaccine in prevention of Plasmodium falciparum disease in Mozambican children: single-blind extended follow-up of a randomised controlled trial. Lancet 2005 Dec 10; 366 (9502): 2012–8
Bojang KA, Olodude F, Pinder M, et al. Safety and immunogenicty of RTS,S/ AS02A candidate malaria vaccine in Gambian children. Vaccine 2005 Jul 14; 23 (32): 4148–57
Macete E, Aponte JJ, Guinovart C, et al. Safety and immunogenicity of the RTS,S/ AS02A candidate malaria vaccine in children aged 1-4 in Mozambique. Trop Med Int Health 2007 Jan; 12 (1): 37–46
Stoute JA, Kester KE, Krzych U, et al. Long-term efficacy and immune responses following immunization with the RTS,S malaria vaccine. J Infect Dis 1998 Oct; 178 (4): 1139–44
Doherty JF, Pinder M, Tornieporth N, et al. A phase I safety and immunogenicity trial with the candidate malaria vaccine RTS,S/SBAS2 in semi-immune adults in The Gambia. Am J Trop Med Hyg 1999 Dec; 61 (6): 865–8
Bojang KA, Milligan PJ, Pinder M, et al. Efficacy of RTS,S/AS02 malaria vaccine against Plasmodium falciparum infection in semi-immune adult men in The Gambia: a randomised trial. Lancet 2001 Dec 8; 358 (9297): 1927–34
Lopez JA, Weilenman C, Audran R, et al. A synthetic malaria vaccine elicits a potent CD8 (+) and CD4 (+) T lymphocyte immune response in humans, implications for vaccination strategies. Eur J Immunol 2001 Jul; 31 (7): 1989–98
Meraldi V, Audran R, Romero JF, et al. OM-174, a new adjuvant with a potential for human use, induces a protective response when administered with the synthetic C-terminal fragment 242-310 from the circumsporozoite protein of Plasmodium berghei. Vaccine 2003 Jun 2; 21 (19-20): 2485–91
Nardin EH, Oliveira GA, Calvo-Calle JM, et al. Phase I testing of a malaria vaccine composed of hepatitis B virus core particles expressing Plasmodium falciparum circumsporozoite epitopes. Infect Immun 2004 Nov; 72 (11): 6519–27
Walther M, Dunachie S, Keating S, et al. Safety, immunogenicity and efficacy of a pre-erythrocytic malaria candidate vaccine, ICC-1132 formulated in Seppic ISA 720. Vaccine 2005 Jan 4; 23 (7): 857–64
Oliveira GA, Wetzel K, Calvo-Calle JM, et al. Safety and enhanced immunogenicity of a hepatitis B core particle Plasmodium falciparum malaria vaccine formulated in adjuvant Montanide ISA 720 in a phase I trial. Infect Immun 2005 Jun; 73 (6): 3587–97
Parker SE, Monteith D, Horton H, et al. Safety of a GM-CSF adjuvant-plasmid DNA malaria vaccine. Gene Ther 2001 Jul; 8 (13): 1011–23
Hillier CJ, Ware LA, Barbosa A, et al. Process development and analysis of liverstage antigen 1, a preerythrocyte-stage protein-based vaccine for Plasmodium falciparum. Infect Immun 2005 Apr; 73 (4): 2109–15
Moorthy VS, Pinder M, Reece WH, et al. Safety and immunogenicity of DNA/ modified vaccinia virus Ankara malaria vaccination in African adults. J Infect Dis 2003 Oct 15; 188 (8): 1239–44
Moorthy VS, Imoukhuede EB, Keating S, et al. Phase 1 evaluation of 3 highly immunogenic prime-boost regimens, including a 12-month reboosting vaccination, for malaria vaccination in Gambian men. J Infect Dis 2004 Jun 15; 189 (12): 2213–9
Bejon P, Mwacharo J, Kai OK, et al. Immunogenicity of the candidate malaria vaccines FP9 and modified vaccinia virus Ankara encoding the pre-erythrocytic antigen ME-TRAP inl-6year old children in a malaria endemic area. Vaccine 2006 May 29; 24 (22): 4709–15
Ophorst OJ, Radosevic K, Havenga MJ, et al. Immunogenicity and protection of a recombinant human adenovirus serotype 35-based malaria vaccine against Plasmodium yoelii in mice. Infect Immun 2006 Jan; 74 (1): 313–20
Bejon P, Andrews L, Andersen RF, et al. Calculation of liver-to-blood inocula, parasite growth rates, and preerythrocytic vaccine efficacy, from serial quantitative polymerase chain reaction studies of volunteers challenged with malaria sporozoites. J Infect Dis 2005 Feb 15; 191 (4): 619–26
BenMohamed L, Gras-Masse H, Tartar A, et al. Lipopeptide immunization without adjuvant induces potent and long-lasting B, T helper, and cytotoxic T lymphocyte responses against a malaria liver stage antigen in mice and chimpanzees. Eur J Immunol 1997 May; 27 (5): 1242–53
Pattnaik P, Shakri AR, Singh S, et al. Immunogenicity of a recombinant malaria vaccine based on receptor binding domain of Plasmodium falciparum EBA-175. Vaccine 2007 Jan 15; 25 (5): 806–13
Ockenhouse CF, Angov E, Kester KE, et al. Phase I safety and immunogenicity trial of FMP1/AS02A, a Plasmodium falciparum MSP-1 asexual blood stage vaccine. Vaccine 2006 Apr 5; 24 (15): 3009–17
Langermans JA, Hensmann M, van Gijlswijk M, et al. Preclinical evaluation of a chimeric malaria vaccine candidate in montanide ISA 720: immunogenicity and safety in rhesus macaques. Hum Vaccin 2006 Sep 11; 2 (5): 222–6
Qian F, Pan W. Construction of a tetR-integrated Salmonella enterica serovar Typhi CVD908 strain that tightly controls expression of the major merozoite surface protein of Plasmodium falciparum for applications in human vaccine production. Infect Immun 2002 Apr; 70 (4): 2029–38
Toebe CS, Clements JD, Cardenas L, et al. Evaluation of immunogenicity of an oral Salmonella vaccine expressing recombinant Plasmodium berghei merozoite surface protein-1. Am J Trop Med Hyg 1997 Feb; 56 (2): 192–9
Matsumoto S, Yukitake H, Kanbara H, et al. Recombinant Mycobacterium bovis bacillus Calmette-Guerin secreting merozoite surface protein 1 (MSP1) induces protection against rodent malaria parasite infection depending on MSP1-stimulated interferon gamma and parasite-specific antibodies. J Exp Med 1998 Sep 7; 188 (5): 845–54
Matsumoto S, Yukitake H, Kanbara H, et al. Long-lasting protective immunity against rodent malaria parasite infection at the blood stage by recombinant BCG secreting merozoite surface protein-1. Vaccine 1999 Dec 10; 18 (9–10): 832–4
Audran R, Cachat M, Lurati F, et al. Phase I malaria vaccine trial with a long synthetic peptide derived from the merozoite surface protein 3 antigen. Infect Immun 2005 Dec; 73 (12): 8017–26
Theisen M, Soe S, Brunstedt K, et al. A Plasmodium falciparum GLURP-MSP3 chimeric protein: expression in Lactococcus lactis, immunogenicity and induction of biologically active antibodies. Vaccine 2004 Mar 12; 22 (9–10): 1188–98
Carvalho LJ, Alves FA, Bianco Jr C, et al. Immunization of Saimiri sciureus monkeys with a recombinant hybrid protein derived from the Plasmodium falciparum antigen glutamate-rich protein and merozoite surface protein 3 can induce partial protection with Freund and Montanide ISA720 adjuvants. Clin Diagn Lab Immunol 2005 Feb; 12 (2): 242–8
Genton B, Betuela I, Feiger I, et al. A recombinant blood-stage malaria vaccine reduces Plasmodium falciparum density and exerts selective pressure on parasite populations in a phase l-2b trial in Papua New Guinea. J Infect Dis 2002 Mar 15; 185 (6): 820–7
Genton B, Al-Yaman F, Betuela I, et al. Safety and immunogenicity of a threecomponent blood-stage malaria vaccine (MSP1, MSP2, RES A) against Plasmodium falciparum in Papua New Guinean children. Vaccine 2003 Dec 8; 22 (1): 30–41
Arizono N, Nakanihsi K, Horii T, et al. Progress in the molecular biology of malaria and the immunology of nematode infections. Trends Parasitol 2007 Apr; 23 (4): 175–81
Zou L, Miles AP, Wang J, et al. Expression of malaria transmission-blocking vaccine antigen Pfs25 in Pichia pastoris for use in human clinical trials. Vaccine 2003 Apr 2; 21 (15): 1650–7
Wu Y, Przysiecki C, Flanagan E, et al. Sustained high-titer antibody responses induced by conjugating a malarial vaccine candidate to outer-membrane protein complex. Proc Natl Acad Sci U S A 2006 Nov 28; 103 (48): 18243–8
Acosta CJ, Galindo CM, Schellenberg D, et al. Evaluation of the SPf66 vaccine for malaria control when delivered through the EPI scheme in Tanzania. Trop Med Int Health 1999 May; 4 (5): 368–76
Alonso PL, Sacarlal J, Aponte JJ, et al. Efficacy of the RTS,S/AS02A vaccine against Plasmodium falciparum infection and disease in young African children: randomised controlled trial. Lancet 2004 Oct 16-22; 364 (9443): 1411–20
Good MF, Pombo D, Quakyi IA, et al. Human T-cell recognition of the circumsporozoite protein of Plasmodium falciparum: immunodominant T-cell domains map to the polymorphic regions of the molecule. Proc Natl Acad Sci U S A 1988 Feb; 85 (4): 1199–203
Taylor RR, Smith DB, Robinson VJ, et al. Human antibody response to Plasmodium falciparum merozoite surface protein 2 is serogroup specific and predominantly of the immunoglobulin G3 subclass. Infect Immun 1995 Nov; 63 (11): 4382–8
Tetteh KK, Cavanagh DR, Corran P, et al. Extensive antigenic polymorphism within the repeat sequence of the Plasmodium falciparum merozoite surface protein 1 block 2 is incorporated in a minimal polyvalent immunogen. Infect Immun 2005 Sep; 73 (9): 5928–35
Polley SD, Tetteh KK, Cavanagh DR, et al. Repeat sequences in block 2 of Plasmodium falciparum merozoite surface protein 1 are targets of antibodies associated with protection from malaria. Infect Immun 2003 Apr; 71 (4): 1833–42
Franks S, Baton L, Tetteh K, et al. Genetic diversity and antigenic polymorphism in Plasmodium falciparum: extensive serological cross-reactivity between allelic variants of merozoite surface protein 2. Infect Immun 2003 Jun; 71 (6): 3485–95
McNeil SA, Halperin SA, Langley JM, et al. Safety and immunogenicity of 26-valent group a streptococcus vaccine in healthy adult volunteers. Clin Infect Dis 2005 Oct 15; 41 (8): 1114–22
Enosse S, Dobano C, Quelhas D, et al. RTS,S/AS02A Malaria vaccine does not induce parasite CSP T cell epitope selection and reduces multiplicity of infection. PLoS Clin Trials 2006 May; 1 (1): e5
Druilhe P, Spertini F, Soesoe D, et al. A malaria vaccine that elicits in humans antibodies able to kill Plasmodium falciparum. PLoS Med 2005 Nov; 2 (11): e344
Croteau G, Doyon L, Thibeault D, et al. Impaired fitness of human immunodeficiency virus type 1 variants with high-level resistance to protease inhibitors. J Virol 1997 Feb; 71 (2): 1089–96
vanDijk MR, Douradinha B, Franke-Fayard B, et al. Genetically attenuated, P36pdeficient malarial sporozoites induce protective immunity and apoptosis of infected liver cells. Proc Natl Acad Sci U S A 2005 Aug 23; 102 (34): 12194–9
Mueller AK, Labaied M, Kappe SH, et al. Genetically modified Plasmodium parasites as a protective experimental malaria vaccine. Nature 2005 Jan 13; 433 (7022): 164–7
Good MF. Genetically modified Plasmodium highlights the potential of whole parasite vaccine strategies. Trends Immunol 2005 Jun; 26 (6): 295–7
Butler D. Mosquito production mooted as fast track to malaria vaccine. Nature 2003 Oct 2; 425 (6957): 437
Gardner MJ, Hall N, Fung E, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002 Oct 3; 419 (6906): 498–511
Hall N, Karras M, Raine JD, et al. A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science 2005 Jan 7; 307 (5706): 82–6
Fraunholz MJ. Systems biology in malaria research. Trends Parasitol 2005 Sep; 21 (9): 393–5
Ecker A, Moon R, Sinden RE, et al. Generation of gene targeting constructs for Plasmodium berghei by a PCR-based method amenable to high throughput applications. Mol Biochem Parasitol 2006 Feb; 145 (2): 265–8
Duraisingh MT, Triglia T, Cowman AF. Negative selection of Plasmodium falciparum reveals targeted gene deletion by double crossover recombination. Int J Parasitol 2002 Jan; 32 (1): 81–9
Cowman AF, Crabb BS. Invasion of red blood cells by malaria parasites. Cell 2006 Feb 24; 124 (4): 755–66
Cooke BM, Buckingham DW, Glenister FK, et al. A Maurer’s cleft-associated protein is essential for expression of the major malaria virulence antigen on the surface of infected red blood cells. J Cell Biol 2006 Mar 13; 172 (6): 899–908
Rug M, Prescott SW, Fernandez KM, et al. The role of KAHRP domains in knob formation and cytoadherence of P. falciparum-infected human erythrocytes. Blood 2006 Jul 1; 108 (1): 370–8
Crabb BS, Cooke BM, Reeder JC, et al. Targeted gene disruption shows that knobs enable malaria-infected red cells to cytoadhere under physiological shear stress. Cell 1997 Apr 18; 89 (2): 287–96
Balu B, Shoue DA, Fraser Jr MJ, et al. High-efficiency transformation of Plasmodium falciparum by the lepidopteran transposable element piggyBac. Proc Natl Acad Sci U S A. 2005 Nov 8; 102 (45): 16391–6
Nkrumah LJ, Muhle RA, Moura PA, et al. Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nat Methods 2006 Aug; 3 (8): 615–21
Crabb BS, Gilson PR. A new system for rapid plasmid integration in Plasmodium parasites. Trends Microbiol 2007 Jan; 15 (1): 3–6
Meissner M, Krejany E, Gilson PR, et al. Tetracycline analogue-regulated transgene expression in Plasmodium falciparum blood stages using Toxoplasma gondii transactivators. Proc Natl Acad Sci U S A 2005 Feb 22; 102 (8): 2980–5
Tediosi F, Hutton G, Maire N, et al. Predicting the cost-effectiveness of introducing a pre-erythrocytic malaria vaccine into the expanded program on immunization in Tanzania. Am J Trop Med Hyg 2006 Aug; 75 (2 Suppl.): 131–43
Acknowledgments
We are grateful to Dr Quinton Fivelman, Dr Louisa McRoberts, and Dr Marie Polley for constructive comments on this review.
Dr Tetteh is funded by the Wellcome Trust (Program Grant 074695) and Dr Polley is funded by the European Commission (FP6; Signal, grant reference 073021/2/03/2). The authors have no conflicts of interest that are directly relevant to the content of this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tetteh, K.K., Polley, S.D. Progress and Challenges towards the Development of Malaria Vaccines. BioDrugs 21, 357–373 (2007). https://doi.org/10.2165/00063030-200721060-00004
Published:
Issue Date:
DOI: https://doi.org/10.2165/00063030-200721060-00004