
Abstract
Cardiac hypertrophy is the heart’s response to a variety of extrinsic and intrinsic stimuli, some of which might finally lead up to a maladaptive state. An integral part of the pathogenesis of the hypertrophic cardiomyopathy disease (HCM) is the activation of the rat sarcoma (RAS)/RAF/MEK (mitogen-activated protein kinase kinase)/MAPK (mitogen-activated protein kinase) cascade. Therefore, the molecular signaling involving RAS has been the subject of intense research efforts, particularly after the identification of the RASopathies. These constitute a class of developmental disorders caused by germline mutations affecting proteins contributing to the RAS pathway. Among other phenotypic features, a subset of these syndromes is characterized by HCM, prompting researchers and clinicians to delve into the chief signaling constituents of cardiac hypertrophy. In this review, we summarize current advances in the knowledge of the molecular signaling events involved in the pathogenesis of cardiac hypertrophy through work completed on patients and on genetically manipulated animals with HCM and RASopathies. Important insights are drawn from the recognition of parallels between cardiac hypertrophy and cancer. Future research promises to further elucidate the complex molecular interactions responsible for cardiac hypertrophy, possibly pointing the way for the identification of new specific targets for the treatment of HCM.
Similar content being viewed by others


Introduction
Rat sarcoma (RAS) belongs to the family of small G proteins, low molecular weight (20–40 kDa) guanine nucleotide-binding proteins. These proteins were identified in 1980 in a sarcoma virus (1) and constitute molecular switches involved in transmitting extracellular stimuli downstream into the cell. Small G proteins exist in two conformations: an inactive form, in which they bind guanosine diphosphate (GDP), and an active form, interacting with guanosine triphosphate (GTP). Extracellular stimuli induce dissociation of GDP and binding of GTP, which prompts a conformational change and consequent binding with downstream effectors. Since both the spontaneous exchange GDP/GTP and the hydrolysis of GTP are inefficient processes, regulatory proteins are needed for them to proceed: guanine nucleotide exchange factors (GEFs) activate the small G protein, while GTPase activating proteins (GAPs) (such as neurofibromin 1 and RASA1 [RAS p21 protein activator (GTPase activating protein) 1]) stimulate the hydrolysis of GTP. Induction of GEF activity is promoted by growth factors, which activate tyrosine kinase receptors (RTKs). Activation of RTKs may be negatively regulated by casitas b-lineage lymphoma (CBL) ubiquitin ligase. Phosphotyrosines are binding sites for adapter proteins, like SHC/growth factor receptor-bound 2 (GRB2) complex, which bind the cytosolic protein son of sevenless (SOS), the most important GEF of RAS. SH2 domain-containing protein tyrosine phosphatase (SHP2) is also a RAS regulator; in fact it interacts with RTKs and adapter proteins like GRB2 and GRB2-associated-binding protein (GAB), inducing RAS signaling. RAS activates, in turn, a cascade of downstream kinases: RAF, mitogen-activated protein kinase kinase (MEK) and mitogen-activated protein kinase (MAPK). Scaffold proteins, like SHOC2, link the RAS to the RAF family. RAF activity can be prevented by SPRED1 (sprouty-related, EVH1 domain containing 1) binding. RAFs, when induced, phosphorylate MEK, which finally activates MAPKs. The molecular targets of the MAPK family are usually nuclear transcription factors, cytoplasmic factors responsible for the initiation of translation and apoptosis regulators (2,3).
Hypertrophic Cardiomyopathy
The response of the heart to meet the change in demands of circulation is dependent primarily on myocardial growth. The heart enhances its contractile function in response to increased workload by increasing cardiomyocyte size and force production (4). For example, physiological hypertrophy is often developed by athletes: the increase of heart muscle mass reduces ventricular wall stress and compensates for the increased hemodynamic demand, improving heart contractility. In contrast, pathological signals may lead to maladaptive hypertrophy, with diminished cardiac output and enhanced risk of sudden death, arrhythmia and heart failure. Pathological signals may be extrinsic, like arterial hypertension, myocardial infarction and aortic stenosis, or intrinsic to the cardiomyocyte, like genetic hypertrophic cardiomyopathy (HCM). Usually the genetic causes of HCM are autosomal dominant mutations on proteins of the sarcomere. A secondary form of HCM may arise as part of a series of dysfunctions in a congenital syndrome. Irrespective of the origin of the pathological hypertrophy, typical features are reexpression of fetal genes and sarcomeric remodeling. In most cases, ventricular hypertrophy is detectable as ventricular wall thickening; however, hypertrophy can also lead to dilation of the ventricular chambers and cause contractile dysfunction and apoptosis.
RAS Pathway Mutations in RASopathies Associated with HCM
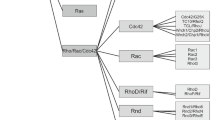
Over the past three decades, much has been learned about both RAS GT-Pase activity and the consequences of somatic mutations in RAS signaling, which occur in cancer (5), with RAS and cancer accounting for more than 20,000 publications in public web libraries. Only recently, the appreciation that RAS activation has important effects on development has emerged: it was the discovery of germline mutations in the genes neurofibromin 1 (NF1) (6) and protein tyrosine phosphatase, non-receptor type 11 (PTPN11) (which encodes SHP2) (7). These genes provided the first indication that aberrant RAS signaling might contribute to the pathogenesis of human developmental diseases. Germline mutations in genes encoding molecules in the RAS/RAF/MEK/MAPK cascade were shown to cause RASopathies, many of these mutations affect SHP2, SOS1, RAS, RAF and MEK proteins (Figure 1). RA-Sopathies include a number of phenotypically similar diseases such as Noonan syndrome (NS, MIM #163950), Costello syndrome (CS, #218040), cardio-facio-cutaneous syndrome (CFC, #115150) and Noonan syndrome with multiple lentigines, also known as LEOPARD syndrome (acronym for lentigines [multiple brown-black spots on the skin], electrocardiographic conduction defects, ocular hypertelorism, pulmonary stenosis, abnormalities of genitals, retarded growth resulting in short stature and deafness or hearing loss due to inner ear malfunction) (LS, #151100, #611554). All of these mutations perturb the intrinsic biochemical properties of the encoded proteins. Germline mutations are usually dominant gain-of-function mutations; LS seems to be an exception, because it is caused by autosomal dominant-negative PTPN11 mutations (8). However, gain-of-function mutations of v-raf-1 murine leukemia viral oncogene homolog 1 (RAF1) have been identified in a small percentage of LS patients (9).

Schematic representation of RAS pathway and its correlation with RASopathies, A growth factor activates its RTK causing phosphorylation of tyrosines, which are bound by adapter proteins, SHC/GRB2 and GAB. These interact with the activators of RAS, SOS1 and SHP2, and induce the dissociation of GDP and binding of GTP. RAS-GTP activates a cascade of downstream kinases: the MAPK pathway. On the other hand, the inhibition of RAS signaling is mediated by GAPs: RASA1 and neurofibromin 1. The RASopathies are developmental pathologies due to mutations on genes encoding proteins of the RAS pathway. HCM is uncommon in NS and CFC patients. However, nearly all NS patients with mutated RAF1 develop HCM. PTPN11 (encoding SHP2) loss-of-function mutations were observed in the majority of LS patients who developed HCM. Mutations in RAF1 have also been identified in a small percentage of LS with HCM. CS patients harboring HRAS mutations consistently show cardiac hypertrophy. Light gray shading indicates the mutation-specific syndromes accompanied by HCM; dark gray shading indicates those syndromes in which cardiac hypertrophy is less common.
The common underlying pathogenetic mechanism involved in RASopathies brings about a significant overlap in phenotype, which includes craniofacial dysmorphology and heart defects. The cardiac abnormalities in CS, NS and CFC syndromes are quite similar, although they vary in severity and frequency. Congenital cardiac defects include pulmonary valve and aortic stenosis, atrial and ventricular septal defects and mitral insufficiency. Among the RASopathies, congenital heart defects are the most common in NS (PTPN11 or SOS1). They also occur in CFC syndrome (v-raf murine sarcoma viral oncogene homolog B1 [BRAF]) and NS (RAF1). Conduction abnormalities in patients with an RAF1 mutation, CS and LS are also well described. Vascular abnormalities (that is, aortic dilation, coronary artery dilation and peripheral aneurysms) have also been reported in patients with NS and LS (10). HCM occurs with similar frequency in CS (10,11) and LS (12), whereas it is less common in CFC syndrome (13–15) and NS patients (16–19). In approximately one-third of CS patients, HCM is chronically severe, worsened or stable. However, the resolution or regression of HCM on echocardiographic follow-up was also reported (10).
The fundamental role of several RTKs, like epidermal growth factor receptor (EGFR) (also known as ErbB), fibroblast GFR and vascular endothelial GFR, has been described in heart development, from the induction of cardiac mesoderm to the formation of cardiac cushions in the atrioventricular (AV) canal and the outflow tract (OFT) and the epithelial-to-mesenchymal transition process leading to valve formation (20). The RAS signaling mutations causing defective AV and OFT development and valvulogenesis (that is, atrioventricular septal defects and pulmonary valvular stenosis) in a subset of RASopathies (21) probably affect multiple populations of cell progenitors. Indeed, various cell lineages contribute at different stages to OFT and AV channels, including the so-called second heart field (22,23). Despite the clear involvement of RAS signaling in the prenatal development of the heart, important effects of this signaling should also be considered in the postnatal response to stress. In this review, we emphasize the role of the RAS pathway in HCM. The age at which HCM is diagnosed ranges from infancy, in patients with a severe form of the rare neonatal phenotype, to childhood, the more common age of presentation (10). In studies focused on fetuses, investigators failed to diagnose prenatal onset of HCM, at least in CS (24,25). So far, hypertrophy has seemed to develop as a counteraction of the cardiomyocyte to increased hemodynamic demands. However, it remains to be established whether the pathological phenotype may result from RAS signaling pathway alterations in fetal cardiomyocytes and, if so, how this occurs.
Pathophysiological Signaling: Experimental Relevance of the RAS/RAF/MEK/MAPK Pathway in HCM
The discovery of mutated RAS-related genes in RASopathy patients has underlined the relevance of this signaling to HCM (Figure 1). In addition to genetic lesions, pathological stimuli, like biomechanical stress and neurohumoral factors, lead to a hypertrophic state of the heart (26–28). The altered stimulation of membrane receptors, such as RTKs, is a crucial initiating step leading to the final activation of MAPKs, which thereby induce the hypertrophic response (27,29–33). By altering the levels and activities of cardiac transcription factors (for example, GATA binding protein 4 [GATA4], myocyte enhancer factor 2 [MEF2] and nuclear factor of activated T cells [NFAT]), the RAS/RAF/MEK/MAPK pathway leads to reexpression of fetal cardiac genes (34). In particular, the upregulation of β-MHC (β myosin heavy chain) and atrial natriuretic factor and the downregulation of SERCA (sarcoplasmic reticulum Ca2+ ATPase) genes occur in the pathological hypertrophy, with consequent loss of efficient contraction and calcium cycling (30,33,34). Such transcriptional remodeling correlates with the loss of cardiac function; conversely, the improvement of cardiac function in response to drugs or implantation of an assist device is accompanied by the normalization of gene expression (35,36). Studies using genetically engineered mice have suggested that direct targeting of the hypertrophic response itself is beneficial and may provide a suitable therapeutic option in such cases (4,37,38). Thus, strategies to normalize cardiac gene expression by controlling its upstream signaling offer an attractive approach for HCM therapy.
To this aim, a considerable number of animal models, mainly murine, have been generated to reproduce a hyper-trophic phenotype due to hyperactivation of RTKs, RAS and RAS-downstream proteins. The attenuation of hypertrophic responses by the use of inhibitors of MEK1/2, dominant-negative RAF1 or MEK1 and antisense oligonucleotides against extracellular signal-regulated kinase 1 and 2 (ERK1/2) definitely substantiated the important role of this cascade for cardiomyocyte hypertrophy (27,30,33). All of the findings accumulated over the past couple of decades underscore the importance of both RAS-downstream ERK-mediated and ERK-independent pathways in cardiac hypertrophy. The earliest evidence of a correlation between RAS and hypertrophy came from experiments on cultured myocytes (39,40). Then, a link between RAS expression levels and the severity of hypertrophy was given by RAS mRNA measurements among HCM patients (41). However, the in vivo proof of its role in hypertrophy derives from transgenic mice in which oncogenic RAS is expressed in the cardiac ventricular chamber (42). Mutations on the v-Ha-ras Harvey rat sarcoma viral oncogene homolog (HRAS) gene have been found in the majority of patients affected by CS with signs of HCM (11). A mouse and a zebra-fish model for HRAS-related CS have been produced so far. However, although the mouse model showed signs of cardiac hypertrophy (43), mutant fish did not (44), revealing an inconsistency with the human phenotype. The involvement of aberrant RTK signaling in the pathogenesis of cardiac hypertrophy has been recently demonstrated by our group with the use of a transgenic conditional mouse model expressing the constitutively active Met tyrosine kinase (45). RTK activation engenders RAS signaling to be turned on through GRB2, which has been implicated in the development of hypertrophy (46). In turn, GRB2 mediates the activation of SOS1, which is mutated in NS patients and mouse models leading to HCM (47). Also, SHP2 is a key component of multiple RTK, cytokine receptor and integrin signaling cascades. The animal model carrying an NSassociated Ptpn11 mutation does not develop cardiac hypertrophy (48), in line with the observation that HCM occurs in less than 5%–10% of NS patients with PTPN11 mutations (19). In contrast, a dominant-negative mutation with catalytic loss of function affecting SHP2 leads to HCM in both LS patients and mice (49). As expected, the LS mice show reduced agonist-evoked ERK/MAPK signaling, with a concomitant enhancement of protein kinase B (AKT)-mammalian target of rapamycin (mTOR) activity.
The importance of excessive phosphoinositide 3-kinase (PI3K)-AKT-mTOR signaling in the pathogenesis of HCM in LS was further proved by rapamycin treatment, which reverses LS cardiac defects. Indeed, even though direct mutations on PI3K or AKT have not been found, it is known that the prolonged constitutive activation of PI3K in the heart results in hypertrophy (50). In contrast, temporally controlled overexpression of cardiac PI3K in adult transgenic mice does not induce hypertrophy, but results in increased contractility (51). In fact, in the cardiac context, PI3K plays important roles in cardiac growth and exercise-induced hypertrophy and exerts protective effects against pathological stimuli (52–54). Similarly, short-term activation of AKT in cardiomyocytes results in physiological adaptive hypertrophy, whereas chronic activation of AKT leads to pathological hypertrophy (55). Thus, the PI3K-AKT pathway shows a high degree of complexity, with different roles in regulating cardiac hypertrophy. RAS activation results in the stimulation of RAF family members, which have been implicated in the development of hypertrophy (56,57) and of HCM in RASopathies (9,13,14). RAF1 mutations have been more frequently found to be associated with HCM, and nearly all NS patients with RAF1 mutations exhibit HCM (58). Like NS patients, mice heterozygous for the NS-associated RAF1 mutation exhibit eccentric cardiac hypertrophy (59). A mouse model of a constitutively active form of BRAF has also been created (60). However, mutant mice show an increased total number of cardiomyocytes, rather than alterations in size, in contrast to data from CFC patients with mutated BRAF who do develop HCM (61).
RAF activity results in the subsequent phosphorylation of MEKs. Mutated MEK1 was found in a few CFC patients with signs of HCM (13). Evidence from cultured cardiac myocytes exposed to a MEK1-specific inhibitor demonstrated the critical contribution of the ERK pathway to hypertrophy (62,63). In vivo, inhibition of MEK attenuated cardiac growth in both induced and genetic models of hypertrophy (48,59,64,65). Final activation of MAPK has also been documented in different heart diseases (66,67). Sprouty1, an endogenous inhibitor of ERK, was reported to be induced in human hearts during hypertrophy regression after implantation of an assist device (68). These observations are in agreement with reports about MAPK phosphatase-1 inhibiting effects on cardiac hypertrophy, in vitro and in vivo (69). However, mice lacking ERK1 and one ERK2 allele show a normal hypertrophic response to pressure overload and exercise (70), suggesting that ERK1/2 may not be mandatory for cardiomyocyte growth. Consistent results were raised in mice with cardiac-specific inhibition of ERK1/2 activity (70–72). These proofs underpin the hypothesis that cardiac hypertrophy can develop independently of ERK1/2.
Recently, a new regulatory mechanism for ERK2 in cardiac hypertrophy was described: Thr188 autophosphorylation on ERK2 directs it to the nucleus, leading to phosphorylation of nuclear targets, without an overall increase in the activity of ERK1/2 (73). Thus, Thr188 phosphorylation might represent a specific switch toward hypertrophic signaling, uncoupled from physiological signaling, and hence embody a good target for therapy. Moreover, because overall ERK1/2 activity remains unchanged, ERK1/2 functions other than the hypertrophic one might be unaffected, reducing massive side effects.
Cooperative Effects on the RAS Pathway: Cross-Links in Hypertrophy
A first suggestion that RAS is in front of a network rather than in a top-down pathway actually comes from genetic syndromes with HCM. For example, whereas both RAS and MEK1 gain-of-function approaches result in a hyper-trophic response, only MEK activation has no cardiotoxic effects (74). This discrepancy might be explained by ERK-independent RAS pathways or by activated V12H RAS recruiting of modulatory proteins, for example, Gβγ-subunits, which could influence its downstream cascade (75). Accordingly, the autophosphorylation of ERK2 at Thr188 requires the integrated activation of the RAF/MEK/ERK1/2 cascade, as well as that of Gαq-coupled receptors (73). Also, activation of β-adrenergic receptors regulates the activity of small GTPases involved in cardiac hypertrophy (76,77): a novel family of exchange proteins directly activated by cyclic AMP (EPAC) (78) has been shown to link Gprotein-coupled receptors (GPCRs) and RAS signaling (Figure 2). EPAC acts as a GEF on RAS-like small GTPases Rap1/2 (79,80), participating in multiple cellular events initiated by GPCRs (76,81,82). In turn, Rap1 activates BRAF and has been involved in cardiac hypertrophy (77). In addition, signaling via GPCRs promotes cardiac hypertrophy via ERK (83,84). Likewise, other GPCRs, including α-adrenergic receptors (28,85), angiotensin receptors (86) and endothelin receptors (63,87), signal through ERK to promote cardiomyocyte hypertrophy. Moreover, nuclear targeted α-adrenergic receptors might activate ERK located in caveolae (88).

RAS Network: cross-links in hypertrophy. The cardiac hypertrophic response implicates signal transduction pathways initiated by ligand-stimulated membrane-bound receptors (RTKs, GPCRs) and biomechanical stress sensors (integrins). Various signaling effectors interact with the RAS/MEK/MAPK pathway. GPCR receptors activate RAS proteins through EPAC, induce release of internal Ca2+ stores and elicit pathological hypertrophy through calcineurin/NFAT. GPCRs also act through ERK activation. GSK3 kinase negatively regulates NFAT and, in turn, is inactivated by the PI3K-AKT pathway stimulated by RAS. Stimuli acting on integrins prompt cardiac hypertrophy through FAK activation and considerable cross-talk with RTK-mediated signaling. In addition, PAK regulates RAF1 activity. All these pathways converge on the modulation of transcriptional factors (MEF2, JUN and GATA4), which induce the expression of genes of the hypertrophic program.
Several studies have also implicated focal adhesion kinase (FAK), a nonreceptor tyrosine kinase, in the first phase of the hypertrophic response to stretch in cardiomyocytes (89) by regulating the activation of MEF2 and JUN N-terminal kinase (JNK)-JUN pathways, which are early activators of the hypertrophic genetic program (90).
Irrespective of the original stimulus, these pathways converge to the activation in the nucleus of transcriptional factors, which have earlier roles during development, including GATA4 and MEF2 (Figure 2). In the normal adult myocardium, only basal MEF2 activity is required for the maintenance of contractile properties, whereas stress stimuli, through MAPK, stimulate its activity (Figure 2) (91,92). In the adult, these factors induce a fetal gene program, which might be beneficial in adapting to stress, in principle, but then leads to altered contractility, uncontrolled calcium transients and inadequate energetic and, finally, maladaptative changes (93–95).
Gene expression profiling from temporally regulated V12H RAS transgenic hearts has suggested that induction of early response genes, loss of mitochondrial function and altered ionic channel proteins are the likely culprits of the pathological changes in extracellular matrix remodeling, cardiac output and electrophysiological parameters (96). Recent work has suggested that the selective induction of Gai in the RAS transgenic heart contributes to impaired sarcoplasmic reticulum calcium cycling (97), and a number of other studies have led to descriptions of RAS-induced alterations in calcium transients (98–100). Interestingly, abnormalities in delicate calcium homeostasis can trigger cardiac hypertrophy through mechanisms that have not been fully elucidated (101). The major link between calcium signaling and expression of fetal genes is represented by NFATs. NFATs are activated by calcineurin through its dephosphorylating action, then migrate into the nucleus, where they interact with MEF2 and GATA4 (102,103). Moreover, GSK3 mediates export of NFATs from the nucleus and termination of transcription. GSK3 is a known effector of the PI3K-AKT pathway, and activation of AKT leads to inhibition of GSK3, thus crossing calcineurin, RAS and PI3K pathways (Figure 2). Indeed, the involvement of PI3K signaling was underlined in cardiac pathogenesis (50) and hypertrophy (51,57). Finally, phosphorylation of RAF1 through PI3K and P21-activated kinase (PAK, Figure 2) provides a costimulatory signal, which, together with RAS, leads to strong activation of RAF1 kinase (104).
What is clear, in light of this exciting progress, is that an intricate web of interconnected signaling modules exists around the hub of RAS, in which RAS and fluctuations in calcium concentrations interplay and regulate each other.
Lessons from Cancer to Cardiac Hypertrophy
Interesting insights from the role of RAS mutations in tumorigenesis might provide a new perspective for considering the role of hyperactive RAS in the development of HCM in RAS syndromes. Parallels between cancer and hypertrophy have already been suggested, concerning both leading causes and therapy (105). Inadequately activated RTKs are implicated in many proliferative disorders and have therefore received considerable attention. More recently, the same pathway was shown to be involved in the pathogenesis of cardiac hypertrophy, prompting scientists to take advantage of the experience gained in cancer for the cardiovascular field. For example, an unanticipated role for mutated CBL in the pathogenesis of a clinically variable condition with features fitting or partially overlapping NS was suggested (17): CBL is a small E3 ubiquitin ligase that negatively regulates intracellular signaling downstream of RTKs. Moreover, cardiac-specific expression of Tpr-Met, the oncogenic fusion protein of hepatocyte growth factor receptor, was shown to result in concentric hypertrophy and congestive heart failure (45).
A first important suggestion comes from the analysis of the strength in degree and duration of the activity of mutated proteins. Activating RAS mutations occur in ~30% of human cancers with a pattern skewed with respect to tissue type and isoform. Interestingly, the somatic mutations triggering cancer tend to be more malignant and less variable than the germline mutations found in RASopathies. Indeed, RAS-related developmental disorders seem to be caused by moderately hyperactivated proteins that can be tolerated in the germline, whereas some, if not most, of the mutations found in cancer are incompatible with development, as suggested by G12D v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) lethality during embryogenesis (106). This might be true also in the case of HCM, which develops in the presence of germline mutants that can be generally considered mild hypermorphs, whereas strong gain-of-function proteins would lead to lethal malformations of the heart, or even cancer, as happens in the Von Hippel-Lindau knock-out mouse (107).
Spontaneous regression is another interesting feature of RAS syndromes: the diagnosis of RASopathies primarily depends on clinical features, but the prevalence of the different characteristics among affected individuals greatly depends on age. The facial features, for example, generally become more difficult to detect in later adolescence and adulthood. The example of juvenile myelomonocytic leukemia is particularly intriguing in this respect, because myeloproliferative disorders that occur in NS infants frequently regress without treatment. Consistently, in some cases (14% of CS patients with HCM and one patient with mutated KRAS) a natural regression of HCM occurs with time (10). Elucidating the mechanisms of spontaneous regression in cancer, still not well understood, might be very useful to stimulate cardiomyocytes to escape from HCM as well.
Interestingly, the tissues that are perturbed in CS (the nervous and musculoskeletal systems) greatly overlap with the types of malignancies that are observed (rhabdomyosarcoma, neuroblastoma, ganglioneuroblastoma and bladder cancer). Because HRAS mutations account only for <1% of all cancer-associated RAS mutations, emerging considerations are that HRAS is transforming only in those tissues in which it exerts a major role and/or the gene is expressed at low levels in most types of cancer-initiating cells. The concept of tumor addiction might be easily transferred to RASopathies as well: specific tissues depend on the sustained activity of specific proteins to grow and survive, as it has been described for Met kinase (108). Accordingly, HRAS, which is mutated in CS, may create a dependence on cardiac cells for their proliferation and survival, so CS patients develop HCM more frequently than patients with other RASopathies. Moreover, for each tissue, or even cell type, there might be a threshold in the response to gain and loss in RAS signaling, dictated by the fact that oncogenes may convey both prosurvival and proapoptotic signals. This idea might explain why either decreasing or increasing SHP2 phosphatase activity has deleterious developmental consequences (109). Finally, cardiomyocytes might be less sensitive to a mutation for which the valves are more sensitive. This may explain why 40% of patients with mutated RAS do not develop HCM. For this reason, efforts still have to be addressed to study the role of each causative molecule not only in pathological but also in physiological conditions.
It is also notable that the kinase activities of some of the mutant BRAF proteins found in CFC syndrome are comparable to BRAF oncoproteins; yet the germline CFC mutations do not predispose to tumor formation at all. These observations suggest that the relatively low risk of cancer in many of these syndromes is not entirely due to the degree of biochemical activity of the mutant protein. Indeed, the observation that many individuals with CS do not develop cancer provides evidence that cooperating mutations or concurrent microenvironmental cues are needed for tumorigenesis. This observation should be kept in mind in analysis of models designed to mimic hypertrophy in RASopathies: these syndromes are diseases of complex systems, in which the paradigm one gene one phenotype might be far from being valid. In fact, in humans as well as in mice, different ERK1/2 functions are selectively switched on (for example, the protective and antiapoptotic functions of ERK1/2 more than their hypertrophic functions) in response to a combination of upstream signals that altogether would differently influence the cardiac phenotype. Furthermore, the constitutive activation of ERK1/2 in these transgenic models may be distinct from the carefully tuned activation pattern of endogenous kinases (33,71), even if one of them carries an activating mutation.
Considering all of these findings, why would germline mutations affecting single genes in RAS signaling be sufficient to confer an overt clinical pheno-type? Once again, cancer docet. The phenomenon of drug resistance in target therapy has been extensively described, that is to say that cancer cells find new pathways to bypass a signaling blockade. Thus, because the germline mutations are present throughout development, there is a substantial amount of time for body cells to adapt to them, activating regulatory feedback loops as well. However, not all the cancers are able to find an escape and those that do not can be eradicated. Correspondingly, the tissues that are perturbed in RAS syndromes may be not only highly sensitive to the RAS/RAF/MEK/MAPK pathway to specify their cell fates, but also relatively incapable of adjusting to overthreshold quantitative variations. It is as if hypertrophic cardiomyocytes were like cancer cells that could not find an escape.
Future Directions for Therapy
Treatment of cardiac manifestations is generally the same in RAS syndrome patients as in the general population. Surgical and pharmacologic treatment (like verapamil, an inhibitor of calcium channels, and β-blockers, inhibitors of adrenergic receptors) are generally used to alleviate severe cardiac hypertrophy. The RAS/MAPK pathway has long been a drug target because of its involvement in malignant tumors (110). As pointed out in this review, efficient therapeutic strategies that directly target the RAF-MEK-ERK1/2 cascade might be the best tool against cardiac hypertrophy (30,33,111). Possibly, advanced therapies could be based on antisense oligonucleotides that inhibit the translation of the altered genetic portion, or on gene-targeted therapies.
Direct inhibitors of the pathway, which have already been identified in anticancer studies, might represent an alternative. Unfortunately, anticancer drugs designed to specifically target molecules in the RAS pathway have shown considerable side effects. Selective inhibitors of RAF, such as PLX4032, have remarkable clinical activity in patients with melanomas who carry mutated BRAF (112). However, as for other inhibitors of oncogenic kinases (113,114), response to PLX4032 is profound but often temporary, because of the loss of addiction and the onset of resistance (115). Moreover, the functional role of MAPKs in the heart itself presents a potential dilemma to any heart disease therapies targeting this pathway: the physiological role of RAS signaling should not be forgotten. In cultured myocytes, MAPK-inducing agents have protective effects against starvation, hypoxia or reoxygenation injury (116,117). MEK transgenic hearts are protected from ischemia/reperfusion and apoptosis; similarly, inactivation of ERK2 promotes damage and death in response to ischemia/reperfusion (118), whereas inhibition of RAF1 promotes myocyte apoptosis and heart failure (58,119). In fact, the RAS pathway modulates the activity of regulators of apoptosis (120–123), as well as of protein kinase C#x03B5 and p53 (124). Thus, although activation of RAS signaling may trigger HCM, inhibiting the pathway could render hearts more vulnerable to stress-induced myocyte death, considerably increasing the risk of worsening heart disease.
Clinical efficacy and feasibility would certainly be implemented whether maladaptive hypertrophic signals could be selectively blocked, without affecting the physiological positive effects. It follows that only detailed knowledge about the differential molecular mechanisms of ERK1/2 activation could lead to realistic therapeutic opportunities. The identification of a Thr188-phosphorylation site on ERK2 might be a first step in this direction: this phosphorylation can selectively activate hypertrophic ERK1/2 functions and, therefore, may be targeted without dangerous side effects. Mutant mice that lack this phosphorylation site show preserved cardiac structure and function and attenuated hypertrophic response, indeed demonstrating that ERK1/2 can be selectively targeted, at least in the adult patient (59).
The latest recommended goals for treating hypertrophy may require multiple drugs. Lower doses in combination may render more efficacy and safety than highest doses of single agents. The choice of added agents should greatly depend on both cost and compliance. Currently, generic statins offer several cost benefits. These inhibitors of the posttranscriptional lipid modifications of RAS are likely to be effective, because they have been shown to exert antitumoral action in different cancer cells with mutations on RAS (125,126). Indeed, statins reduced cardiac hypertrophy in a transgenic model of HCM (127,128). A combination of low doses of statins and specific inhibitors of pathological ERK1/2 signaling represents, at the moment, a pretty exciting possibility still to be explored.
Conclusion
The RAS/RAF/MEK/MAPK pathway is involved in both proliferation and differentiation of different cellular lines, the cardiac lineage included, and thus plays a pivotal role during both adulthood and embryogenesis; this characteristic finally excludes the possibility of completely inhibiting this signaling cascade in young patients (for example, pediatric patients with RASopathy-associated HCM). Although the need of introducing novel drugs in the pediatric clinics is evident, the balance between risk and benefit should be carefully considered. Finally, the dose used to treat children should be reduced as much as possible, considering the possible adverse impact of even a modest degree of toxicity. We suggest that the severity and degree of cardiovascular impairment in pediatric patients with RASopathies should be subjected to accurate stratification; this could enable the minimization of drug dosage to spare patients with mild manifestations of HCM from treatment-related morbidity and mortality. On the other hand, the most severe cases should be treated with targeted and rationally designed therapies aimed at chronicizing a disease that would be otherwise lethal.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Shih TY, Papageorge AG, Stokes PE, Weeks MO, Scolnick EM. (1980) Guanine nucleotide-binding and autophosphorylating activities associated with the p21src protein of Harvey murine sarcoma virus. Nature. 287:686–91.
Raman M, Chen W, Cobb MH. (2007) Differential regulation and properties of MAPKs. Oncogene. 26:3100–12.
Yoon S, Seger R. (2006) The extracellular signalregulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 24:21–14.
Hill JA, Olson EN. (2008) Mechanisms of disease: cardiac plasticity. New. Engl. J. Med. 358:1370–80.
Takai Y, Sasaki T, Matozaki T. (2001) Small GTP-binding proteins. Physiol. Rev. 81:153–208.
Xu GF, et al. (1990) The Neurofibromatosis Type-1 gene encodes a protein related to GAP. Cell. 62:599–608.
Tartaglia M, et al. (2001) Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 29:465–8.
Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG. (2006) PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J. Biol. Chem. 281:6785–92.
Pandit B, et al. (2007) Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat. Genet. 39:1007–12.
Lin AE, et al. (2011) Clinical, pathological, and molecular analyses of cardiovascular abnormalities in Costello Syndrome: a Ras/MAPK pathway syndrome. Am. J. Med. Genet. A. 155A:486–507.
Gripp KW, et al. (2006) HRAS mutation analysis in Costello syndrome: genotype and phenotype correlation. Am. J. Med. Genet. A. 140A:1–7.
Sarkozy A, Digilio MC, Dallapiccola B. (2008) Leopard syndrome. Orphanet J. Rare Dis. 3:13.
Gripp KW, et al. (2007) Further delineation of the phenotype resulting from BRAF or MEK1 germline mutations helps differentiate cardiofacio-cutaneous syndrome from Costello syndrome. Am. J. Med. Genet. A. 143A:1472–80.
Niihori T, et al. (2006) Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat. Genet. 38:294–6.
Rodriguez-Viciana P. et al. (2006) Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 311:1287–90.
Cirstea IC, et al. (2010) A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat. Genet. 42:27–9.
Martinelli S, et al. (2010) Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am. J. Hum. Genet. 87:250–7.
Roberts AE, et al. (2007) Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat. Genet. 39:70–4.
Tartaglia M, et al. (2002) PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am. J. Hum. Genet. 70:1555–63.
Rose BA, Force T, Wang YB. (2010) Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol. Rev. 90:1507–46.
Yutzey KE, Colbert M, Robbins J (2005) Ras-related signaling pathways in valve development: ebb and flow. Physiol. 20:390–7.
Rochais F, Mesbah K, Kelly RG (2009) Signaling pathways controlling second heart field development. Circ. Res. 104:933–42.
Snarr BS, Kern CB, Wessels A (2008) Origin and fate of cardiac mesenchyme. Dev. Dyn. 237:2804–19.
Lin AE, et al. (2009) Prenatal features of Costello syndrome: ultrasonographic findings and atrial tachycardia. Prenatal Diag. 29:682–90.
Smith LP, Podraza J, Proud VK. (2009) Polyhydramnios, fetal overgrowth, and macrocephaly: prenatal ultrasound findings of Costello syndrome. Am. J. Med. Genet. A. 149A: 779–84.
Dorn GW II, Brown JH. (1999) Gq signaling in cardiac adaptation and maladaptation. Trends Cardiovasc. Med. 9:26–34.
Heineke J, Molkentin JD. (2006) Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell. Biol. 7:589–600.
Xiao L, et al. (2001) MEK1/2-ERK1/2 mediates alpha(1)-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J. Mol. Cell. Cardiol. 33:779–87.
Dwyer JP, et al. (2008) Myocardial gene expression associated with genetic cardiac hypertrophy in the absence of hypertension. Hypertens. Res. 31:941–55.
Muslin AJ. (2008) MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin. Sci. 115:203–18.
Onan D, Pipolo L, Yang E, Hannan RD, Thomas WG. (2004) Urotensin II promotes hypertrophy of cardiac myocytes via mitogen-activated protein kinases. Mol. Endocrinol. 18:2344–54.
Samarel AM et al. (2001) Src and multiple MAP kinase activation in cardiac hypertrophy and congestive heart failure under chronic pressure-overload: comparison with acute mechanical stretch. J. Mol. Cell. Cardiol. 33:1637–48.
Wang YB. (2007) Mitogen-activated protein kinases in heart development and diseases. Circulation. 116:1413–23.
Olson EN, Schneider MD. (2003) Sizing up the heart: development redux in disease. Genes Dev. 17:1937–56.
Blaxall BC, Tschannen-Moran BM, Milano CA, Koch WJ. (2003) Differential gene expression and genomic patient stratification following left ventricular assist device support. J. Am. Coll. Cardiol. 41:1096–06.
Lowes BD, et al. (2002) Myocardial gene expression in dilated cardiomyopathy treated with beta-blocking agents. New Engl. J. Med. 346:1357–65.
Esposito G, et al. (2002) Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation. 105:85–92.
Frey N, Katus HA, Olson EN, Hill JA. (2004) Hypertrophy of the heart: a new therapeutic target? Circulation. 109:1580–9.
Fuller SJ, Finn SG, Downward J, Sugden PH. (1998) Stimulation of gene expression in neonatal rat ventricular myocytes by Ras is mediated by Ral guanine nucleotide dissociation stimulator (Ral.GDS) and phosphatidylinositol 3-kinase in addition to Raf. Biochem. J. 335:241–6.
Thorburn A, et al. (1993) Hras-dependent pathways can activate morphological and genetic-markers of cardiac-muscle cell hypertrophy. J. Biol. Chem. 268:2244–9.
Kai H, et al. (1998) Expression of proto-oncogenes and gene mutation of sarcomeric proteins in patients with hypertrophic cardiomyopathy. Circ. Res. 83:594–601.
Hunter JJ, Tanaka N, Rockman HA, Ross J, Chien KR. (1995) Ventricular expression of A Mlc-2V-Ras fusion gene induces cardiac-hypertrophy and selective diastolic dysfunction in transgenic mice. J. Biol. Chem. 270:23173–8.
Schuhmacher AJ, et al. (2008) A mouse model for Costello syndrome reveals an Ang II-mediated hypertensive condition. J. Clin. Invest. 118:2169–79.
Santoriello C, et al. (2009) Expression of H-RASV12 in a zebrafish model of Costello syndrome causes cellular senescence in adult proliferating cells. Dis. Model Mech. 2:56–67.
Leo C, et al. (2011) Activated Met signalling in the developing mouse heart leads to cardiac disease. Plos One. 6:e14675.
Zhang SS, et al. (2003) The role of the Grb2-p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J. Clin. Invest. 111:833–41.
Chen PC, et al. (2010) Activation of multiple signaling pathways causes developmental defects in mice with a Noonan syndrome-associated Sos1 mutation. J. Clin. Invest. 120:4353–65.
Araki T, et al. (2004) Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat. Med. 10:849–57.
Marin TM, et al. (2011) Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J. Clin. Invest. 121:1026–43.
Oudit GY, Penninger JM. (2009) Cardiac regulation by phosphoinositide 3-kinases and PTEN. Cardiovasc. Res. 82:250–60.
Yano N, et al. (2008) Temporally controlled overexpression of cardiac-specific PI3K alpha induces enhanced myocardial contractility-a new transgenic model. Am. J. Physiol. Heart Circ. Physiol. 295:H1690–4.
McMullen JR, et al. (2003) Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc. Natl. Acad. Sci. U. S. A. 100:12355–60.
McMullen JR, et al. (2007) Protective effects of exercise and phosphoinositide 3-kinase(p110 alpha) si3naling in dilated and hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A. 104:612–7.
Ruan HM, et al. (2009) Inducible and cardiac specific PTEN inactivation protects ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 46:193–200.
Shiojima I, et al. (2005) Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Invest. 115:2108–18.
Harris IS, et al. (2004) Raf-1 kinase is required for cardiac hypertrophy and cardiomyocyte survival in response to pressure overload. Circulation. 110:718–23.
Klein M et al. (2008) Combined tyrosine and serine/threonine kinase inhibition by sorafenib prevents progression of experimental pulmonary hypertension and myocardial remodeling. Circulation. 118:2081–90.
Kobayashi T, et al. (2010) Molecular and clinical analysis of RAF1 in Noonan syndrome and related disorders: dephosphorylation of serine 259 as the essential mechanism for mutant activation. Hum. Mutat. 31:284–94.
Wu X. (2011) MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1L613V mutation. J. Clin. Invest. 121:1009–25.
Urosevic J, et al. (2011) Constitutive activation of B-Raf in the mouse germ line provides a model for human cardio-facio-cutaneous syndrome. Proc. Natl. Acad. Sci. U. S. A. 108:5015–20.
Allanson JE et al. (2011) Cardio-Facio-Cutaneous syndrome: does genotype predict phenotype? Am. J. Med. Genet. C. Semin. Med. Genet. 157:129–35.
Clerk A, Aggeli IKS, Stathopoulou K, Sugden PH. (2006) Peptide growth factors signal differentially through protein kinase C to extracellular signal-regulated kinases in neonatal cardiomyocytes. Cell. Signal. 18:225–35.
Kennedy RA, Kemp TJ, Sugden PH, Clerk A. (2006) Using U0126 to dissect the role of the extracellular signal-regulated kinase 1/2 (ERK1/2) cascade in the regulation of gene expression by endothelin-1 in cardiac myocytes. J. Mol. Cell. Cardiol. 41:236–47.
Sanada S, et al. (2003) Long-acting Ca2+ blockers prevent myocardial remodeling induced by chronic NO inhibition in rats. Hypertension. 41:963–7.
Yue TL, et al. (2000) Extracellular signal-regulated kinase plays an essential role in hypertrophic agonists, endothelin-1 and phenylephrine-induced cardiomyocyte hypertrophy. J. Biol. Chem. 275: 37895–901.
Armstrong SC. (2004) Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc. Res. 61:427–36.
Takeishi Y, et al. (2002) Activation of mitogen-activated protein kinases and p90 ribosomal S6 kinase in failing human hearts with dilated cardiomyopathy. Cardiovasc. Res. 53:131–7.
Huebert RC, et al. (2004) Identification and regulation of Sprouty1, a negative inhibitor of the ERK cascade, in the human heart. Physiol. Genomics. 18:284–9.
Bueno OF, et al. (2001) The dual-specificity phosphatase MKP-1 limits the cardiac hypertrophic response in vitro and in vivo. Circ. Res. 88:88–96.
Purcell NH, et al. (2007) Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc. Natl. Acad. Sci. U. S. A. 104:14074–9.
Luttrell DK, Luttrell LM. (2003) Signaling in time and space: G protein-coupled receptors and mitogen-activated protein kinases. Assay Drug Dev. Techn. 1:327–38.
Owens DM, Keyse SM. (2007) Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene. 26:3203–13.
Lorenz K, Schmitt JP, Schmitteckert EM, Lohse MJ. (2009) A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat. Med. 15:75–83.
Bueno OF, et al. (2000) The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. Embo. J. 19:6341–50.
Slupsky JR, et al. (1999) Binding of G beta gamma subunits to cRaf1 downregulates G-protein-coupled receptor signalling. Curr. Biol. 9:971–4.
Metrich M, et al. (2008) Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ. Res. 102:959–65.
Morel E, et al. (2005) cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ. Res. 97:1296–304.
Ponsioen B, et al. (2004) Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. Embo. Rep. 5:1176–80.
de Rooij J, et al. (1998) Epac is a Rap1 guaninenucleotide-exchange factor directly activated by cyclic AMP. Nature. 396:474–77.
Kawasaki H, et al. (1998) A family of cAMP-binding proteins that directly activate Rap1. Science. 282:2275–9.
Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG. (2006) Cell physiology of cAMP sensor Epac. J. Physiol. 577:5–15.
Schmidt M, Sand C, Jakobs KH, Michel MC, Weernink PA. (2007) Epac and the cardiovascular system. Curr. Opin. Pharmacol. 7:193–200.
Barki-Harrington L, Perrino C, Rockman HA. (2004) Network integration of the adrenergic system in cardiac hypertrophy. Cardiovasc. Res. 63:391–402.
Salazar NC, Chen J, Rockman HA. (2007) Cardiac GPCRs: GPCR signaling in healthy and failing hearts. Biochim. Biophys. Acta. 1768:1006–18.
Kuster GM, et al. (2005) Alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes is mediated via thioredoxin-1-sensitive oxidative modification of thiols on Ras. Circulation. 111:1192–8.
Aoki H, Richmond M, Izumo S, Sadoshima J. (2000) Specific role of the extracellular signalregulated kinase pathway in angiotensin II-induced cardiac hypertrophy in vitro. Biochem. J. 347:275–84.
Cullingford TE, et al. (2008) Temporal regulation of expression of immediate early and second phase transcripts by endothelin-1 in cardiomyocytes. Genome Biol. 9:R32.
Wright CD, et al. (2008) Nuclear alpha 1-adrenergic receptors signal activated ERK localization to caveolae in adult cardiac myocytes. Circ. Res. 103:992–1000.
Torsoni AS, Constancio SS, Nadruz W, Hanks SK, Franchini KG. (2003) Focal adhesion kinase is activated and mediates the early hyper-trophic response to stretch in cardiac myocytes. Circ. Res. 93:140–7.
Nadruz W, Corat MAF, Marin TM, Pereira GAG, Franchini KG. (2005) Focal adhesion kinase mediates MEF2 and c-Jun activation by stretch: role in the activation of the cardiac hypertrophic genetic program. Cardiovasc. Res. 68:87–97.
Passier R, et al. (2000) CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J. Clin. Invest. 105:1395–406.
McKinsey TA, Zhang CL, Olson EN. (2002) MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem. Sci. 27:40–7.
Miyata S, Minobe W, Bristow MR, Leinwand LA. (2000) Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ. Res. 86:386–90.
Abraham WT, et al. (2002) Coordinate changes in myosin heavy chain isoform gene expression are selectively associated with alterations in dilated cardiomyopathy phenotype. Mol. Med. 8:750–60.
Braunwald E, Bristow MR. (2000) Congestive heart failure: fifty years of progress. Circulation. 102:14–23.
Mitchell S, et al. (2006) Distinct gene expression profiles in adult mouse heart following targeted MAP kinase activation. Physiol. Genomics. 25:50–9.
Ruan HM, et al. (2007) Gi alpha 1-mediated cardiac electrophysiological remodeling and arrhythmia in hypertrophic cardiomyopathy. Circulation. 116:596–605.
Chen J, et al. (1998) Selective requirement of myosin light chain 2v in embryonic heart function. J. Biol. Chem. 273:1252–6.
Ho PD, et al. (2001) Ras reduces L-type calcium channel current in cardiac myocytes — corrective effects of L-channels and SERCA2 on [Ca2+](i) regulation and cell morphology. Circ. Res. 88:63–9.
Zheng MZ, et al. (2004) Sarcoplasmic reticulum calcium defect in Ras-induced hypertrophic cardiomyopathy heart. Am. J. Physiol. Heart Circ. Physiol. 286: H424–33.
Frey N, McKinsey TA, Olson EN. (2000) Decoding calcium signals involved in cardiac growth and function. Nat. Med. 6:1221–7.
Crabtree GR, Olson EN. (2002) NFAT signaling: choreographing the social lives of cells. Cell. 109: S67–79.
Molkentin JD, et al. (1998) A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 93:215–28.
Chaudhary A, et al. (2000) Phosphatidylinositol 3-kinase regulates Raf1 through Pak phosphorylation of serine 338. Curr. Biol. 10:551–4.
Grimminger F, Schermuly RT, Ghofrani HA. (2010) Targeting non-malignant disorders with tyrosine kinase inhibitors. Nat. Rev. Drug. Discov. 9:956–70.
Tuveson DA, et al. (2004) Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 5:375–87.
Lei L et al. (2008) Hypoxia-inducible factor-dependent degeneration, failure, and malignant transformation of the heart in the absence of the von Hippel-Lindau protein. Mol. Cell. Biol. 28:3790–803.
Comoglio PM, Giordano S, Trusolino L. (2008) Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat. Rev. Drug. Discov. 7:504–16.
Grossmann KS. (2011) The tyrosine phosphatase Shp2 in development and cancer. Adv. Cancer Res. 106:53–89.
Young A, et al. (2009) Ras signaling and therapies. Adv Cancer Res. 102:1–17.
Gutkind JS, Offermanns S. (2009) A new Gq-initiated MAPK signaling pathway in the heart. Dev. Cell. 16:163–4.
Flaherty KT, et al. (2010) Inhibition of mutated, activated BRAF in metastatic melanoma. New Engl. J. Med. 363:809–19.
Garrett JT, Arteaga CL. (2011) Resistance to HER2-directed antibodies and tyrosine kinase inhibitors. Cancer Biol. Ther. 11:793–800.
Poulikakos PI, Solit DB. (2011) Resistance to MEK inhibitors: should we co-target upstream? Sci. Signal. 4:pe16.
Solit DB, Rosen N. (2011) Resistance to BRAF inhibition in melanomas. New Engl. J. Med. 364:772–4.
Bueno OF, Molkentin JD. (2002) Involvement of extracellular signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ. Res. 91:776–81.
Sugden PH & Clerk A. (2006) Oxidative stress and growth-regulating intracellular signaling pathways in cardiac myocytes. Antioxid. Redox Signal. 8:2111–24.
Lips DJ, et al. (2004) MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation. 109:1938–41.
Yamaguchi O, et al. (2004) Cardiac-specific disruption of the c-raf-1 gene induces cardiac dysfunction and apoptosis. J. Clin. Invest. 114:937–43.
Le Mellay V, Troppmair J, Benz R, Rapp UR. (2002) Negative regulation of mitochondrial VDAC channels by C-Raf kinase. BMC Cell Biol. 3:14.
O’Neill E. (2011) Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science. 306:2267–70.
Rapp UR, Rennefahrt U, Troppmair J. (2004) Bcl-2 proteins: master switches at the intersection of death signaling and the survival control by Raf kinases. Biochim Biophys Acta 1644:149–58.
Tian S, et al. (2006) Interaction and stabilization of X-linked inhibitor of apoptosis by Raf-1 protein kinase. Int. J. Oncol. 29:861–7.
Cox AD, Der CJ. (2003) The dark side of Ras: regulation of apoptosis. Oncogene 22:8999–9006.
Park IH, Kim JY, Jung JI, Han JY. (2010) Lovastatin overcomes gefitinib resistance in human non-small cell lung cancer cells with K-Ras mutations. Invest. New Drugs. 28:791–9.
Perchellet JP, et al. (2009) Novel synthetic inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase activity that inhibit tumor cell proliferation and are structurally unrelated to existing statins. Int. J. Mol. Med. 24:633–43.
Patel R, et al. (2001) Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 104:317–24.
Senthil V, et al. (2005) Prevention of cardiac hypertrophy by atorvastatin in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circ. Res. 97:285–92.
Acknowledgments
We kindly acknowledge the constant support of the Association Francaise contre les Myopathies (AFM) and the Seventh Framework Programme. V Sala was a Fellow of Università Italo Francese in 2011 (UIF, Cap. III Progetto Vinci 2008). The fellowships of V Sala and C Leo were granted by the FP7-2010-ICT-GC „EM-SAFETY“ project no. 265772. We gratefully thank GB Ferrero for helpful discussion.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (http://creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Sala, V., Gallo, S., Leo, C. et al. Signaling to Cardiac Hypertrophy: Insights from Human and Mouse RASopathies. Mol Med 18, 938–947 (2012). https://doi.org/10.2119/molmed.2011.00512
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2011.00512