
Abstract
Inside eukaryotic cells, small RNA duplexes, called small interfering RNAs (siRNAs), activate a conserved RNA interference (RNAi) pathway which leads to specific degradation of complementary target mRNAs through base-pairing recognition. As with other viruses, studies have shown that replication of the HIV-1 in cultured cells can be targeted and inhibited by synthetic siRNAs. The relative ease of siRNA design and the versatility of RNAi to target a broad spectrum of mRNAs have led to the promise that drug discovery in the RNAi pathway could be effective against pathogens.
This review discusses the current experimental principles that guide the application of RNAi against HIV and describes challenges and limitations that need to be surmounted in order for siRNAs to become practical antiviral drugs. The practical use of RNAi therapy for HIV infection will depend on overcoming several challenges, including the ability to establish long-term expression of siRNA without off-target effects and the capacity to counteract mutant escape viruses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
RNA interference (RNAi) has emerged as a common in vitro tool for silencing gene expression.[1,2] The introduction of small RNA duplexes of 19 to 21 nucleotides into cells can elicit specific degradation of complementary gene sequences through base-pairing.[3] It is thought that the RNAi pathway serves as part of the innate immune defense of eukaryotes against invasion by exogenous nucleic acids.[4] Hence, in plants and Drosophila, when a cell is infected by a virus, an RNAi response is triggered by the foreign double-stranded RNA (dsRNA) molecules that originate from the virus.[5] It has been shown recently that retroviruses such as human T-cell leukemia virus (HTLV) and HIV generate dsRNAs that can potentially be engaged into the RNAi pathway of mammalian cells.[6,7]
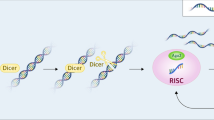
An early step in the RNAi response enlists an RNase III enzyme called Dicer to bind and cleave long dsRNAs into small duplexes of 19 to 21 nucleotides, termed small interfering RNA (siRNA). Dicer-siRNA complex is then recognized by trans activation response region (TAR) RNA-binding protein (TRBP) and protein activator of PKR (PACT), two cellular dsRNA binding proteins. The multi-protein entity is shuttled through interaction with the argonaute 2 (Ago2) protein into an effector complex, the RNA-induced silencing complex (RISC).[8,9] One of the two strands of the siRNA duplex is loaded onto RISC to serve as the guide strand while the second strand, the passenger RNA, is degraded.[10] RISC uses its guide RNA for base-pairing-mediated recognition of target messenger RNA (mRNA). Once this specific hybridization is achieved, the PIWI domain of the Ago2 protein within the RISC complex degrades and silences the targeted substrate mRNA.[11] Because siRNAs are easy to design and synthesize and because they have sequence specificity for silencing mRNAs, these small RNAs potentially represent a future class of antiviral drugs.
1. Inhibition of HIV Replication in Tissue Culture Using Small Interfering RNA (siRNA)
In a relatively short period of time, RNAi has been shown in cultured cells to be efficacious against various viruses including respiratory syncytial virus (RSV),[12] influenza virus,[13] poliovirus,[14] and hepatitis C virus (HCV).[15] For HIV, a variety of siRNAs have also been reported to be effective in interrupting viral infection. Two siRNA strategies have been considered for HIV; the first is to target essential viral genes and the second is to target cellular genes required by HIV for replication (table I).

Examples of siRNAs directed against human immunodeficiency virus
To date, HIV sequences that have been targeted include the structural gag gene,[16] the infectivity factors vif and nef,[3] tat, rev, env, and TAR RNA.[21,37,38] In each instance, sequence-specific silencing of viral RNA and transient suppression of HIV replication over a period of 3 to 4 days in single round infection of cultured cells have been achieved. However, it is important to note that virion-associated HIV-1 genomic RNA that infects cells appears to be resistant to RNAi-mediated degradation.[39] This finding argues that if a goal is to prevent the genesis of integrated provirus then, by using siRNA, one should target cellular factors required for early steps of HIV-1 replication rather than viral sequences.
siRNA directed to several HIV-1 relevant cellular factors have indeed been tested for antiviral efficacy. These include siRNA targeted to:
-
1.
viral entry (e.g. siRNA against HIV coreceptors chemokine [C-C motif] receptor 5 [CCR5] and chemokine [C-X-C motif] receptor 4 [CXCR4][25,26])
-
2.
HIV transcription (e.g. siRNA against cyclin T1, cyclindependent kinase 9 [CDK9], or suppressor of Ty5 homolog [SUPT5H; SPT5][28,30,40])
-
3.
export of HIV-1 RNA from the nucleus into the cytoplasm (e.g. siRNA against RPA interacting protein [RPAIN; hRIP][31] or Src-associated protein in mitosis [Sam68][32])
-
4.
virus assembly and budding (e.g. targeting LYST interacting protein 5 [LIP5], a cellular protein involved in multivesicular body formation,[33] and targeting tumor susceptibility gene 101 [TSG101], also called vacuolar protein-sorting protein 23, involved in endocytic trafficking[36])
-
5.
other cellular proteins used by HIV-1 for replication such as peptidylprolyl isomerase A (cyclophilin A),[34] the RAS oncogene family member RAB9,[35] and CDK2.[29]
In many of the above examples, it should be pointed out that instead of directly introducing siRNA duplexes into cells the effect of which is transient, in many cases, RNAi was expressed using a DNA plasmid expressing a short hairpin RNA (shRNA). It is recognized that the cell’s Dicer enzyme can remove the hairpin loop from shRNA molecules, processing them to their siRNA counterparts. Currently, a myriad of systems are available for expressing shRNAs (e.g. under the control of an RNA polymerase III U6 or H1 promoter or an RNA polymerase II CMV promoter) in several vector systems (e.g. adenoviral or lentiviral vector[17,41]), allowing for long-term silencing even in nondividing cells.
2. Escape and Evasion of HIV from RNA Interference
HIV-1 is a notoriously mutable virus. To be effective, antiviral drugs based on sequence hybridization must take into account that HIV-1 mutates its sequence easily. Indeed, HIV-1 can escape siRNAs through nucleotide mutations. For example, Das et al.[42] observed that an siRNA targeted to nef rapidly elicited the emergence of siRNA-resistant viruses with point mutations in the nef gene. Alternatively, the deletation of an siRNA recognition site can also allow the virus to evade restriction.[43] Moreover, there is evidence that HIV-1 can undergo nucleotide substitutions that induce an alternative RNA folding, thus shielding a previously targeted sequence from access by the siRNA.[42]
To minimize the mutational escape of HIV, one strategy is to simultaneously use multiple siRNAs to target discrete sequences. The likelihood that a single viral RNA molecule would mutate all targeted sequences becomes increasingly small as the number of targets is escalated.[44] This multi-targeting strategy is aided by computational modeling, which can predict how HIV-1 might evolve in cells that express multiple antiviral siRNAs.[45] Another way to circumvent viral escape is to target, not virus sequences, but cellular genes needed by HIV-1. An example of dual-specific shRNAs that efficiently inhibited viral entry[23] by simultaneously targeting HIV-1 cell surface receptors CXCR4 and CD4 or CCR5 and CXCR4 have been reported.[46]19
In addition to evolving siRNA-escape mutations, viruses can also encode suppressor factors that attenuate the cell’s RNAi response.[47,48] While much remains to be elucidated, HIV-1 appears to have multifaceted ways to suppress the cell’s RNAi machinery.[49] The viral Tat protein appears to work as a protein suppressor of RNAi.[50] In addition, the viral RNA, TAR,[51] is also used by HIV-1 as a suppressor of RNAi.[52,53] If siRNA-based therapy of HIV-1 and other viruses is to be usefully contemplated, one needs to consider strategies that address virus-encoded RNAi suppressors. Despite virally encoded suppressors, there is ample evidence for RNAi-mediated inhibition of HIV-1 replication. It is possible that the suppressors are either synthesized too late or in amounts too small to counter the effect of a pre-existing siRNA or shRNA.
3. Toxicity and Off-Target Effects from sh(si)RNAs
The siRNA machinery used in eukaryotic cells is the same as that used to process a class of endogenous small RNAs called microRNAs (miRNAs). miRNAs are important regulators of at least 30% of cellular genes, including those involved in development, signal transduction, apoptosis, cell proliferation, and tumorigenesis.[54–57] To date, 474 human miRNAs have been described, with more likely to be discovered (see miRNA database[58]).[59]
The mechanism of miRNA action and the relevance of miRNAs to HIV-1 infection have been reviewed elsewhere[47,60] and will not be elaborated further here. What needs to be emphasized is the fact that the shared factors for shRNA, siRNA, and miRNA processing are saturable entities. Because miRNAs are essential to cellular metabolism and viability, if the machinery for miRNA genesis is diverted to handle exogenous siRNA or shRNA, then a dearth of miRNAs can potentially manifest as cellular toxicity. This hitherto unexpected finding was indeed demonstrated in a recent study of in vivo overexpression of shRNA in mice.[61] In that study, severe liver toxicity and the death of 150 mice occurred within 2 weeks of shRNA treatment. The explanation for the fatalities was the saturation by shRNA of the pathway normally used in the liver to process miRNAs. Hence, the high shRNA expression in the mouse liver overwhelmed the export pathway (i.e. exportin-5) needed to transport precursor miRNAs from the nucleus into the cytoplasm. The interruption of proper transport of miRNA precursors led to reduced levels of mature miRNAs with resulting cellular toxicity.
Apart from the problem of pathway saturation, off-target effects of siRNAs raise additional concerns. On-target effects emanate from perfect complementarity between guide-RNA and target mRNA (i.e. 19 of 19 matched nucleotide base-pairings), while off-target effects can arise from as few as 7 nucleotides of fortuitous base-pairing between siRNA and mRNA.[62] Considered this way, a particular siRNA could potentially suppress the expression of its intended target and several unintended mRNAs.[63] This off-target phenomenon can be a significant drawback should siRNAs be envisioned for in vivo use as human drugs. One anticipates that methods and modifications that minimize off-target hybridization would need to be greatly improved to enable further siRNA drug development.[64]
Another unwelcome adverse effect of siRNA use is the potential to inappropriately elicit interferon and inflammatory cytokines. Normally, RNA duplexes shorter than 30 nucleotides in length would not be expected to trigger the interferon-linked protein kinase R pathway. However, recently it was found that some GU-rich sequences, such as 5′-UGUGU-3′and 5′-GUC-CUUCAA-3′, which are toll-like receptor (TLR) immunostimulatory motifs, can trigger an interferon response[65] even when presented in duplexes shorter than 30 nucleotides. To minimize nonspecific interferon responses, judicious care must be exercised in the sequence design of siRNA drugs.
4. Development of Antiviral siRNA Drugs: Strategies and Limitations
Recent optimism regarding the possibility of siRNA drugs arises from studies that indicate that intravenous injections of an siRNA directed against Fas can protect mice from liver injury and fibrosis in two models of autoimmune hepatitis[66] and from findings that intranasal administrations of separate siRNAs protected mice from RSV infection[67] and Rhesus macaques from SARS coronavirus infection.[68] siRNA application to the mouse genital tract has also been found to be protective against lethal herpes simplex virus type 2 infection.[69] These preliminary positive findings have prompted phase I studies to be initiated in humans. To date, three clinical trials have been completed in humans and show no significant siRNA toxicity.[70] siRNAs are currently in clinical investigation for two diseases: age-related macular degeneration (AMD) caused by an overexpression of the vascular endothelial growth factor (VEGF) and respiratory infection caused by the RSV.
At this time, there is no siRNA drug for HIV-1. As outlined above, there are several issues unique to the targeting of viral genes which do not apply to the targeting of cellular genes. However, a question that is relevant to all siRNA applications is how best to deliver siRNA into virus-infected cells. In this regard, several methods are being considered. For example, in HCV infections, a procedure that couples siRNA to cholesterol[71] allows for efficient targeting of hepatocytes, the cells preferentially infected by virus. Elsewhere, a novel strategy using antibody-mediated siRNA delivery has been proposed for HIV-1-infected cells.[18] Whether these biochemical delivery techniques can distribute siRNAs effectively to in vivo compartments awaits further verification.
There are also virus-based approaches to deliver shRNA; however, this type of gene therapy approach does raise issues related to biosafety.[72] A major focus for HIV-1-infected cells has been the use of lentiviral vectors, which have the ability to infect nondividing cells, such as resting T-cells, macrophages, and progenitor hematopoietic cells (CD34+ cells). Examples of progress in vector design include an HIV-1 inducible vector with an anti-Rev cassette, which was shown to be effective in inhibiting HIV replication[22] and a doxycycline-dependent lentivector.[73] Going forward, a major challenge is to perfect vectors that are wholly silent for shRNA expression unless specifically triggered by HIV-1 infection.
Despite the optimized delivery of siRNA and shRNA into cells, the ability of viruses to mutate and escape from RNAi is a significantly daunting problem. A multi-modal approach may have to be adopted in order to minimize viral escape.[44] For example, shRNA targeted to Rev can be used at the same time as a trans dominant-negative Rev protein (RevM1O) to inhibit HIV-1 infection.[74] When tested in tissue culture, RevM1O protected shRNA-transduced human hematopoietic cells from HIV-1 infection; no shRNA-resistant virus emerged during a 70-day infection period. Separately, a combination of three different RNA technologies has been proposed for HIV-1 therapy.[75] Here, the strategy is to simultaneously use an shRNA against Rev and Tat proteins, a TAR RNA decoy to suppress HIV-1 transcription, and an anti-CCR5 ribozyme to attenuate HIV-1 co-receptor on the cell surface. In tissue culture, this approach protected CD34+ stem cells from HIV-1 infection without the emergence of viral escape mutants.[75]
5. Conclusion
The principles for developing siRNAs as anti-HIV-1 drugs are sound and compelling. The practical challenges of long-term expression/delivery of siRNA or shRNA, the propensity of virus to escape and evade RNAi, and off-target and toxicity issues are real and difficult (figure 1). The realization of efficacious and practical siRNA drugs for HIV-1 will require further technological breakthroughs and much additional time and effort.

Challenges and limitations that need to be surmounted in order for small interfering RNAs (siRNAs) to become practical antiviral drugs. Ago2 = argonaute 2; miRNA = microRNA; mRNA = messenger RNA; RISC = RNA-induced silencing complex; RNAi = RNA interference; shRNA = short hairpin RNA; siRNA = small interfering RNA; TRBP = TAR RNA-binding protein.
References
Sharp PA. RNAi and double-strand RNA. Genes Dev 1999 Jan 15; 13(2): 139–41
Elbashir SM, Harborth J, Lendeckel W, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001 May 24; 411(6836): 494–8
Jacque JM, Triques K, Stevenson M. Modulation of HIV-1 replication by RNA interference. Nature 2002 Jul 25; 418(6896): 435–8
Voinnet O. Induction and suppression of RNA silencing: insights from viral infections. Nat Rev Genet 2005 Mar; 6(3): 206–20
Fritz JH, Girardin SE, Philpott DJ. Innate immune defense through RNA interference. Sci STKE 2006 Jun 13; 2006(339): pe27
Cavanagh MH, Landry S, Audet B, et al. HTLV-I antisense transcripts initiating in the 3t?LTR are alternatively spliced and polyadenylated. Retrovirology 2006; 3: 15
Ludwig LB, Ambrus JL Jr, Krawczyk KA, et al. Human immunodeficiency virustype 1 LTR DNA contains an intrinsic gene producing antisense RNA and protein products. Retrovirology 2006 Nov 8; 3(1): 80
Chendrimada TP, Gregory RI, Kumaraswamy E, et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005 Aug 4; 436(7051): 740–1
Lee Y, Hur I, Park SY, et al. The role of PACT in the RNA silencing pathway. Embo J 2006 Feb 8; 25(3): 522–32
Matranga C, Tomari Y, Shin C, et al. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell 2005 Nov 18; 123(4): 607–20
Meister G, Landthaler M, Patkaniowska A, et al. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell 2004 Jul 23; 15(2): 185–97
Barik S. Control of nonsegmented negative-strand RNA virus replication by siRNA. Virus Res 2004 Jun 1; 102(1): 27–35
Ge Q, McManus MT, Nguyen T, et al. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc Natl Acad Sci U S A 2003 Mar 4; 100(5): 2718–23
Gitlin L, Karelsky S, Andino R. Short interfering RNA confers intracellular antiviral immunity in human cells. Nature 2002 Jul 25; 418(6896): 430–4
Randall G, Grakoui A, Rice CM. Clearance of replicating hepatitis C virus replicon RNAs in cell culture by small interfering RNAs. Proc Natl Acad Sci USA 2003 Jan 7; 100(1): 235–40
Novina CD, Murray MF, Dykxhoorn DM, et al. siRNA-directed inhibition of HIV-1 infection. Nat Med 2002 Jul; 8(7): 681–6
Lee SK, Dykxhoorn DM, Kumar P, et al. Lentiviral delivery of short hairpin RNAs protects CD4 T cells from multiple clades and primary isolates of HIV. Blood 2005 Aug 1; 106(3): 818–26
Song E, Zhu P, Lee SK, et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol 2005 Jun; 23(6): 709–17
Chang LJ, Liu X, He J. Lentiviral siRNAs targeting multiple highly conserved RNA sequences of human immunodeficiency virus type 1. Gene Ther 2005 Jul; 12(14): 1133–44
Park WS, Hayafune M, Miyano-Kurosaki N, et al. Specific HIV-1 env gene silencing by small interfering RNAs in human peripheral blood mononuclear cells. Gene Ther 2003 Nov; 10(24): 2046–50
Coburn GA, Cullen BR. Potent and specific inhibition of human immunodeficiency virus type 1 replication by RNA interference. J Virol 2002 Sep; 76(18): 9225–31
Unwalla HJ, Li MJ, Kim JD, et al. Negative feedback inhibition of HIV-1 by TAT-inducible expression of siRNA. Nat Biotechnol 2004 Dec; 22(12): 1573–8
Anderson J, Banerjea A, Akkina R. Bispecific short hairpin siRNA constructs targeted to CD4, CXCR4, and CCR5 confer HIV-1 resistance. Oligonucleotides 2003; 13(5): 303–12
Han W, Wind-Rotolo M, Kirkman RL, et al. Inhibition of human immunodeficiency virus type 1 replication by siRNA targeted to the highly conserved primer binding site. Virology 2004 Dec 5; 330(1): 221–32
Anderson J, Banerjea A, Planelles V, et al. Potent suppression of HIV type 1 infection by a short hairpin anti-CXCR4 siRNA. AIDS Res Hum Retroviruses 2003 Aug; 19(8): 699–706
Zhou N, Fang J, Mukhtar M, et al. Inhibition of HIV-1 fusion with small interfering RNAs targeting the chemokine coreceptor CXCR4. Gene Ther 2004 Dec; 11(23): 1703–12
Banerjea A, Li MJ, Bauer G, et al. Inhibition of HIV-1 by lentiviral vector-transduced siRNAs in T lymphocytes differentiated in SCID-hu mice and CD34+ progenitor cell-derived macrophages. Mol Ther 2003 Jul; 8(1): 62–71
Li Z, Xiong Y, Peng Y, et al. Specific inhibition of HIV-1 replication by short hairpin RNAs targeting human cyclin T1 without inducing apoptosis. FEBS Lett 2005 Jun 6; 579(14): 3100–6
Ammosova T, Berro R, Kashanchi F, et al. RNA interference directed to CDK2 inhibits HIV-1 transcription. Virology 2005 Oct 25; 341(2): 171–8
Ping YH, Chu CY, Cao H, et al. Modulating HIV-1 replication by RNA interference directed against human transcription elongation factor SPT5. Retrovirology 2004 Dec 27; 1(1): 46
Yu Z, Sanchez-Velar N, Catrina IE, et al. The cellular HIV-1 Rev cofactor hRIP is required for viral replication. Proc Natl Acad Sci U S A 2005 Mar 15; 102(11): 4027–32
Modem S, Badri KR, Holland TC, et al. Sam68 is absolutely required for Rev function and HIV-1 production. Nucleic Acids Res 2005; 33(3): 873–9
Ward DM, Vaughn MB, Shiflett SL, et al. The role of LIP5 and CHMP5 in multivesicular body formation and HIV-1 budding in mammalian cells. J Biol Chem 2005 Mar 18; 280(11): 10548–55
Sokolskaja E, Sayah DM, Luban J. Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J Virol 2004 Dec; 78(23): 12800–8
Murray JL, Mavrakis M, McDonald NJ, et al. Rab9 GTPase is required for replication of human immunodeficiency virus type 1, filoviruses, and measles virus. J Virol 2005 Sep; 79(18): 11742–51
Garrus JE, von Schwedler UK, Pornillos OW, et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001 Oct 5; 107(1): 55–65
Capodici J, Kariko K, Weissman D. Inhibition of HIV-1 infection by small interfering RNA-mediated RNA interference. J Immunol 2002 Nov 1; 169(9): 5196–201
Boden D, Pusch O, Ramratnam B. HIV-1-specific RNA interference. Curr Opin Mol Ther 2004 Aug; 6(4): 373–80
Westerhout EM, Ter Brake O, Berkhout B. The virion-associated incoming HIV-1 RNA genome is not targeted by RNA interference. Retrovirology 2006 Sep 4; 3(1): 57
Chiu YL, Cao H, Jacque JM, et al. Inhibition of human immunodeficiency virus type 1 replication by RNA interference directed against human transcription elongation factor P-TEFb (CDK9/CyclinT1). J Virol 2004 Mar; 78(5): 2517–29
Lee NS, Dohjima T, Bauer G, et al. Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat Biotechnol 2002 May; 20(5): 500–5
Das AT, Brummelkamp TR, Westerhout EM, et al. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J Virol 2004 Mar; 78(5): 2601–5
Westerhout EM, Ooms M, Vink M, et al. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res 2005; 33(2): 796–804
Ter Brake O, Konstantinova P, Ceylan M, et al. Silencing of HIV-1 with RNA Interference: a multiple shRNA approach. Mol Ther 2006; 14(6): 883–92
Leonard JN, Schaffer DV. Computational design of antiviral RNA interference strategies that resist human immunodeficiency virus escape. J Virol 2005 Feb; 79(3): 1645–54
Anderson J, Akkina R. CXCR4 and CCR5 shRNA transgenic CD34+ cell derived macrophages are functionally normal and resist HIV-1 infection. Retrovirology 2005 Aug 18; 2: 53
Yeung ML, Bennasser Y, Le SY, et al. siRNA, miRNA and HIV: promises and challenges. Cell Res 2005 Nov-Dec; 15(11–12): 935–46
Saumet A, Lecellier CH. Anti-viral RNA silencing: do we look like plants? Retrovirology 2006; 3: 3
Bennasser Y, Yeung ML, Benkirane M, et al. RNA interference and HIV-1: hits and misses. Curr Opin HIV AIDS 2006 May 2006; 1(3): 208–11
Bennasser Y, Le SY, Benkirane M, et al. Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity 2005 May; 22(5): 607–19
Berkhout B, Jeang KT. Detailed mutational analysis of TAR RNA: critical spacing between the bulge and loop recognition domains. Nucleic Acids Res 1991 Nov 25; 19(22): 6169–76
Bennasser Y, Yeung ML, Jeang KT. HIV-1 tar RNA subverts RNA interferencein transfected cells through sequestration of tar RNA binding protein, TRBP. J Biol Chem 2006; 281(38): 27674–8
Gatignol A, Laine S, Clerzius G. Dual role of TRBP in HIV replication and RNA interference: viral diversion of a cellular pathway or evasion from antiviral immunity? Retrovirology 2005 Oct 27; 2: 65
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005 Jan 14; 120(1): 15–20
Kim VN. Small RNAs: classification, biogenesis, and function. Mol Cells 2005 Feb 28; 19(1): 1–15
Croce CM, Calin GA. miRNAs, cancer, and stem cell division. Cell 2005 Jul 15; 122(1): 6–7
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004 Feb 23; 116(2): 281–97
miRBase Targets Version 3.0 [online]. Available from URL: http://microrna.sanger.ac.uk/targets/v3/[Accessed 2007 Jan 9]
John B, Enright AJ, Aravin A, et al. Human microRNA targets. PLoS Biol 2004 Nov; 2(11): e363
Yeung ML, Bennasser Y, Jeang KT. miRNAs in the biology of cancers and viral infections. Curr Med Chemistry. In press
Grimm D, Streetz KL, Jopling CL, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006 May 25; 441(7092): 537–41
Brennecke J, Stark A, Russell RB, et al. Principles of microRNA-target recognition. PLoS Biol 2005 Mar; 3(3): e85
Jackson AL, Burchard J, Schelter J, et al. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 2006 Jul; 12(7): 1179–87
Jackson AL, Burchard J, Leake D, et al. Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA 2006 Jul; 12(7): 1197–205
Judge AD, Sood V, Shaw JR, et al. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol 2005 Apr; 23(4): 457–62
Song E, Lee SK, Wang J, et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med 2003 Mar; 9(3): 347–51
Bitko V, Musiyenko A, Shulyayeva O, et al. Inhibition of respiratory viruses by nasally administered siRNA. Nat Med 2005 Jan; 11(1): 50–5
Li BJ, Tang Q, Cheng D, et al. Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in Rhesus macaque. Nat Med 2005 Sep; 11(9): 944–51
Palliser D, Chowdhury D, Wang QY, et al. An siRNA-based microbicide protects mice from lethal herpes simplex virus 2 infection. Nature 2006 Jan 5; 439(7072): 89–94
Behlke MA. Progress towards in vivo use of siRNAs. Mol Ther 2006 Apr; 13(4): 644–70
Soutschek J, Akinc A, Bramlage B, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004 Nov 11; 432(7014): 173–8
Baum C, Schambach A, Bohne J, et al. Retrovirus vectors: toward the plentivirus? Mol Ther 2006 Jun; 13(6): 1050–63
Westerhout EM, Vink M, Haasnoot PC, et al. A conditionally replicating HIV-based vector that stably expresses an antiviral shRNA against HIV-1 replication. Mol Ther 2006 Aug; 14(2): 268–75
Unwalla HJ, Li HT, Bahner I, et al. Novel Pol II fusion promoter directs human immunodeficiency virus type 1-inducible coexpression of a short hairpin RNA and protein. J Virol 2006 Feb; 80(4): 1863–73
Li MJ, Kim J, Li S, et al. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-CCR5 ribozyme, and a nucleolar-localizing TAR decoy. Mol Ther 2005 Nov; 12(5): 900–9
Acknowledgments
We thank members of the Jeang Laboratory for their critical readings of the manuscript. Our work is supported by intramural funding from National Institute of Allergy and Infectious Diseases and the National Institutes of Health.
The authors declare that they have no conflict of interest directly relevant to the contents of the article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bennasser, Y., Yeung, M.L. & Jeang, KT. RNAi Therapy for HIV Infection. BioDrugs 21, 17–22 (2007). https://doi.org/10.2165/00063030-200721010-00003
Published:
Issue Date:
DOI: https://doi.org/10.2165/00063030-200721010-00003