
Abstract
Treatment of acute lung injury (ALI) and its most severe form, acute respiratory distress syndrome (ARDS), remain unsolved problems of intensive care medicine. ALI/ARDS are characterized by lung edema due to increased permeability of the alveolar-capillary barrier and subsequent impairment of arterial oxygenation. Lung edema, endothelial and epithelial injury are accompanied by an influx of neutrophils into the interstitium and broncheoalveolar space. Hence, activation and recruitment of neutrophils are regarded to play a key role in progression of ALI/ARDS. Neutrophils are the first cells to be recruited to the site of inflammation and have a potent antimicrobial armour that includes oxidants, proteinases and cationic peptides. Under pathological circumstances, however, unregulated release of these microbicidal compounds into the extracellular space paradoxically can damage host tissues. This review focuses on the mechanisms of neutrophil recruitment into the lung and on the contribution of neutrophils to tissue damage in ALI.
Similar content being viewed by others

Introduction
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are characterized by an increased permeability of the alveolar-capillary barrier resulting in lung edema with protein-rich fluid, thus resulting in impairment of arterial oxygenation. ALI/ARDS is defined as a lung disease with acute onset, non-cardiac, diffuse bilateral pulmonary infiltrates and a paO2/FiO2 ≤ 300 for ALI or a paO2/FiO2 ≤ 200 for ARDS. The age-adjusted incidence of ALI/ARDS is estimated with 86.2 per 100,000 person-years (1). Despite all innovations in intensive care medicine, the mortality of ARDS remains up to 40% (2). Whereas pneumonia or sepsis can undoubtedly cause ALI and ARDS, several noninfectious causes also may trigger ALI/ARDS, for example, acid aspiration, hyperoxia, high pressure ventilation, pulmonary contusion, reperfusion or bleomycin (3). While these agents induce lung damage by direct exposure to the lung, similar lung damage can arise indirectly. In particular, trauma, pancreatitis or transfusion can initiate an inflammatory response called systemic inflammatory response syndrome (SIRS) that may lead to ALI or ARDS (4).
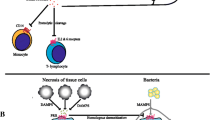
The alveolar epithelium contains two different cell types. The flat type I cells build the structure of an alveolar wall, accounting for only 20% of the epithelial cells but covering 80% of the alveolar surface area. The cuboidal type II cells, which account for 80% of the alveolar cells, secrete pulmonary surfactant to lower the surface tension and regulate fluid balance across the epithelium alveolar. As progenitor cells, alveolar type II cells may regenerate type I cells after injury (Figure 1A).

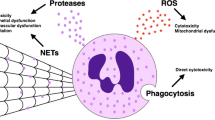
Neutrophil-mediated inflammatory processes in acute lung injury. (A) Normal alveolus. (B) Recruitment of neutrophils into the lung. (C) Tissue damage in acute lung injury.
Recent animal studies have revealed that endothelial injury appears within minutes to hours after ALI induction and results in intercellular gaps of the endothelium. Formation of intercellular gaps can be regarded as the basis for increased microvascular permeability (4). In addition, the contribution of epithelial injury to progression of ALI/ARDS has become increasingly obvious. Decreases in epithelial cell barrier function facilitate influx of protein rich fluid and other macromolecules into alveolar space. Furthermore, epithelial injury leads to impaired cell fluid transport and reduced production of surfactant (5).
Lung edema, endothelial and epithelial injury are accompanied by an influx of neutrophils into the interstitium and broncheoalveolar space. Neutrophils are considered to play a key role in the progression of ALI and ARDS (6), as activation and transmigration of neutrophils is a hallmark event in the progression of ALI and ARDS. Proof for the importance of neutrophils in ALI comes from clinical data and animal models. In patients with ARDS, the concentration of neutrophils in the bronchoalveolar lavage (BAL) fluid correlates with severity of ARDS and outcome (7–9), whereas the severity of lung injury has been reduced by neutrophil depletion in mice (10). Furthermore, after blocking interleukin-8 (IL-8), a major chemoattractant for neutrophils, rabbits have been protected from acid aspiration-induced lung injury (11). Although neutrophils can migrate into the alveolar space without damaging the alveolar-capillary barrier (12), recruitment of neutrophils into the lung is an important step in ALI. In addition, ALI/ARDS can occur in children and adults with neutropenia (13,14,15) indicating that, under specific conditions, neutrophil-independent mechanisms alone allow for development of ALI. Despite that, a multitude of experimental and clinical data point at the causative role of neutrophils in lung injury. Although neutrophil activation is vital for the host defense, overzealous activation leads to tissue damage by release of cytotoxic and immune cell-activating agents such as proteinases, cationic polypeptides, cytokines, and reactive oxygen species (ROS). In this review, we aim to highlight mechanisms by which neutrophils are recruited into the lung, and by which neutrophils contribute to the onset and progression of ALI by means of infiltrating the lung tissue and releasing preformed granule proteins or vast amounts of ROS. The majority of this review is based on experimental data derived from various animal models of ALI (Tables 1 and 2). When looking at the role of individual proteins in a complex pathophysiological process such as ALI (Tables 1 and 2) one should keep in mind that there is likely a publication bias against studies with negative outcome.
Neutrophil Dependency of Acute Lung Injury
Although neutrophil recruitment into the lung is a hallmark of ALI, there are neutrophil-dependent (for example, LPS administration, acid-induced ALI, transfusion-related ALI) and neutrophil-independent (for example, oleic acid, hyperoxia) models of ALI (3). Administration of LPS, either via an intravenous or an intra-alveolar route, induces changes in neutrophil deformability and the entrapment of neutrophils in the pulmonary capillaries. The neutrophil entrapment is followed by permeability changes and edema formation (16,17). Similarly, acid aspiration is a neutrophil-dependent form of ALI characterized by injury of the alveolar epithelium and also to the capillary endothelium (11,18). Moreover, mechanical ventilation can induce lung injury and inflammation. The ventilator-induced lung injury (VILI) damages tissue owing to mechanical stretch and activation of specific intracellular pathways. Beside this mechanical damage, both an inflammatory response and neutrophils are needed for the development of increased permeability and hyaline membranes in VILI (19).
After intravenous application of oleic acid, ALI is induced by direct damage to the capillary endothelium resulting in neutrophil accumulation. This model is neutrophil independent, indicating that a direct effect of oleic acid on the endothelium is the crucial step (20). It is generally accepted that hyperoxia-induced ALI is mediated by free radicals. Although hyperoxia increases NF-κB translocation in lung mononuclear cells, release of proinflammatory mediators and accumulation of neutrophils in the lung, neutrophil depletion does not prevent hyperoxia-induced ALI (21,22).
Neutrophil Recruitment into the Lung
The acute phase of ALI and ARDS is distinguished by the influx of protein-rich edema fluid into the air spaces as a consequence of increased permeability of the alveolar-capillary barrier. This increased permeability is facilitated by widespread injury and activation of both lung and systemic endothelium. Furthermore, epithelial injury—which leads to reduced surfactant production and reduced fluid transport—aggravates the pulmonary edema. Another histological hallmark is the recruitment of neutrophil into the lung. However, neutrophils can migrate into the lungs of humans without causing injury. Neutrophil emigration into the lungs is not sufficient to cause ALI; neutrophil activation is also required.
Neutrophils are the earliest immune cells to be recruited to the site of injury or inflammation. After activation, neutrophils are able to egress from the vasculature and migrate through the interstitium into the alveolar space. Extravasation of neutrophils normally takes place in postcapillary venules in a cascade-like sequence of capture (or tethering) and rolling, which is selectinmediated. Rolling is followed by chemokine-dependent activation of integrins ultimately leading to adhesion on the endothelium. Subsequently, neutrophils transmigrate into the tissue using a paracellular or transcellular route (23) (Figure 1B).
In contrast to many other organs, neutrophil recruitment into the lung takes place in the small capillaries in a sequence of activation, sequestration from blood to interstitium and transepithelial (from interstitium to alveolar airspace) migration. In particular, the microcirculation of the lung has to be recognized, because the neutrophils (ranging from 6 to 10 µm) have to change their shape to pass through the small pulmonary capillaries (diameter from 2 to 15µm) (24). In this context, Doerschuk et al. has shown in an animal model that the specific architecture of the lung brings about a prolonged transit time of the neutrophils (25). Considering the specific condition of the lung, recent studies have focused on the mechanical properties of the neutrophils after activation. Inflammatory stimuli, primarily those which bind to seven-transmembrane-spanning G-protein linked receptors, induce changes in the cytoskeleton of neutrophils that reduce the capability to deform (26–29). Furthermore, the reduced deformability of neutrophils, induced by increases in F-actin at the cell periphery, leads to an increase of neutrophil sequestration in the interstitium (30).
Before neutrophils migrate along the endothelium into the interstitium and alveolar airspace, they tether and roll on the endothelium. Rolling which is mediated by selectins is followed by slow rolling and arresting on the endothelium, which is mediated by integrins and chemokines. Whereas neutrophils penetrate mainly interendothelial junctions at bicellular or tricellular corners, there also is a transcellular alternative route (31). In particular, neutrophils that adhere to the endothelium affect the endothelial cytoskeleton, inducing remodeling of tight junctions (for example, platelet endothelial cell adhesion molecule [PECAM]-1, CD99, VE-cadherin, JAMSs). Consequently, this transient remodeling of tight junctions of the endothelium facilitates transmigration of neutrophils (32,33).
Selectins
Selectins are a family of transmembrane molecules expressed on the surface of leukocytes and activated endothelial cells, and are essential for initiating the rolling process of neutrophils on the endothelium. This initial step in leukocyte adhesion is capture, which is governed by interactions between L-, E- and P-selectins and P-selectin glycoprotein ligand (PSGL1). L-selectin is expressed by leukocytes, P-selectin is expressed by inflamed endothelium and platelets, E-selectin is expressed by inflamed endothelium and PSGL1 is expressed by leukocytes and endothelium (34,35). However, the role of selectins for neutrophil transmigration in the lung is not completely understood and depends on the inflammatory stimulus. Neu-trophil emigration induced by Streptococcus pneumoniae was not reduced in mice deficient of E-selectin and P-selectin and additional inhibition of L-selectin (36). Furthermore, blocking all three selectins does not attenuate the LPS-induced migration of neutrophils into the alveolar space (37). In addition, inhibition of P-selectin does not decrease transfusion-related ALI (38,39). In contrast, all selectins are involved in the progress of lung injury caused by activation of complement or intratracheal application of IgG (40,41). Moreover, severity of ALI in the rat caused by infusion of cobra venom factor (VCF) or LPS correlates with P-selectin plasma levels (42). Experimental data suggest that inhibition or deletion of one or more selectins protects from ALI (43–45). In VCF-induced ALI, anti-P-selectin antibodies (Abs) have inhibited neutrophil transmigration and lung edema in wild type mice, but P-selectin−/− mice have not been protected from ALI (46). Zarbock et al. have shown that acid aspiration-induced P-selectin-dependent plateletneutrophil interactions in blood and in lung capillaries. Reducing circulating platelets or blocking P-selectin has halted the development of ALI. Interestingly, Zarbock et al. have displayed platelet-derived rather than endothelial-derived P-selectin as the relevant adhesion molecule facilitating neutrophil sequestration (47).
Integrins
Subsequent to rolling of neutrophils is slow rolling and arrest on the endothelial surface involving b1 and b2 integrins. Integrins, membrane-linked proteins composed of a and b chains, play an important role in cell-to-cell adhesions and for cell adhesion to the extracellular matrix. Moreover, integrins are involved in neutrophil migration, phagocytosis, ROS release and cytokine production (48). Integrins mediate cell adhesion by interaction with adhesion molecules (for example, intercellular adhesion molecule [ICAM]-1, ICAM-2, vascular cell adhesion molecule [VCAM]-1). Integrins are able to activate intracellular pathways with transmembrane connections to the cytoskeleton (49). In particular, neutrophil adhesion is commonly mediated through β2-integrins (50). Deficiency of β2-integrin in humans is known as leukocyte adhesion deficiency (LAD) and causes recurrent bacterial infections (that is, pneumonia, gingivitis, abscesses or peritonitis). Interestingly, in acute lung injury, neu-trophils adhere in β2-integrin-dependent or in β2-integrin-independent pathways, depending on the stimulus and kind of lung injury model. Stimuli, like Pseudomonas aeruginosa, IgG immune complexes, Escherichia coli, and IL-1 facilitate neutrophil emigration in a β2-integrin-mediated fashion (51–53). Inhibition of β2-integrins attenuated neutrophil recruitment (54–56). Most stimuli inducing β2-integrin-dependent neutrophil migration enhance endothelial ICAM-1 expression. In VCF-induced ALI, anti-ICAM-1 antibodies reduce ALI in wild type mice but do not reduce ALI in ICAM-1−/− mice. However, mutant mice (ICAM-1−/− and double knockout P-selectin−/− and ICAM-1−/−) are not protected from acute lung injury (Table 2). Whereas neutrophils utilize other alternative adhesion pathways in these mutant mice, antibodies that block ICAM-1 may inhibit neutrophil transmigration and ALI through other mechanism than simply adhesion blockade. (46) (Table 1).
Staphylococcus aureus, Streptococcus pneumonia, group B Streptococcus, hydrochloric acid, hyperoxia or C5a are stimuli that initiate neutrophil emigration in a β2-integrin-independent fashion (57–59). In contrast to the β2-integrin-dependent pathway, neutrophils emigrate in the absence of increased ICAM-1 expression. Here, β1-integrins (very late antigen [VLA]-4 and VLA-5) interact with VCAM-1 (31). This pathway of neutrophil emigration may not require β2-integrins, because recent studies blocking these adhesion molecules have failed to decrease neutrophil migration (38,60). Yet another interesting feature regarding neutrophil emigration in ALI is the frequently observed divergence of studies using knock-out mice or blocking strategies (46). Some of this discrepancy may be attributed to the fact that when deleting a gene throughout growth and development, alternative pathways may have evolved, which is not the case when acutely applying an antagonist.
Chemokines
Neutrophil infiltration of the lung is controlled by a complex network of chemokines that are released by a variety of cell types. Alveolar macrophages are a major source of chemokines in the alveolar space (Figure 1A) and produce IL-8, growth-regulated oncogene (GRO)-related peptides and CXCL5 (also known as epithelial neutrophil-activating protein [ENA]-78) (61). High concentrations of IL-8 in BAL fluid from ARDS patients are associated with increased neutrophil influx into the airspace (62,63). Recent studies have revealed that IL-8 in BAL fluid is bound to IL-8 autoantibodies (anti-IL-8:IL-8 complexes) (64,65) and BAL fluid concentrations of these complexes correlate with development and outcome of ALI (66,67). In particular, this complex exhibits chemotactic and pro-inflammatory activity (68). Moreover, intratracheal application of IL-8 induces lung injury which can be attenuated by inhibition in different models of ALI (11,69–73) (Table 1).
In rodents, the most relevant chemokines for neutrophil recruitment into the lung are keratinocyte-derived chemokine (KC, also named CXCL1) or cytokine-induced neutrophil chemoat-tractant (CINC; the rat homolog to KC) and macrophage inflammatory protein-2 (MIP-2, also named CXCL2) (74). Similar to IL-8, CXCL1, CXCL2, lipopolysaccha-ride-induced CXC chemokine (LIX, also named CXCL5) and lungkine (CXCL15) bind to CXCR2. Inhibition or knockout of CXCR2 receptor diminishes neutrophil influx into the lung (75–82). In contrast to the multiple CXC chemokines only two CXC chemokines receptors, CXCR1 and CXCR2, have been shown to mediate the response to CXC chemokines in human neutrophils. Whereas human CXCR1 binds to CXCL6 and CXCL8 (IL-8) with a high affinity, human CXCR2 binds also to CXCL6 and IL-8 as well as several CXC chemokines (GRO-α, GRO-β, GRO-γ, CXCL1, CXCL2, CXCL3), ENA-78 (CXCL5) and (CXCL7) (83). Clinical studies have revealed a reduced expression of CXCR2 but not CXCR1 in septic shock (84,85). Thus, therapy to limit neutrophil accumulation at sites of inflammation should be directed at CXCR1, because CXCR2 is downregulated while CXCR1 is upregulated.
Whereas CXCL1 and CXCL2 have been suggested to be the most important chemokines for neutrophil recruitment, CXCL5, which is derived from platelets and alveolar type II cells, was found as a regulator of chemokine scavenging and pulmonary host defense to bacterial infection. CXCL5−/− mice showed decreased mortality in a model of E. coli-induced pneumonia. CXCL5 increased plasma concentrations of CXCL1 and CXCL2 by binding to erythrocyte duffy antigen receptor (DARC) (86). DARC has a high affinity for CC and CXC chemokines and acts as chemokine sink. In a model of ALI induced by LPS inhalation, DARC of red blood cells prevented excessive infiltration of neutrophils into the lung by scavenging chemokines, because DARC−/− mice displayed increased neutrophil infiltration in this model ALI (87).
Hydrogen sulfide (H2S), which is generated in many types of mammalian cells during cysteine metabolism, functions as neurotransmitter, vasodilator and inflammatory mediator. In this context, inhibition of cystathionine g-lysase which produces endogenous H2S during cysteine metabolism also has been shown to be protective against ALI associated with septic peritonitis (88,89) (Tables 1 and 2).
Cytokines
Cytokines are small proteins secreted by cells of the immune system, thereby transmitting signals between cells on other cells. After an acute insult such as trauma or acid aspiration and when the inciting event is not in the lung (for example, extrapulmonary sepsis, nonpul-monary trauma) there is systemic release of mediators including LPS; cytokines such as TNF, IL-1 and IL-6; and lipid mediators such as PAF and eicosanoids that have diverse effects on endothelium, epithelium and on circulating as well as resident immune cells. Moreover, apoptotic epithelium, which appears early in ALI, contributes to recruitment of neutrophils. Perl et al. have demonstrated that the apoptosis of epithelium and release of cytokines and chemokines is mediated by activation Fas receptor (also known as CD95) driven pathway in indirect ALI (90). Tumor necrosis factor (TNF) and IL-1 stimulate other cells locally, such as macrophages, fibroblasts, endothelial cells and epithelial cells to discharge other proinflammatory chemokines, such as IL-8, which is known as a potent chemotactic factor for neutrophils. TNF and IL-1 are early response cytokines in acute lung injury (91). While proinflammatory cytokines IL-1β and TNF have been identified in BAL fluids from patients with ARDS, their specific antagonists IL-1RA and soluble TNF receptors (sTNFR I and II) also were present (92). IL-1RA competitively inhibits binding of IL-1β to its primary cell-to-surface receptor. Whereas healthy volunteers revealed a concentration ratio IL-1β:IL-1RA of 1:1 in the BAL fluid, patients with persistent ARDS displayed an average ratio of 10:1 (93). Although there are divergent results of absolute values of IL-1β depending on different immunoassays and measurements (92), concentrations of IL-1β and its antagonist correlate with severity of disease and outcome of patients with ARDS (94). TNF is elevated in BAL fluid of patient with ARDS, although levels of TNF do not predict clinical outcome. Interestingly, severity of illness is related to the ratio of TNF and soluble TNF receptors (sTNFR) (94). Whereas inhibition of TNF-converting enzyme reduced ALI in rats after intratracheal application of LPS (95) (Table 1), TNF-deficient mice (TNF−/−) or TNF receptor deficient mice (TNF R1/2−/−) are not protected from intestinal ischemia/reperfusion-induced lung inflammation (96). Mice with deficiency for Caspase-1, which is identified as IL-1β-converting enzyme, develop less ALI in comparison to wild type mice (97) (Table 2). Cytokines display diverse effects in acute lung injury resulting in activation of endothelium and circulating and resident leukocytes.
Granule Proteins in Acute Lung Injury
Neutrophils contain four granule subsets: azurophilic (also known as primary) granules, specific (also known as secondary), gelatinase granules (also known as tertiary) and secretory vesicles. Granule proteins of neutrophils are synthesized at different stages of myelopoiesis and targeted to granule subsets (98). Different granule subsets show distinct propensities for release. Secretory vesicles are mobilized following initial neutrophil to en-dothelial cell contact and tertiary granules are released during subsequent neutrophil transendothelial migration. Primary and secondary granules are discharged once the neutrophil has entered the tissue. Secretory vesicles are rich in membrane-bound receptors, but granules released at later stages mainly contain proteases and antimicrobial polypeptides (99). Recent evidence suggests an important role for neutrophil-derived granule proteins during the onset of acute lung injury induced by Streptococcus pyogenes (100). While depletion of neutrophils completely abolished the lung damage in this model, injection of the supernatant from activated neutrophils into neutropenic mice restored the deleterious effect of neutrophils, indicating an important role for neutrophil granule proteins during ALI.
Neutrophil-Derived Serine Proteases
Besides its important antimicrobial functions, the serine protease neutrophil elastase holds an evident role during the pathogenesis of lung injury. For example, elastase levels are increased in BAL fluid (101, 102) and plasma (103) of patients with ALI and ARDS. These elevated levels correlate with the severity of the lung injury (103,104). Moreover, small clinical trials provided evidence for the protective effect of elastase inhibition (105–108). Furthermore, administration of elastase induces lung damage in murine models of ALI (109–111) whereas elastase inhibition attenuated the development of ALI (112–116) (Table 1). Neutrophil elastase deficient mice display impaired host defense against Gram-negative but not Gram-positive bacteria (117). In addition, deficiency of elastase is accompanied by increased susceptibility to fungal infections, but decreased susceptibility to endotoxin-triggered ALI (Table 2).
Although neutrophil elastase plays an important role in the development of ALI, it remains unclear whether it is due to degrading the basement membrane or due to damage to the epithelium or en-dothelium. Ginzberg et al. revealed in vitro an elastase-induced increase in epithelial permeability by alteration of the actin skeleton. As a consequence of the increased permeability, accelerated transmigration of neutrophils and circular defects in the epithelial monolayer were observed (118). Proteolytic cleavage of E-cadherin (118) and endothelial VE-cadherin (119) is yet another mechanism by which elastase increases permeability. Recent studies have revealed that the neutrophil serine proteases proeinase-3, cathepsin G and elastase can degrade surfactant protein D and surfactant protein A (120,121). Surfactant proteins A and D belong to the collectin family and participate in the clearance of apoptotic neutrophils (122). Thus, neutrophil-derived serine proteases can prolong inflammation by degradation of antiin-flammatory proteins. Moreover, Massberg et al. have shown that neu-trophil elastase and cathepsin G, along with externalized nucleosomes, promote coagulation and intravascular thrombus formation in vivo, which also may relate to permeability increases (123).
Neutrophil elastase and other proteinases also may act through binding to cell surface receptors and activating signal transduction pathways (124). In particular, elastase stimulates lung epithelium to release growth factors and proinflammatory cytokines (125–127). Furthermore, neutrophil elastase induced lung epithelial apoptosis via PAR-1 and the subsequent activation of the NF-κB pathway (128). Cathepsin G, yet another serine protease stored in primary granules, belongs to the group of lysosomal proteinases. They participate in a broad range of functions in neutrophils including clearance of internalized pathogens, proteolytic modification of cytokines and chemokines, activation as well as shedding of cell surface receptors and apoptosis (129,130). Activation of neutrophil cathepsin G and other serine proteases depends on two separate amino-terminal processing steps. The second step of activation occurs during or before transport to the granules and requires activity of Cathepsin C (also known as dipeptidyl peptidase I (DPPI)) (129). Mice deficient of serine proteases highlight the important role of these proteases for intracellular clearance of certain bacteria or protection against fungal infections (131). However, recent studies reveal that neutrophil serine proteases also act as key regulators of immune responses by proteolytic modification of cytokines, chemokines and growth factors as well as by activating specific receptors (129). In addition, cathepsin G may induce tissue damage and permeability changes directly in ALI. In vitro experiments displayed increased permeability of cultured type II pneumocytes (132). Whereas administration of Cathepsin G und neutrophil elastase induce lung emphysema (133), inhaled Cathepsin G induces airway hyperresponsiveness a characteristic attribute of asthma (134). Furthermore, human urinary trypsin inhibitor, which exhibits widespread inhibitory effects on serine proteases, reduces superantigen-induced lung injury (135).
In contrast to neutrophil elastase, proteinase 3 is presented at the plasma membrane of nonactivated neutrophils (136). Proteinase 3 can kill bacteria and fungi via inhibition of protein synthesis and oxygen metabolism. Moreover, the serine proteases are also involved in non-infectious inflammatory disease (129,137). In humans, proteinase 3 is elucidated as the main target antigen of neutrophil cytoplasm autoantibodies (c-ANCA) in Wegener granulomatosis, a vasculitis which affects lungs, kidneys and the skin (138). Proteinase 3 interacts with specific intracellular protein substrates during proliferation and apoptosis (137). Recent studies showed that proteinase 3 can activate proinflammatory cytokines, such as TNF and IL-1β (139).
Taken together, neutrophil serine proteases are major constituents of neutrophils and are released at the site of inflammation. Besides their traditional antimicrobial function, neutrophil serine proteases can activate proinflammatory cytokines and playing an important role in regulating the innate immune response. Thus, serine protease may present an interesting target in inflammatory diseases such as ALI and newly developed protease inhibitors may deserve careful evaluation as antiinflammatory agents (140).
Matrix Metalloproteinases (MMPS)
Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases that are produced by a variety of cell types that occupy a central role in embryogenesis and in normal physiological conditions, such as proliferation, cell motility, remodeling, wound healing, angiogenesis and key reproductive events. MMP-2 (gelatinase A) and MMP-9 (gelatinase B) stored in tertiary granules of neutrophils and MMP-8 (collagenase 2) from secondary granules of neutrophils are the most extensively studied MMPs in the context of ALI. Although MMPs can be released by resident cells, recent studies demonstrated the pathogenetic role of MMP-released from neutrophils in ALI (141,142). In particular, BAL fluid (143–145) and plasma (146,147) of patients with ALI/ARDS displayed elevated levels of MMPs which correlated with clinical severity (148). Moreover, inhibition of MMP-9 attenuates ventilator-induced lung injury in rats (149), although there are conflicting results in the literature. In ozone-induced airway inflammation, MMP-9−/− mice display increased lung permeability, neutrophils in the lung, and higher BAL levels of keratinocyte-derived chemokine (KC) and macrophage inflammatory protein-1a (MIP-1a) when compared with MMP-9+/+ mice (150). These findings are confirmed by similar results in ventilator-induced lung injury (151,152). Increased levels of MMP-9 in BAL fluid in ALI may contribute to modulation of inflammation by affecting cytokine and chemokine levels as well as their activities (150). Furthermore, MMP-8−/− mice display a two-fold increase of neutrophils in the BAL fluid after intratracheal lipopolysaccharide application (153,154). Interestingly, levels of MIP-1a are elevated in wild type mice in comparison with MMP-8−/−, indicating that MMP-8 reduced ALI by inactivating MIP-1a (154). In addition to these results in ALI, deficiency of MMP-8 is protective in a model of acute liver injury (155) but can delay the resolution of inflammation in skin (156).
In summary, these examples illustrate that MMPs mediate both beneficial and deleterious effects in acute lung injury. Besides their functions on extracellular matrix (that is, degradation, turnover and remodeling), MMPs modulate inflammation and neutrophil influx as well as epithelial and endothelial integrity. Because different MMPs may have opposite functions in inflammation and tissue repair, unspecific inhibition of MMPs does not seem reasonable as a therapeutic approach.
Neutrophil-Derived Cationic Polypeptides
Upon activation, neutrophils release a wide array of cationic polypeptides, which are acknowledged primarily for their antimicrobial activity. However, some of these polypeptides potently activate neighboring cells, thus giving them the name alarmins (157, 158). Lactoferrin, which belongs to the family of iron-binding proteins, is stored in secondary granules of neutrophils and exhibits antibacterial, antiviral and antifungal activity (159). Neutrophils are a major source of lactoferrin. Normal lactoferrin levels in the blood are very low with 1 µg/mL, but under septic conditions these can rise up to 200 µg/mL and are likely to be higher at the inflammatory site itself (160). Besides its antimicrobial activity, lactoferrin exhibits immune-modulating activities. Lactoferrin set free from apoptotic cells inhibits migration of neutrophils and eosinophils (161,162). In contrast, lactoferrin acts as a chemoattractant for monocytes (162). Moreover, lactoferrin may induce production of proinflammatory cytokines, such as MIP-1a and MIP-2 (163). While the chemotactic effect of lactoferrin is mediated by a so-far unknown G-protein-coupled receptor, much of the immune cell activating effect is mediated via ligation of TLR4 (158). In contrast to its proinflammatory effects, lactoferrin also can decrease LPS-induced mitochondrial dysfunction in cultured cells and in an animal endotoxemia model (164). In summary, besides its antimicrobial activity, lactoferrin also modulates local inflammatory processes. However, at this point, data from animal models are missing that point at the importance of lactoferrin in ALI.
The antimicrobial polypeptide LL-37 is released from neutrophil secondary granules in its inactive pro-form hCAP18. Activation occurs upon secretion by proteolytic modification by proteinase-3. In addition to its broad antimicrobial activity, LL-37 can promote inflammatory responses by activation of monocytes, neutrophils and T-lymphocytes (165,166). LL-37 activates monocytes and macrophages directly and promotes their migration via ligation of formyl-peptide receptors (167). In the context of ARDS, LL-37 is elevated significantly in the BAL fluid of these patients in comparison with normal controls (168). When instilled into murine lungs, enhanced levels of MCP-1 and TNF can be retrieved, likely based on the activation of macrophages and epithelial cells (169,170). In addition, LL-37 forms complexes with self-DNA which potently activate the immune system (171). Necrotic cells, which are abundant in ALI, are a common source of such DNA. Interestingly, LL-37 in itself exerts cytotoxic and proapoptotic effects toward endothelial cells and epithelial cells (172). In contrast, LL-37 inhibits apoptosis in neutrophils themselves (173,174), contributing to enhanced accumulation of neutrophils at the site of inflammation. At this point, no data from ALI models of mice lacking CRAMP, the murine homologue of LL-37, are readily available. Nevertheless, as LL-37 is a potent proin-flammatory granule protein, it may hold significant roles in ALI.
Defensins are small, arginine-rich cationic peptides that are divided in two subgroups, α-defensins and β-defensins. Human α-defensins 1–4 (HNPs 1–4) are produced principally by neutrophils and stored in primary (azurophilic) granules (175). High concentrations of α-defensins have been found in BAL fluids from patients with ARDS correlating with the severity of disease (176). Since neutrophils are the major source of HNPs, it appears likely that many HNPs are in fact neutrophil-borne. Besides their microbicidal function, α-defensins act as an effector of cytokine production. In this context it has been shown that HNPs activate macrophages to induce the release of TNF and interferon (IFN)-γ and to promote a phenotypic switch towards a more proinflammatory phenotype (177). In acute lung injury, α-defensins also induce IL-8, a chemokine that potently attracts neutrophils (178). Moreover, α-defensins increase the permeability of the epithelial monolayer in vitro (179–182). HNPs also exert chemotactic effects on immature dendritic cells, T cells and mast cells (183–185). Since murine neutrophils lack α-defensins it is difficult to address their role in murine models of ALI. Thus, only a transgenic mouse (α-defensins+/+) model is available, which displays increased ALI and disrupted capillary to epithelial barrier (186).
Azurocidin (also known as CAP37 or HBP) is stored in secretory vesicles and primary granules of neutrophils and is released upon neutrophil adhesion and during neutrophil extravasation (187,188). Its positive charge allows for immobilization on the endothelial cell surface where it induces adhesion of inflammatory monocytes, but also promotes permeability changes (188,189). In fact, seminal studies suggest an almost exclusive role of azurocidin in neutrophil-dependent permeability changes (189,190). In vivo experiments studying the lung damage induced by S. pyogenes revealed an important role for neutrophil granule proteins (100,191). Therein, M1 protein shed from the surface of S. pyogenes forms complexes with fibrinogen. These induce neutrophil activation and degranulation via ligation of β2-integrins, and experimental data indicate a prominent role for azurocidin in the M1 protein-induced lung edema formation and tissue destruction. At this point, no data from patient samples are available that would strengthen the perception of a role of azurocidin in ALI. However, antibodies which are found in TRALI have been shown to trigger discharge of azurocidin from neutrophils (192). In addition, in circumstances that may lead to ALI such as severe burns or sepsis, circulating azurocidin levels are increased significantly, allowing speculation on their potential role in lung damage (193–195).
Oxidants And Ros-Formation
Neutrophils produce vast quantities of reactive oxygen (ROS) and nitrogen (RNS) species like O•−2 and NO• through their oxidant-generating systems such as the phagocyte NADPH oxidase and nitric oxide synthase (NOS) respectively. The controlled enzymatic generation of ROS, which includes superoxide anion, hydrogen peroxide, hydroxyl radical and others by neutrophils, is an integral component of the innate immune system. During ingestion of invading pathogens into phagosomes, ROS generated at the phagosome membrane are released directly into the phagosome. Besides the well-known antimicrobicidal function of ROS during phagocytosis, low-level formation of ROS acts as intracellular signaling, so called “redox signaling” (196). ROS is released into the cytosol where they alter the redox state of the cell and modify other cell contents, such as proteins and lipids by oxidation (197).
The membrane-bound multicomponent enzyme complex NADPH oxidase, which is dormant in resting cells and can be activated rapidly by chemoattractant peptides or chemokines, generates much ROS after activation. The oxidase consists of the catalytic subunit gp91phox (otherwise known as NOX2), the regulatory subunits p22phox, p47phox, p40phox, p67phox and the small GTPase RAC. NOX2, which is found predominately in phagocytes, belongs to the family of NOX and dual oxidases (DUOX) (198). Furthermore, the myeloperoxidase, which is found in the neutrophil granules, catalyzes the production of additional ROS species, that is, the hydroxyl radical (*OH) and hypochlorous acid (HOCl). Activated neutrophils produce prostaglandine E and F using the arachinodic acid metabolism and this metabolism produces ROS, which is able to regulate other signaling pathways in neutrophils directly or indirectly. Deficiency of NADPH oxidase in humans, known as a chronic granulomatous disease, causes recurrent infections because phagocytic cells fail to produce ROS and to kill engulfed foreign organisms.
Exposure of the lung to inhaled or instilled oxidants induces lung injury (199). The pathogenetic role of oxidants is highlighted by elevated levels of plasma and lung oxidants in patients with ALI/ARDS. In addition, these levels of oxidants correlate with severity of the disease. In animal models of ALI, neutrophil-derived ROS and RNS caused lung injury as shown by histological examination and permeability measurements (200, 201). A recent study revealed that ROS can disrupt intercellular tight junctions of the endothelium by phosphorylation of focal adhesion kinase (202). In vitro, ROS induced cell apoptosis and necrosis of alveolar type II cells during oxygen exposure (203). In vivo, NOX-1−/− mice, but not NOX-2−/− mice, are protected from hyperoxia induced ALI; these results reveal that NADPH oxidase 1 plays a crucial role in hyperoxia-induced ALI (204). A previous study with NADPH oxidase−/− mice revealed no protection from CVF-induced ALI, indicating that oxygen radical production and lung injury may occur through alternative pathways in mice with genetic deletion of NADPH oxidase (205). Furthermore inhibition of NADPH or nitric oxide synthase (NOS) has decreased sepsis-induced ALI (206) and respectively LPS induced ALI (207).
ROS may prolong inflammation by modulating neutrophil function. After oxidation of acid spingomyelinase, ROS delays neutrophil apoptosis in a caspase-8-dependent way (208). Neutrophil apoptosis also can be accelerated by MPO through CD11b/CD18 integrins (209). Consequently, neutrophil-derived oxidants (203) are regarded to present a major role in neutrophil-mediated tissue injury, including ALI/ARDS.
Conclusions
Activation and migration of neutrophils into the lung is one of the key events in ALI. We have highlighted the contribution of neutrophils and their secretory products to ALI. Up to now, no pharmacological therapy has emerged for the treatment of all patients with ALI/ARDS, because the patients and the causes underlying the disease are very heterogeneous. Whereas inhibition of adhesion molecules has reduced ALI in several animal models, the translation into clinical trial has proven difficult owing to the redundancy of the molecules involved (210). Hence, the inflammatory response induced by neutrophils can be controlled by inhibition of degranulation to avoid host tissue damage. Furthermore, antioxidants may reduce the ROS-related tissue damage in acute lung injury. Although neutrophil recruitment into the lung is necessary for host defense in case of bacterial infection, regulation of neutrophil activity might be a possible therapeutic approach.
Disclosures
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Bernard GR, et al. (1994) The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am. J. Respir. Crit. Care Med. 149:818–24.
Ware LB, Matthay MA. (2000) The acute respiratory distress syndrome. N. Engl. J. Med. 342:1334–9.
Matute-Bello G, Frevert CW, Martin TR. (2008) Animal models of acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 295:L379–99.
Ware LB. (2006) Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin. Respir. Crit. Care Med. 27:337–49.
Manicone AM. (2009) Role of the pulmonary epithelium and inflammatory signals in acute lung injury. Expert Rev. Clin. Immunol. 5:63–75.
Abraham E. (2003) Neutrophils and acute lung injury 619. Crit. Care Med. 31:S195–9.
Matthay MA, Eschenbacher WL, Goetzl EJ. (1984) Elevated concentrations of leukotriene D4 in pulmonary edema fluid of patients with the adult respiratory distress syndrome. J. Clin. Immunol. 4:479–83.
Parsons PE, Fowler AA, Hyers TM, Henson PM. (1985) Chemotactic activity in bronchoalveolar lavage fluid from patients with adult respiratory distress syndrome. Am. Rev. Respir. Dis. 132:490–3.
Steinberg KP, et al. (1994) Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 150:113–22.
Abraham E, Carmody A, Shenkar R, Arcaroli J. (2000) Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 279:L1137–5.
Folkesson HG, Matthay MA, Hebert CA, Broad-dus VC. (1995) Acid aspiration-induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J. Clin. Invest. 96:107–16.
Martin TR, Pistorese BP, Chi EY, Goodman RB, Matthay MA. (1989) Effects of leukotriene B4 in the human lung. Recruitment of neutrophils into the alveolar spaces without a change in protein permeability. J. Clin. Invest. 84:1609–19.
Laufe MD, Simon RH, Flint A, Keller JB. (1986) Adult respiratory distress syndrome in neutropenic patients. Am. J. Med. 80:1022–6.
Ognibene FP, et al. (1986) Adult respiratory distress syndrome in patients with severe neutropenia. N. Engl. J. Med. 315:547–51.
Sivan Y, Mor C, al-Jundi S, Newth CJ. (1990) Adult respiratory distress syndrome in severely neutropenic children. Pediatr. Pulmonol. 8:104–8.
Wiener-Kronish JP, Albertine KH, Matthay MA. (1991) Differential responses of the endothelial and epithelial barriers of the lung in sheep to Es-cherichia coli endotoxin. J. Clin. Invest. 88:864–75.
Wiggs BR, et al. (1994) Contributions of capillary pathway size and neutrophil deformability to neutrophil transit through rabbit lungs. J. Appl. Physiol. 77:463–70.
Knight PR, Druskovich G, Tait AR, Johnson KJ. (1992) The role of neutrophils, oxidants, and proteases in the pathogenesis of acid pulmonary injury. Anesthesiology. 77:772–8.
Kawano T, et al. (1987) Effect of granulocyte depletion in a ventilated surfactant-depleted lung. J. Appl. Physiol. 62:27–33.
Julien M, Hoeffel JM, Flick MR. (1986) Oleic acid lung injury in sheep. J. Appl. Physiol. 60:433–40.
O’Brien-Ladner AR, Nelson ME, Cowley BD Jr., Bailey K, Wesselius LJ. (1995) Hyperoxia amplifies TNF-alpha production in LPS-stimulated human alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 12:275–9.
Shea LM, et al. (1996) Hyperoxia activates NF-kappaB and increases TNF-alpha and IFN-gamma gene expression in mouse pulmonary lymphocytes. J. Immunol. 157:3902–8.
Ley K, Laudanna C, Cybulsky MI, Nourshargh S. (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7:678–89.
Doerschuk CM. (2000) Leukocyte trafficking in alveoli and airway passages. Respir. Res. 1:136–40.
Doerschuk CM, et al. (1987) Marginated pool of neutrophils in rabbit lungs. J. Appl. Physiol. 63:1806–15.
Tanaka H, Nishino M, Dahms TE. (2002) Physiologic responses to small emboli and hemody-namic effects of changes in deformability of poly-morphonuclear leukocytes in isolated rabbit lung. Microvasc. Res. 63:81–90.
Doerschuk CM, Mizgerd JP, Kubo H, Qin L, Kumasaka T. (1999) Adhesion molecules and cellular biomechanical changes in acute lung injury: Giles F. Filley Lecture. Chest. 116:37–43S.
Worthen GS, Schwab B III, Elson EL, Downey GP. (1989) Mechanics of stimulated neutrophils: cell stiffening induces retention in capillaries. Science. 245:183–6.
Drost EM, MacNee W. (2002) Potential role of IL-8, platelet-activating factor and TNF-alpha in the sequestration of neutrophils in the lung: effects on neutrophil deformability, adhesion receptor expression, and chemotaxis. Eur. J. Immunol. 32:393–403.
Motosugi H, et al. (1996) Changes in neutrophil actin and shape during sequestration induced by complement fragments in rabbits. Am. J. Pathol. 149:963–73.
Burns AR, Smith CW, Walker DC. (2003) Unique structural features that influence neutrophil emigration into the lung. Physiol. Rev. 83:309–36.
Furuse M, et al. (1994) Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. J. Cell. Biol. 127:1617–26.
Su WH, Chen HI, Jen CJ. (2002) Differential movements of VE-cadherin and PECAM-1 during transmigration of polymorphonuclear leukocytes through human umbilical vein endothelium. Blood. 100:3597–603.
Carlow DA, et al. (2009) PSGL-1 function in immunity and steady state homeostasis. Immunological Reviews 230:75–96.
Laszik Z, et al. (1996) P-selectin glycoprotein ligand-1 is broadly expressed in cells of myeloid, lymphoid, and dendritic lineage and in some nonhematopoietic cells. Blood. 88:3010–21.
Mizgerd JP, et al. (1996) Selectins and neutrophil traffic: margination and Streptococcus pneumoniae-induced emigration in murine lungs. J. Exp. Med. 184:639–45.
Burns JA, Issekutz TB, Yagita H, Issekutz AC. (2001) The alpha 4 beta 1 (very late antigen (VLA)-4, CD49d/CD29) and alpha 5 beta 1 (VLA-5, CD49e/CD29) integrins mediate beta 2 (CD11/CD18) integrin-independent neutrophil recruitment to endotoxin-induced lung inflammation. J. Immunol. 166:4644–9.
Looney MR, et al. (2009) Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J. Clin. Invest. 119:3450–61.
Chandra A, et al. (2003) P-selectin blockade fails to improve acute lung injury in sheep. Clin. Sci. (Lond). 104:313–21.
Mulligan MS, Miyasaka M, Ward PA. (1996) Protective effects of combined adhesion molecule blockade in models of acute lung injury. Proc. Assoc. Am. Physicians. 108:198–208.
Mulligan MS, et al. (1993) Protective effects of oligosaccharides in P-selectin-dependent lung injury. Nature. 364:149–51.
Hirose M, Murai T, Kawashima H. (2007) Elevation of rat plasma P-selectin in acute lung injury. Biochim. Biophys. Acta. 1772:382–9.
Bock D, Aydt EM, Kuebler WM, Wolff G. (2006) The role of selectins during lung inflammation and their potential impact for innovative therapeutic strategies. Curr. Respir. Med. Rev. 2:339–54.
Hayashi H, Koike H, Kurata Y, Imanishi N, Tojo SJ. (1999) Protective effects of sialyl Lewis X and anti-P-selectin antibody against lipopolysaccharide-induced acute lung injury in rabbits. Eur. J. Pharmacol. 370:47–56.
Ridings PC, et al. (1995) Sepsis-induced acute lung injury is attenuated by selectin blockade following the onset of sepsis. Arch. Surg. 130:1199–208.
Doerschuk CM, et al. (1996) The role of P-selectin and ICAM-1 in acute lung injury as determined using blocking antibodies and mutant mice. J. Immunol. 157:4609–14.
Zarbock A, Singbartl K, Ley K. (2006) Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J. Clin. Invest. 116:3211–9.
Abram CL, Lowell CA. (2009) The ins and outs of leukocyte integrin signaling. Annu. Rev. Immunol. 27:339–62.
Hynes RO. (2002) Integrins: bidirectional, allosteric signaling machines. Cell. 110:673–87.
Kim S, Schein AJ, Nadel JA. (2005) E-cadherin promotes EGFR-mediated cell differentiation and MUC5AC mucin expression in cultured human airway epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 289: L1049–60.
Ganter MT, et al. (2008) Interleukin-1ta causes acute lung injury via {alpha}v{beta}5 and {alpha}v{beta}6 integrin-dependent mechanisms. Circ. Res. 102:804–12.
Song Y, et al. (2008) Role of integrin {alpha}vta6 in acute lung injury induced by pseudomonas aeruginosa. Infect. Immun. 76:2325–32.
Xu J, et al. (2008) Nonmuscle myosin light-chain kinase mediates neutrophil transmigration in sepsis-induced lung inflammation by activating [beta]2 integrins. Nat. Immunol. 9:880–6.
Moreland JG, Fuhrman RM, Pruessner JA, Schwartz DA. (2002) CD11b and intercellular adhesion molecule-1 are involved in pulmonary neutrophil recruitment in lipopolysaccharide-induced airway disease. Am. J. Respir. Cell Mol. Biol. 27:474–80.
Mulligan M, et al. (1992) Roles of beta 2 integrins of rat neutrophils in complement- and oxygen radical-mediated acute inflammatory injury. J. Immunol. 148:1847–57.
Xu N, Rahman A, Minshall RD, Tiruppathi C, Malik AB. (2000) ta2-Integrin blockade driven by E-selectin promoter prevents neutrophil sequestration and lung injury in mice. Circ. Res. 87:254–60.
Hellewell PG, Young SK, Henson PM, Worthen GS. (1994) Disparate role of the beta 2-integrin CD18 in the local accumulation of neutrophils in pulmonary and cutaneous inflammation in the rabbit. Am. J. Respir. Cell Mol. Biol. 10:391–8.
Mizgerd JP, et al. (1997) Neutrophil emigration in the skin, lungs, and peritoneum: different requirements for CD11/CD18 revealed by CD18-deficient mice. J. Exp. Med. 186:1357–64.
Winn RK, et al. (1993) Role of protein synthesis and CD11/CD18 adhesion complex in neutrophil emigration into the lung. Exp. Lung Res. 19:221–35.
Doerschuk CM, Winn RK, Coxson HO, Harlan JM. (1990) CD18-dependent and -independent mechanisms of neutrophil emigration in the pulmonary and systemic microcirculation of rabbits. J. Immunol. 144:2327–33.
Puneet P, Moochhala S, Bhatia M. (2005) Chemokines in acute respiratory distress syndrome. Am. J. Physiol. Lung Cell. Mol. Physiol. 288:L3–15.
Miller EJ, Cohen AB, Matthay MA. (1996) Increased interleukin-8 concentrations in the pulmonary edema fluid of patients with acute respiratory distress syndrome from sepsis. Crit. Care Med. 24:1448–54.
Miller EJ, et al. (1992) Elevated levels of NAP-1/ interleukin-8 are present in the airspaces of patients with the adult respiratory distress syndrome and are associated with increased mortality. Am. Rev. Respir. Dis. 146:427–32.
Fudala R, Krupa A, Stankowska D, Allen TC, Kurdowska AK. (2008) Anti-interleukin-8 autoantibody:interleukin-8 immune complexes in acute lung injury/acute respiratory distress syndrome. Clin. Sci. (Lond). 114:403–12.
Allen TC, Fudala R, Nash SE, Kurdowska A. (2007) Anti-interleukin 8 autoantibody:interleukin 8 immune complexes visualized by laser confocal microscopy in injured lung. Arch. Pathol. Lab. Med. 131:452–6.
Kurdowska A, et al. (2002) Anti-interleukin-8 au-toantibodies in patients at risk for acute respiratory distress syndrome. Crit. Care Med. 30:2335–7.
Kurdowska A, et al. (2001) Anti-interleukin 8 au- toantibody: interleukin 8 complexes in the acute respiratory distress syndrome. Relationship between the complexes and clinical disease activity. Am. J. Respir. Crit. Care Med. 163:463–8.
Krupa A, Kato H, Matthay MA, Kurdowska AK. (2004) Proinflammatory activity of anti-IL-8 au-toantibody:IL-8 complexes in alveolar edema fluid from patients with acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 286:L1105–13.
Yang XD, Corvalan JR, Wang P, Roy CM, Davis CG. (1999) Fully human anti-interleukin-8 monoclonal antibodies: potential therapeutics for the treatment of inflammatory disease states. J. Leukoc. Biol. 66:401–10.
Osman MO, et al. (1998) A monoclonal antiinterleukin 8 antibody (WS-4) inhibits cytokine response and acute lung injury in experimental severe acute necrotising pancreatitis in rabbits. Gut. 43:232–9.
Bao Z, Ye Q, Gong W, Xiang Y, Wan H. (2010) Humanized monoclonal antibody against the chemokine CXCL-8 (IL-8) effectively prevents acute lung injury. Int. Immunopharmacol. 10:259–63.
Laffon M, Pittet J-F, Modelska K, Matthay MA, Young DM. (1999) Interleukin-8 mediates injury from smoke inhalation to both the lung endothelial and the alveolar epithelial barriers in rabbits. Am. J. Respir. Crit. Care Med. 160:1443–9.
Sekido N, et al. (1993) Prevention of lung reperfusion injury in rabbits by a monoclonal antibody against interleukin-8. Nature. 365:654–7.
Olson TS, Ley K. (2002) Chemokines and chemokine receptors in leukocyte trafficking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 283:R7–28.
Zarbock A, Allegretti M, Ley K. (2008) Therapeutic inhibition of CXCR2 by Reparixin attenuates acute lung injury in mice. Br. J. Pharmacol. 155:357–64.
Bhatia M, Hegde A. (2007) Treatment with an- tileukinate, a CXCR2 chemokine receptor antagonist, protects mice against acute pancreatitis and associated lung injury. Regul. Pept. 138:40–8.
Lomas-Neira JL, Chung CS, Grutkoski PS, Miller EJ, Ayala A. (2004) CXCR2 inhibition suppresses hemorrhage-induced priming for acute lung injury in mice. J. Leukoc. Biol. 76:58–64.
Goncalves AS, Appelberg R. (2002) The involvement of the chemokine receptor CXCR2 in neutrophil recruitment in LPS-induced inflammation and in Mycobacterium avium infection. Scand. J. Immunol. 55:585–91.
Auten RL, et al. (2001) Nonpeptide CXCR2 antagonist prevents neutrophil accumulation in hyperoxia-exposed newborn rats. J. Pharmacol. Exp. Ther. 299:90–5.
Belperio JA, et al. (2005) CXCR2/CXCR2 lig-and biology during lung transplant ischemia-reperfusion injury. J. Immunol. 175:6931–9.
Belperio JA, et al. (2002) Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J. Clin. Invest. 110:1703–16.
Reutershan J, et al. (2006) Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J. Clin. Invest 116:695–702.
Stillie R, Farooq SM, Gordon JR, Stadnyk AW. (2009) The functional significance behind expressing two IL-8 receptor types on PMN. J. Leukoc. Biol. 86:529–43.
Chishti AD, et al. (2001) Expression of chemokine receptors CXCR1 and CXCR2 during cardiopulmonary bypass. J. Thorac. Cardiovasc. Surg. 122:1162–6.
Cummings CJ, et al. (1999) Expression and function of the chemokine receptors CXCR1 and CXCR2 in sepsis. J. Immunol. 162:2341–6.
Mei J, et al. (2010) CXCL5 regulates chemokine scavenging and pulmonary host defense to bacterial infection. Immunity. 33:106–17.
Reutershan J, Harry B, Chang D, Bagby GJ, Ley K. (2009) DARC on RBC limits lung injury by balancing compartmental distribution of CXC chemokines. Eur. J. Immunol. 39:1597–607.
Zhang H, Zhi L, Moochhala SM, Moore PK, Bhatia M. (2007) Endogenous hydrogen sulfide regulates leukocyte trafficking in cecal ligation and puncture-induced sepsis. J. Leukoc. Biol. 82:894–905.
Zhang H, Zhi L, Moore PK, Bhatia M. (2006) Role of hydrogen sulfide in cecal ligation and puncture-induced sepsis in the mouse. Am. J. Physiol. Lung Cell. Mol. Physiol. 290:L1193–201.
Perl M, et al. (2007) Fas-induced pulmonary apoptosis and inflammation during indirect acute lung injury. Am. J. Respir. Crit. Care Med. 176:591–601.
Nathan CF. (1987) Secretory products of macrophages. J. Clin. Invest. 79:319–26.
Goodman RB, Pugin J, Lee JS, Matthay MA. (2003) Cytokine-mediated inflammation in acute lung injury. Cytokine Growth Factor Rev. 14:523–35.
Goodman RB, et al. (1996) Inflammatory cytokines in patients with persistence of the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 154:602–11.
Park WY, et al. (2001) Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 164:1896–903.
Shimizu M, et al. (2009) Effects of TNF-alpha-converting enzyme inhibition on acute lung injury induced by endotoxin in the rat. Shock. 32:535–40.
Soares AL, et al. (2010) Tumor necrosis factor is not associated with intestinal ischemia/reperfusion-induced lung inflammation. Shock. 34:306–13.
Hoshino T, et al. (2009) Role of proinflammatory cytokines IL-18 and IL-1ta in bleomycin-induced lung injury in humans and mice. Am. J. Respir. Cell Mol. Biol. 41:661–70.
Borregaard N, Cowland JB. (1997) Granules of the human neutrophilic polymorphonuclear leukocyte. Blood. 89:3503–21.
Faurschou M, Borregaard N. (2003) Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 5:1317–27.
Soehnlein O, et al. (2008) Neutrophil degranulation mediates severe lung damage triggered by streptococcal M1 protein 172. Eur. Respir. J. 32:405–12.
Cochrane CG, Spragg RG, Revak SD, Cohen AB, McGuire WW. (1983) The presence of neutrophil elastase and evidence of oxidation activity in bronchoalveolar lavage fluid of patients with adult respiratory distress syndrome. Am. Rev. Respir. Dis. 127:S25–7.
Lee CT, et al. (1981) Elastolytic activity in pulmonary lavage fluid from patients with adult respiratory-distress syndrome. N. Engl. J. Med. 304:192–6.
Donnelly SC, et al. (1995) Plasma elastase levels and the development of the adult respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 151:1428–33.
Zemans RL, Colgan SP, Downey GP. (2009) Transepithelial migration of neutrophils: mechanisms and implications for acute lung injury. Am. J. Respir. Cell Mol. Biol. 40:519–35.
Fujii M, et al. (2010) Effect of a neutrophil elastase inhibitor on acute lung injury after car-diopulmonary bypass. Interact. Cardiovasc. Thorac. Surg. 10:859–62.
Inoue Y, et al. (2009) Development of a highly water-soluble peptide-based human neutrophil elastase inhibitor; AE-3763 for treatment of acute organ injury. Bioorg. Med. Chem. 17:7477–86.
Kawahara Y, et al. (2009) Prospective randomized controlled study on the effects of perioperative administration of a neutrophil elastase inhibitor to patients undergoing video-assisted thoracoscopic surgery for thoracic esophageal cancer. Dis. Esophagus.
Suda K, et al. (2007) Neutrophil elastase inhibitor improves postoperative clinical courses after thoracic esophagectomy. Dis. Esophagus 20:478–486.
Delacourt C, et al. (2002) Protection against acute lung injury by intravenous or intratracheal pretreatment with EPI-HNE-4, a new potent neutrophil elastase inhibitor. Am. J. Respir. Cell Mol. Biol. 26:290–7.
Janusz MJ, Hare M. (1994) Inhibition of human neutrophil elastase and cathepsin G by a biphenyl disulfonic acid copolymer. Int. J. Immunopharmacol. 16:623–32.
Tremblay GM, Vachon E, Larouche C, Bourbonnais Y. (2002) Inhibition of human neutrophil elastase-induced acute lung injury in hamsters by recombinant human pre-elafin (trappin-2). Chest. 121:582–8.
Hagio T, et al. (2008) Inhibition of neutrophil elastase reduces lung injury and bacterial count in hamsters. Pulm. Pharmacol. Ther. 21:884–91.
Hagio T, Matsumoto S, Nakao S, Matsuoka S, Kawabata K. (2005) Sivelestat, a specific neutrophil elastase inhibitor, prevented phorbol myristate acetate-induced acute lung injury in conscious rabbits. Pulm Pharmacol. Ther. 18:285–90.
Kawabata K, et al. (2000) Delayed neutrophil elastase inhibition prevents subsequent progression of acute lung injury induced by endotoxin inhalation in hamsters. Am. J. Respir. Crit. Care Med. 161:2013–8.
Miyahara T, Kubo K, Koizumi T, Kobayashi T, Sekiguchi M. (1996) Specific neutrophil elastase inhibitor does not attenuate acute lung injury induced by air embolism in awake sheep. Exp. Lung Res. 22:613–25.
Sakamaki F, et al. (1996) Effect of a specific neu-trophil elastase inhibitor, ONO-5046, on endotoxin-induced acute lung injury. Am. J. Respir. Crit. Care Med. 153:391–7.
Belaaouaj A, et al. (1998) Mice lacking neu-trophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat. Med. 4:615–8.
Ginzberg HH, et al. (2001) Neutrophil-mediated epithelial injury during transmigration: role of elastase. Am. J. Physiol. Gastrointest. Liver Physiol. 281: G705–17.
Carden D, et al. (1998) Neutrophil elastase promotes lung microvascular injury and proteolysis of endothelial cadherins. Am. J. Physiol. 275: H385–92.
Hirche TO, et al. (2004) Neutrophil serine proteinases inactivate surfactant protein D by cleaving within a conserved subregion of the carbohydrate recognition domain. J. Biol. Chem. 279:27688–98.
Rubio F, Cooley J, Accurso FJ, Remold-O’Donnell E. (2004) Linkage of neutrophil serine proteases and decreased surfactant protein-A (SPA) levels in inflammatory lung disease. Thorax. 59:318–23.
Vandivier RW, et al. (2002) Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J. Immunol. 169:3978–86.
Massberg S, et al. (2010) Reciprocal coupling of coagulation and innate immunity via neu-trophil serine proteases. Nat. Med. 16:887–96.
Soehnlein O. (2009) An elegant defense: how neutrophils shape the immune response. Trends Immunol. 30:511–2.
Buczek-Thomas JA, et al. (2004) Elastase mediates the release of growth factors from lung in vivo. Am. J. Respir. Cell Mol. Biol. 31:344–50.
Chen HC, et al. (2004) Neutrophil elastase induces IL-8 synthesis by lung epithelial cells via the mitogen-activated protein kinase pathway. J. Biomed. Sci. 11:49–58.
Witherden IR, et al. (2004) Primary human alveolar type II epithelial cell chemokine release: effects of cigarette smoke and neutrophil elastase. Am. J. Respir. Cell Mol. Biol. 30:500–9.
Suzuki T, et al. (2009) Leukocyte elastase induces lung epithelial apoptosis via a PAR-1-,NF-kappaB-, and p53-dependent pathway. Am. J. Respir. Cell Mol. Biol. 41:742–55.
Pham CT. (2006) Neutrophil serine proteases: specific regulators of inflammation. Nat. Rev. Immunol. 6:541–50.
Pham CT. (2008) Neutrophil serine proteases fine-tune the inflammatory response. Int. J. Biochem. Cell Biol. 40:1317–33.
Reeves EP, et al. (2002) Killing activity of neu-trophils is mediated through activation of proteases by K+ flux. Nature. 416:291–7.
Rochat T, Casale J, Hunninghake GW, Peterson MW. (1988) Neutrophil cathepsin G increases permeability of cultured type II pneumocytes. Am. J. Physiol. 255:C603–11.
Lucey EC, et al. (1985) Effect of combined human neutrophil cathepsin G and elastase on induction of secretory cell metaplasia and emphysema in hamsters, with in vitro observations on elastolysis by these enzymes. Am. Rev. Respir. Dis. 132:362–6.
Coyle AJ, Uchida D, Ackerman SJ, Mitzner W, Irvin CG. (1994) Role of cationic proteins in the airway. Hyperresponsiveness due to airway inflammation. Am. J. Respir. Crit. Care Med. 150:S63–71.
Onai H, Kudo S. (2001) Suppression of superantigen-induced lung injury and vasculitis by preadministration of human urinary trypsin inhibitor. Eur. J. Clin. Invest. 31:272–80.
Schreiber A, Busjahn A, Luft FC, Kettritz R. (2003) Membrane expression of proteinase 3 is genetically determined. J. Am. Soc. Nephrol. 14:68–75.
Hajjar E, Broemstrup T, Kantari C, Witko-Sarsat V, Reuter N. (2010) Structures of human proteinase 3 and neutrophil elastase–so similar yet so different. FEBS J. 277:2238–54.
Moriceau S, et al. (2009) Coronin-1 is associated with neutrophil survival and is cleaved during apoptosis: potential implication in neutrophils from cystic fibrosis patients. J. Immunol. 182:7254–63.
Meyer-Hoffert U. (2009) Neutrophil-derived serine proteases modulate innate immune responses. Front. Biosci. 14:3409–18.
Zani ML, Baranger K, Guyot N, Dallet-Choisy S, Moreau T. (2009) Protease inhibitors derived from elafin and SLPI and engineered to have enhanced specificity towards neutrophil serine proteases. Protein Sci. 18:579–94.
Plitas G, et al. (2003) Experimental hindlimb ischemia increases neutrophil-mediated matrix metalloproteinase activity: a potential mechanism for lung injury after limb ischemia. J. Am. Coll. Surg. 196:761–7.
Steinberg J, et al. (2003) Metalloproteinase inhibition reduces lung injury and improves survival after cecal ligation and puncture in rats. J. Surg. Res. 111:185–95.
Delclaux C, et al. (1997) Gelatinases in epithelial lining fluid of patients with adult respiratory distress syndrome. Am. J. Physiol. 272:L442–51.
Ricou B, et al. (1996) Matrix metalloproteinases and TIMP in acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 154:346–52.
Torii K, et al. (1997) Higher concentrations of matrix metalloproteinases in bronchoalveolar lavage fluid of patients with adult respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 155:43–6.
Pugin J, Verghese G, Widmer MC, Matthay MA. (1999) The alveolar space is the site of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress syndrome. Crit. Care Med. 27:304–12.
Steinberg J, et al. (2001) Evidence of increased matrix metalloproteinase-9 concentration in patients following cardiopulmonary bypass. J. Extra Corpor. Technol. 33:218–22.
Fligiel SE, et al. (2006) Matrix metalloproteinases and matrix metalloproteinase inhibitors in acute lung injury. Hum. Pathol. 37:422–30.
Kim JH, et al. (2006) Inhibition of matrix metal- loproteinase-9 prevents neutrophilic inflammation in ventilator-induced lung injury. Am. J. Physiol. Lung Cell. Mol, Physiol 291:L580–7.
Yoon HK, Cho HY, Kleeberger SR. (2007) Protective role of matrix metalloproteinase-9 in ozone-induced airway inflammation. Environ. Health Perspect. 115:1557–63.
Albaiceta GM, et al. (2008) Lack of matrix metal-loproteinase-9 worsens ventilator-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 294:L535–43.
Albaiceta GM, et al. (2009) Absence or inhibition of matrix metalloproteinase-8 decreases ventilator-induced lung injury. Am. J. Respir. Cell Mol. Biol. 43:555–63.
Owen CA, Hu Z, Lopez-Otin C, Shapiro SD. (2004) Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinase. J. Immunol. 172:7791–803.
Quintero PA, Knolle MD, Cala LF, Zhuang Y, Owen CA. (2010) Matrix metalloproteinase-8 inactivates macrophage inflammatory protein-1{alpha} to reduce acute lung inflammation and injury in mice. J. Immunol. 184:1575–88.
Van Lint P, et al. (2005) Resistance of collage-nase-2 (matrix metalloproteinase-8)-deficient mice to TNF-induced lethal hepatitis. J. Immunol. 175:7642–9.
Balbin M, et al. (2003) Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat. Genet. 35:252–7.
Soehnlein O, Weber C, Lindbom L. (2009) Neutrophil granule proteins tune monocytic cell function. Trends Immunol. 30:538–46.
Yang D, de la Rosa G, Tewary P, Oppenheim JJ. (2009) Alarmins link neutrophils and dendritic cells. Trends Immunol. 30:531–7.
Levay PF, Viljoen M. (1995) Lactoferrin: a general review. Haematologica. 80:252–67.
Li KJ, et al. (2006) Release of surface-expressed lactoferrin from polymorphonuclear neutrophils after contact with CD4+ T cells and its modulation on Th1/Th2 cytokine production. J. Leukoc. Biol. 80:350–8.
Bournazou I, et al. (2009) Apoptotic human cells inhibit migration of granulocytes via release of lactoferrin. J. Clin. Invest. 119:20–32.
de la Rosa G, Yang D, Tewary P, Varadhachary A, Oppenheim JJ. (2008) Lactoferrin acts as an alarmin to promote the recruitment and activation of APCs and antigen-specific immune responses. J. Immunol. 180:6868–76.
Actor JK, et al. (2002) Lactoferrin immunomodulation of DTH response in mice. Int. Immunophar-macol. 2:475–86.
Kruzel ML, et al. (2010) Lactoferrin decreases LPS-induced mitochondrial dysfunction in cultured cells and in animal endotoxemia model. Innate Immun. 16:67–79.
Doss M, White MR, Tecle T, Hartshorn KL. (2010) Human defensins and LL-37 in mucosal immunity. J. Leukoc. Biol. 87:79–92.
Kai-Larsen Y, Agerberth B. (2008) The role of the multifunctional peptide LL-37 in host defense. Front Biosci. 13:3760–7.
Soehnlein O, et al. (2008) Neutrophil secretion products pave the way for inflammatory monocytes 165. Blood. 112:1461–71.
Fahy RJ, Wewers MD. (2005) Pulmonary defense and the human cathelicidin hCAP-18/LL-37. Immunol. Res. 31:75–89.
Scott MG, Davidson DJ, Gold MR, Bowdish D, Hancock REW. (2002) The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J. Immunol. 169:3883–3891.
van der Does AM, et al. (2010) LL-37 Directs macrophage differentiation toward macrophages with a proinflammatory signature. J. Immunol. 185:1442–9.
Soehnlein O, Lindbom L. (2010) Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 10:427–39.
Aarbiou J, et al. (2006) Mechanisms of cell death induced by the neutrophil antimicrobial peptides alpha-defensins and LL-37. Inflamm. Res. 55:119–27.
Barlow PG, et al. (2006) The human cationic host defense peptide LL-37 mediates contrasting effects on apoptotic pathways in different primary cells of the innate immune system. J. Leukoc. Biol. 80:509–20.
Nagaoka I, Tamura H, Hirata M. (2006) An antimicrobial cathelicidin peptide, human CAP18/LL-37, suppresses neutrophil apoptosis via the activation of formyl-peptide receptorlike 1 and P2X7. J. Immunol. 176:3044–52.
Tecle T, Tripathi S, Hartshorn KL. (2010) Review: defensins and cathelicidins in lung immunity. Innate Immun. 16:151–9.
Ashitani J, et al. (2004) High concentrations of alpha-defensins in plasma and bronchoalveolar lavage fluid of patients with acute respiratory distress syndrome. Life Sci. 75:1123–34.
Soehnlein O, et al. (2008) Neutrophil primary granule proteins HBP and HNP1–3 boost bacterial phagocytosis by human and murine macrophages 168. J. Clin. Invest 118:3491–502.
Sakamoto N, et al. (2005) Differential effects of {alpha}- and ta-defensin on cytokine production by cultured human bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 288:L508–13.
Aarbiou J, et al. (2006) Mechanisms of cell death induced by the neutrophil antimicrobial peptides alpha-defensins and LL-37. Inflamm. Res. 55:119–27.
Nygaard SD, Ganz T, Peterson MW. (1993) Defensins reduce the barrier integrity of a cultured epithelial monolayer without cytotoxicity. Am. J. Respir. Cell Mol. Biol. 8:193–200.
Soong LB, Ganz T, Ellison A, Caughey GH. (1997) Purification and characterization of defensins from cystic fibrosis sputum. Inflamm. Res 46:98–102.
Van WS, Mannesse-Lazeroms SP, Dijkman JH, Hiemstra PS. (1997) Effect of neutrophil serine proteinases and defensins on lung epithelial cells: modulation of cytotoxicity and IL-8 production. J. Leukoc. Biol. 62:217–26.
Chertov O, et al. (1996) Identification of defensin-1, defensin-2, and CAP37/azurocidin as T-cell chemoattractant proteins released from interleukin-8-stimulated neutrophils. J. Biol. Chem. 271:2935–40.
Grigat J, Soruri A, Forssmann U, Riggert J, Zwirner J. (2007) Chemoattraction of macrophages, T lymphocytes, and mast cells is evolutionarily conserved within the human alpha-defensin family. J. Immunol. 179:3958–65.
Yang D, Chen Q, Chertov O, Oppenheim JJ. (2000) Human neutrophil defensins selectively chemoattract naive T and immature dendritic cells. J. Leukoc. Biol. 68:9–14.
Bdeir K, et al. (2010) Neutrophil {alpha}-defensins cause lung injury by disrupting the capillary-epithelial barrier. Am. J. Respir. Crit. Care Med. 181:935–46.
Soehnlein O, Lindbom L. (2009) Neutrophil-derived azurocidin alarms the immune system. J. Leukoc. Biol. 85:344–51.
Soehnlein O, et al. (2005) Neutrophil-derived heparin-binding protein (HBP/CAP37) deposited on endothelium enhances monocyte arrest under flow conditions 164. J. Immunol. 174:6399–405.
Gautam N, et al. (2001) Heparin-binding protein (HBP/CAP37): a missing link in neutrophil-evoked alteration of vascular permeability. Nat. Med. 7:1123–7.
Di Gennaro A, et al. (2009) Leukotriene B4-induced changes in vascular permeability are mediated by neutrophil release of heparin-binding protein (HBP/CAP37/azurocidin). FASEB J. 23:1750–7.
Herwald H, et al. (2004) M protein, a classical bacterial virulence determinant, forms complexes with fibrinogen that induce vascular leakage. Cell. 116:367–79.
Yasui K, et al. (2008) Possible involvement of heparin-binding protein in transfusion-related acute lung injury. Transfusion. 48:978–87.
Johansson J, Lindbom L, Herwald H, Sjoberg F. (2009) Neutrophil-derived heparin binding protein—a mediator of increased vascular permeability after burns? Burns. 35:1185–7.
Linder A, Christensson B, Herwald H, Bjorck L, Akesson P. (2009) Heparin-binding protein: an early marker of circulatory failure in sepsis. Clin. Infect. Dis. 49:1044–50.
Linder A, Soehnlein O, Akesson P. (2010) Roles of heparin-binding protein in bacterial infections. J. Innate Immun. 2:431–8.
Fialkow L, Wang Y, Downey GP. (2007) Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic. Biol. Med. 42:153–64.
Liang B, Petty HR. (1992) Imaging neutrophil activation: analysis of the translocation and utilization of NAD(P)H-associated autofluorescence during antibody-dependent target oxidation. J. Cell Physiol. 152:145–56.
Lambeth JD. (2004) NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 4:181–9.
Johnson KJ, Fantone JC, III, Kaplan J, Ward PA. (1981) In vivo damage of rat lungs by oxygen metabolites. J. Clin. Invest 67:983–93.
Auten RL, Whorton MH, Nicholas Mason S. (2002) Blocking neutrophil influx reduces DNA damage in hyperoxia-exposed newborn rat lung. Am. J. Respir. Cell. Mol. Biol. 26:391–7.
Auten RL Jr., Mason SN, Tanaka DT, Welty-Wolf K, Whorton MH. (2001) Anti-neutrophil chemokine preserves alveolar development in hyperoxia-exposed newborn rats. Am. J. Physiol. Lung Cell Mol. Physiol 281: L336–44.
Chiarugi P, et al. (2003) Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J. Cell Biol. 161:933–44
Roper JM, et al. (2004) In vivo exposure to hyperoxia induces DNA damage in a population of alveolar type II epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 286:L1045–54.
Carnesecchi S, et al. (2009) NADPH oxidase-1 plays a crucial role in hyperoxia-induced acute lung injury in mice. Am. J. Respir. Crit. Care Med. 180:972–81.
Kubo H, et al. (1996) Preservation of complement-induced lung injury in mice with deficiency of NADPH oxidase. J. Clin. Invest. 97:2680–4.
Wang W, Suzuki Y, Tanigaki T, Rank DR, Raffin TA. (1994) Effect of the NADPH oxidase inhibitor apocynin on septic lung injury in guinea pigs. Am. J. Respir. Crit. Care Med. 150:1449–52.
Kristof AS, Goldberg P, Laubach V, Hussain SN. (1998) Role of inducible nitric oxide synthase in endotoxin-induced acute lung injury. Am. J. Respir. Crit. Care Med. 158:1883–9.
Scheel-Toellner D, et al. (2004) Clustering of death receptors in lipid rafts initiates neutrophil spontaneous apoptosis. Biochem. Soc. Trans 32:679–81.
El Kebir D, Jozsef L, Pan W, Filep JG. (2008) Myeloperoxidase delays neutrophil apoptosis through CD11b/CD18 integrins and prolongs inflammation. Circ. Res. 103:352–9.
Ulbrich H, Eriksson EE, Lindbom L. (2003) Leukocyte and endothelial cell adhesion molecules as targets for therapeutic interventions in inflammatory disease. Trends Pharmacol. Sci. 24:640–7.
Miller EJ, Cohen AB, Peterson BT. (1996) Peptide inhibitor of interleukin-8 (IL-8) reduces staphylococcal enterotoxin-A (SEA) induced neutrophil trafficking to the lung. Inflamm. Res. 45:393–7.
Hayashi S, Yatsunami J, Fukuno Y, Kawashima M, Miller EJ. (2002) Antileukinate, a hexapep-tide inhibitor of CXC-chemokine receptor, suppresses bleomycin-induced acute lung injury in mice. Lung. 180:339–48.
Bhatia M, et al. (2000) Treatment with neutralising antibody against cytokine induced neutrophil chemoattractant (CINC) protects rats against acute pancreatitis associated lung injury. Gut. 47:838–44.
Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. (2001) Transient expression of IL-1b induces acute lung injury and chronic repair leading to pulmonary fibrosis. J. Clin. Invest. 107:1529–36.
Gerard C, et al. (1997) Targeted disruption of the beta-chemokine receptor CCR1 protects against pancreatitis-associated lung injury. J. Clin. Invest. 100:2022–7.
Hirche TO, Atkinson JJ, Bahr S, Belaaouaj A. (2004) Deficiency in neutrophil elastase does not impair neutrophil recruitment to inflamed sites. Am. J. Respir. Cell Mol. Biol. 30:576–84.
Tkalcevic J, et al. (2000) Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity. 12:201–10.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Grommes, J., Soehnlein, O. Contribution of Neutrophils to Acute Lung Injury. Mol Med 17, 293–307 (2011). https://doi.org/10.2119/molmed.2010.00138
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2010.00138