
Abstract
Pregnancy-induced hypertension (PIH), also known as preeclampsia, Is one of the major causes of maternal and fetal death. While the precise cause of PIH is not known, aberrant cytokine production and placenta participation are considered to be important factors. Gestational cigarette smoking, which is widely accepted to be harmful to both the mother and fetus, is protective against PIH. Based on the antiinflammatory activity of nicotine, the major component of cigarettes, we examined the effect of nicotine and other cholinergic agonists on placental inflammatory responses ex vivo. We observed that nicotine and other cholinergic agonists significantly suppress placenta cytokine production following stimulation. Placenta cells express the α 7 nicotinic acetylcholine receptor (α7nAChR), and using cholinergic antagonists, we demonstrated that the antiinflammatory effect of nicotine and other cholinergic agonists is, in part, mediated through the nAChR pathway. By contrast, cholinergic stimulation had no effect on the expression of soluble fms-like tyrosine kinase (sFlt), an antiangiogenic substance implicated in maternal vascular dysfunction during PIH. Mechanistic studies reveal that cholinergic agonists exert their antiinflammatory effects through the NFκB pathway. Taken together, our results suggest that cholinergic agonists, including nicotine, may reduce cytokine production by placenta cells via NFκB to protect against PIH.
Similar content being viewed by others

Introduction
Despite the many detrimental effects of smoking on maternal and fetal health, several studies show that maternal smoking is protective against pregnancy-induced hypertension (PIH), also known as preeclampsia (1–6). PIH is one of the leading causes of perinatal morbidity and mortality (7), occurring in 5%–10% of all pregnancies. PIH is characterized by maternal hypertension, proteinuria, and edema during the second half of pregnancy (7,8). Although the pathogenesis of PIH is poorly understood, the role of the placenta in mediating PIH is well accepted, as the condition is resolved upon delivery of the placenta following childbirth. While the association between angiogenic factors and PIH has been recently discovered (9–11), numerous reports highlight the proposed contribution of leukocyte activation and several proinflammatory cytokines to the development of PIH, including TNFα (TNF), IL-6, IL-1 receptor antagonist, and IL-8 (12–18). Further studies implicate a generalized phenomenon of maternal immune cell activation via NFκB, a critical regulator of inflammation (19,20).
The systemic antiinflammatory effect of cholinergic agents including nicotine, the major constituent of cigarettes, has been described in numerous reports (reviewed in 21). Nicotine suppresses inflammation during experimental ulcerative colitis (22), improves survival during experimental sepsis via its effect on the production of proinflammatory mediators (23), and blocks leukocyte recruitment in vivo (24). Nicotine suppresses TNF production by LPS-stimulated macrophages (25) and microglial cells (26) through the alpha7 nicotinic acetylcholine receptor (α7nAChR), and inhibits endothelial cell activation in vitro and in vivo (24). Based on the observations that smoking protects against PIH and nicotine exerts antiinflammatory effects, we examined the effects of cholinergic agonists, including nicotine, on placental inflammatory responses ex vivo.
Materials and Methods
Reagents
LPS, nicotine, and mecamylamine chloride were obtained from Sigma-Aldrich (St Louis, MO, USA). Cholinergic agonists, GTS-21 and CAP55, were provided by Y. Al-Abed (The Feinstein Institute for Medical Research).
Placenta Cell Isolation and Analysis of Cytokine and sFlt Expression
Anonymous human placentas obtained from normal, term pregnancies at North Shore University Hospital (exempt from IRB review) were processed within three to four hours of delivery. For each placenta, five random 1× 1×1 inch placenta pieces were collected, washed in PBS, and minced, and the resulting pooled mixture was passed through a 40 µM mesh to obtain a single cell suspension. Contaminating erythrocytes were removed by hypotonic water lysis, and isolated cells were washed once with PBS. Placental cells were plated on 0.15% gelatin-coated 96-well plates (2×106 cells/mL) in RPMI containing 10% FBS, penicillin, streptomycin, and glutamine at 37°C and 5% CO2. Plated placenta cells were stimulated with lipopolysaccharide (LPS) (isolated from Escherichia coli serotype 0111:B4, 0–1000 ng/mL) and incubated overnight. To examine the effect of cholinergic agonists on cytokine production, placenta cells were pretreated for 0.5 hours with cholinergic agonists (nicotine, GTS-21, CAP55) prior to LPS stimulation (each sample was assayed in triplicate or quadruplicate). Following an overnight incubation, cell-free supernatants were collected and assayed for cytokine production (IL-1β, IL-6, and IL-8) or sFlt production by ELISA (R&D Systems, Minneapolis, MN, USA). TNF production was assayed by ELISA according to Hesse et al (27). Each ELISA sample was assayed in triplicate. The sensitivities of the IL-1β, IL-6, and IL-8 ELISAs were less than 4 pg/mL. The sensitivity of the sFlt ELISA was 10 pg/mL. The sensitivity of the TNF ELISA was 39 pg/mL. The mean intra-and interassay coefficient of variation for all ELISA assays (i.e. precision) was ≤15%. Additional studies employed pre-treatment with the cholinergic antagonist, mecamylamine, prior to treatment with cholinergic agonists and LPS. Cell death was assessed by LDH using the Cyto-Tox96 Cytotoxicity Assay kit (Promega, Madison, Wisconsin, USA) according to the manufacturer’s instructions. Each experiment was repeated at least three to four times (with different placentas).
Analysis of α7nAChR Expression by Placenta Cells
Western blotting. Placenta cells were resuspended in 50 mM Tris-HCl pH 6.8, and 3% SDS and solubilized at 37 °C for one hour. Lysates were separated by SDS-PAGE electrophoresis and transferred to PDVF membrane. Membranes were probed with rabbit antihuman α7nAChR (Chemicon, Temecula, CA, USA) followed by incubation with HRP-conjugated goat antirabbit antibody and ECL development (Amersham Biosciences/GE Healthcare, Piscataway, NJ, USA).
RT-PCR. Total RNA was isolated from placenta cells using the RNeasy mini kit (Qiagen, Valencia, CA, USA). Briefly cDNA was prepared using M-MLV reverse transcriptase (Invitrogen, Carlsbad, CA, USA) from 1µg RNA. We amplified 2.5-µL aliquots of cDNA by PCR using PCR SuperMix (Invitrogen) in a thermal cycler (model 9600; Perkin Elmer, Waltham, MA, USA) using primer sequences for a α7nAChR and αα7nAChR duplicate gene (α7 (F-GGCAGATATCAGTGGC-TATA; R-CTTCATTCGCAGGAACC); α7dup (F-CGGTGCCCCTTGCCATTTTC; R-CAGAGTGCTTTCTGCACCTTTGG). These experiments were repeated twice (with different placentas) with similar results. Representative data are shown.
Analysis of Placenta Cytokine Expression by Quantitative PCR
Placenta cells were processed and plated (in 6-well plates) as described above. Placenta cells were pretreated with cholinergic agonists for 30 min prior to LPS stimulation (100 ng/mL). Two hours later, total RNA from placenta cells treated with cholinergic agonists (or vehicle) ± LPS was isolated using the RNeasy RNA isolation kit (Qiagen). The relative expression of TNF, IL-1β, IL-8, and IL-6 mRNA was assessed by quantitative realtime PCR using TaqMan technology with GAPDH as an internal control. Reactions (performed in duplicate) were completed using 50 ng RNA, Eurogentec quantitative RTqPCR master mix. and the Prism 7700 sequence detection system (Applied Biosystems, Foster City, CA, USA). Results were expressed as fold increase with respect to the vehicle control. Primer sequences were as follows:
TNF F-TCTTCTCGAACCCCGAGTGA, R-CCTCTGATGGCACCACCAG, probe-TAGCCCATGTTGTAGCAAACCCTCAA GCT; IL-8 F-CTAGGACAAGAGCCAGG-AAGAAAC, R-CCACGGCCAGCTTGGA, probe-ACCGGAAGGAACCATCT-CACTGTGTGTAAA; IL-6 F-CTGCA-GAAAAAGGCAAAGAATCTAG, R-CG-TCAGCAGGCTGGCATT, probe-TGCAATAACCACCCCTGACCCAACC; IL-1β F-TGCACCTGTACGATCACT-GAACT, R-TGGACCAGACATCAC-CAAGCT, and probe-CACGCTC-CGGGACTCACAGCA. The respective mRNA levels were calculated using the ΔΔCt method. Expression levels were normalized to the housekeeping gene GAPDH. Data from three experiments (using three different placentas) are presented as mean fold increase over control (mean ± SD).
NFκB Activation Studies
Placenta cells were prepared as described above and plated in T25 flasks. Placenta cultures were treated (in duplicate) with vehicle or nicotine (10−4 M) for 15 min prior to LPS stimulation (0–100 ng/mL). After one hour, placenta cells were collected and nuclear lysates were prepared using a nuclear extraction kit (Active Motif, Carlsbad, CA, USA) according to the manufacturer’s instructions. The nuclear extracts were then analyzed for NFκB activation using the TransAM NFκB p65/NFκB p50 Chemi Transcription Factor Assay Kit (Active Motif), according to the manufacturer’s instructions. This assay is based on the binding of nuclear p65/p50 to an oligonucleotide containing an NFκB consensus site bound to a 96-well plate. Binding of p65/p55 to the oligonucleotide is determined using antibodies specific for the p65/p50 subunits of NFκB followed by chemiluminescent detection. The specificity of the assay was confirmed using the appropriate controls. Data from three separate experiments are presented as mean percent control ± SD.
Statistics
Analysis of variance (ANOVA) followed by the Dunnett’s Test (to make pair-wise comparisons) and the Student t-test (unpaired, 2 tailed) were employed for comparing groups as appropriate. The differences were considered significant if P was less than 0.05.
Results
Nicotine Blocks TNF Production by LPS-Stimulated Placenta Cells Ex Vivo
To mimic inflammation associated with PIH, human placenta cells were treated with LPS to induce TNF production. Under basal conditions, TNF was not detectable in the placenta cell culture supernatants collected after an overnight incubation (Figure 1A). LPS treatment of placenta cells stimulated TNF production, in a dose-dependent manner (Figure 1A). Nicotine treatment (≥10−6M) of the placenta cell cultures significantly reduced LPS-induced TNF production over a wide range of LPS concentrations (Figure 1B), with up to 50%–60% inhibition. Similar results were observed with placenta tissue explants (data not shown). These effects were comparable to the suppressive effects of dexamethasone (Dex, 1–10 µM), a well-known anti-inflammatory agent, on TNF production following LPS stimulation (Figure 1C). Consistent with these observations, nicotine significantly reduced placenta cell TNF mRNA expression induced by LPS, as determined by Q-PCR (Figure 1D). The concentrations of nicotine and Dex used for these experiments were not cytotoxic to the placenta cells (Table 1).

Nicotine reduces TNF expression by LPS-treated placenta cells. (A) LPS stimulation (1-1000 ng/mL) of placenta cells ex vivo induces TNF release. (B) Nicotine or (C) dexamethasone treatment reduces TNF production by LPS stimulated placenta cells. (D) Nicotine (10−4 M) treatment suppresses TNF mRNA expression by LPS-stimulated (100 ng/mL) placenta cells. Data from four separate experiments are shown as mean ± SD. *P < 0.05, **P < 0.01 when compared with appropriate vehicle control.
Nicotine Reduces Cytokine Expression by LPS-Treated Placenta Cells
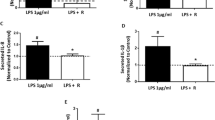
Next, we examined the effect of nicotine on the expression of other proinflammatory cytokines by placenta cells following LPS treatment ex vivo. We found that nicotine significantly reduced IL-1β, IL-8, and IL-6 mRNA expression following LPS stimulation (Figure 2A). Consistent with these observations, we found that nicotine significantly reduced LPS-induced IL-6 production by placenta cells (Figure 2B). However, the inhibitory effects of nicotine on LPS-induced IL-6 mRNA and protein levels were less dramatic than that observed for TNF expression (see Figures 1B and 1D). Similar results were observed for IL-1β and IL-8 protein expression. Nicotine (10−4 M) reduced IL-1β and IL-8 production by placenta cells following LPS stimulation (100 ng/mL) by 33% (±SD) and 34.5% (±5.4%, SD), respectively.

Nicotine inhibits cytokine expression by LPS-treated placenta cells. (A) Nicotine decreases IL-1β, IL-8, and IL-6 mRNA expression by LPS-stimulated (100 ng/mL) placenta cells. Data are shown as fold increase mRNA expression over vehicle control (no LPS). (B) LPS treatment (100 ng/mL) induces IL-6 protein production by placenta cells (inset), which can be suppressed by nicotine. Data from three separate experiments are shown as mean ± SD. *P < 0.05, **P < 0.01 when compared with appropriate control.
Placenta Cells Express the α7nAChR
The well-established role of the α7nAChR in mediating the antiinflammatory effects of nicotine on macrophages, endothelial cells, and microglial cells (24–26) prompted us to examine the expression of the α7nAChR by placenta cells. We found that placenta cells expressed α7nAChR and α7nAChRdup mRNA and protein, as determined by RT-PCR and Western blotting methods (Figures 3A and 3B).

Placenta cells express the α7-nAChR. Placenta cells express α7-nAChR mRNA (A) and protein (B) as determined by RT-PCR and Western blotting, respectively.
α7nAChR Selective Agonists Suppress Placenta Inflammatory Responses
Based on our observations that nicotine reduced proinflammatory cytokine production by placenta cells expressing the α7nAChR, we investigated the effects of GTS-21 [3-(2,4)-dimethoxybenzylidine anabaseine or DMXB], a well-described selective α7nAChR agonist (28–30) and CAP55, a novel α7nAChR agonist (24) on LPS-induced cytokine expression by placenta cells (LPS = 100 ng/mL). Both GTS-21 and CAP55 significantly reduced TNF, IL-6, and IL-8 mRNA expression induced by LPS (Figure 4A). Likewise, both GTS-21 and CAP55, a novel selective α7nAChR agonist, significantly inhibited both LPS-induced TNF (Figure 4B) and IL-6 production (Figure 4C) by placenta cells in a dose-dependent manner. Treatment of placenta cells with GTS-21 (10−5 M) also reduced LPS-induced (100 ng/mL) IL-1β and IL-8 by approximately 35% each. We found GTS-21 to be more effective in reducing cytokine production than nicotine or CAP55 (when used at the same concentrations). Neither GTS-21 nor CAP55 showed any cytotoxic effects on placenta cells at the concentrations used (Table 2).

α7-nAChR-selective cholinergic agonists suppress cytokine expression by placenta cells. (A) GTS-21 inhibits LPS-stimulated (100 ng/mL) TNF, IL-1β, IL-8, and IL-6 mRNA levels. Data are shown as fold increase mRNA expression over vehicle control. GTS-21 and CAP55 suppress (B) TNF and (C) IL-6 protein production induced by placenta cells following LPS stimulation (100 ng/mL). Data from three separate experiments are shown as mean ± SD. *P < 0.05, **P < 0.01, when compared with appropriate vehicle control.
To further demonstrate the role of the nAChR pathway in mediating the antiinflammatory effects of nicotine, GTS-21, and CAP55, we used nAChR antagonists in our model system. Pretreatment of placenta cells with the cholinergic antagonist mecamylamine (1–5 µM) reversed the antiinflammatory effects of nicotine, GTS-21, and CAP55 on LPS-induced TNF production by up to 67%, suggesting that the inhibitory action of these cholinergic agonists is mediated, in part, through the nAChR pathway.
Cholinergic Agonists Do Not Suppress sFlt Production Following LPS Treatment
Several studies suggest that excess soluble fms-like tyrosine kinase (sFlt, also known as soluble VEGR-1), produced by the placenta, contributes to the pathogenesis of PIH (9–11). Therefore, we examined whether LPS (100 ng/mL) induced sFlt expression by placental cells ex vivo and whether sFlt expression was modulated by cholinergic agonists. We found that LPS significantly induced sFlt production by placental cells ex vivo [Figure 5 (inset)]. The induction of sFlt by LPS was not as dramatic as that observed for cytokines such as TNF. In contrast to what we observed for TNF, we did not observe an inhibitory effect of nicotine on sFLT production following LPS stimulation of placental explants (Figure 5).

LPS induces sFlt expression by the placenta but nicotine does not inhibit LPS-induced sFlt production. Placenta cells were treated with either vehicle or nicotine (10−4 M) prior to LPS stimulation (100 ng/mL) or increasing concentrations of LPS alone (inset). After an overnight incubation, cell-free culture supernatants were collected and assayed for sFlt by ELISA. Data from three separate experiments are shown as mean ± SD. *P < 0.05 when compared with vehicle control.
Cholinergic Agonists Mediate Their Antiinflammatory Effects through NFκB
Next, we examined the potential role of the NFκB pathway in mediating the anti-inflammatory effects of cholinergic agonists on placenta cytokine production. To assess NFκB activation, we utilized the quantitative and sensitive TransAM p65/p50 luminescent assay, which is based on the binding of nuclear NFκB (p65/p50) to a target oligonucleotide bound to a 96-well plate, followed by incubation with nuclear extract and antibodies specific for p65/p50. LPS (100 ng/mL) significantly induced NFκB activation by placenta cell explants (Figure 6). We observed that treatment of placenta cells with either nicotine (10−4 M) or GTS-21 (10−5 M) inhibited NFκB activation induced by LPS (100 ng/mL) (Figure 6).

Cholinergic stimulation reduces NFκB activation by LPS-treated placenta cells. Placenta cells were treated with either vehicle, nicotine (10−4 M), or GTS-21 (10−5 M) prior to LPS stimulation (100 ng/mL). Data from three separate experiments are shown as mean percent control ± SD. *P < 0.05, **P < 0.01 (control = vehicle/LPS treatment).
Discussion
In 2002, 11.4% of all women giving birth in the US reported smoking (31). Undoubtedly, smoking during pregnancy may be detrimental to both maternal and fetal health. Smoking is associated with preterm birth and preterm premature rupture of membranes (PPROM), placenta abruption, placenta previa-acc-reta, intrauterine fetal growth retardation, and sudden infant death syndrome (32–36). Despite the adverse effects of smoking on maternal-fetal health, numerous studies demonstrate that smoking is protective against PIH (1-6). Interestingly, a history of cigarette smoking before pregnancy alone is not protective against PIH (37), suggesting that some agent(s) found in cigarettes may mediate the protective effects of smoking during pregnancy. Nicotine, the major constituent of cigarettes, and/or cholinergic stimulation via vagus nerve stimulation inhibit inflammatory cytokine production in vitro and in vivo using several experimental models including sepsis, postoperative ileus, and pancreatitis (23,38,39). In addition, nicotine is effectively used for the treatment of inflammatory bowel disease in the clinical setting (40,41). Our observations showing that nicotine and other cholinergic agents suppress placenta cytokine production ex vivo raise the possibility that nicotine exerts antiinflammatory effects to protect against PIH.
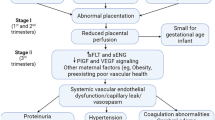
Numerous reports support the hypothesis that aberrant proinflammatory cytokine production, with an excessive maternal inflammatory response in pregnancy, plays a critical role in the development of PIH (42–49). The immune theory of PIH is further supported by several epidemiological studies and research reports demonstrating the deposition of immune complexes and complement activation preceding maternal vascular injury and systemic inflammation during PIH [reviewed in (50)]. PIH is associated with increased maternal circulating proinflammatory factors, including TNF, IL-6, and IL-8 (12–17), as well as enhanced expression of TNF (protein and mRNA) by PIH placentas (14) and increased IL-1β, IL-6, and IL-8 production by peripheral monocytes (49).
While our studies demonstrate the production of cytokines including TNF by LPS-treated placenta cell explants, additional studies will be required to determine which cells within the placenta produce cytokines following stimulation. Several cell types within the placenta, including Hofbauer cells (placenta resident macrophages), syncytiotrophoblasts, and cytotrophoblasts produce cytokines (51–54). Previous studies using experimental model systems reveal the highest expression of TNF by trophoblast cells within the placenta (54).
Using an ex vivo model system with placenta cell explants, we investigated the effects of nicotine and cholinergic stimulation on cytokine production by placenta cells. Our observations are comparable with previous studies demonstrating the production of numerous cytokines by the placenta ex vivo following LPS stimulation and the antiinflammatory effects of glucocorticoids on placenta cell inflammatory responses from cultured normal and PIH placental explants (55). Using this model system, we demonstrate for the first time the antiin-flammatory effects of cholinergic agonists, including nicotine on placental inflammatory responses. Our observations suggest that nicotine contained in cigarettes may confer the protective effect of smoking on PIH by mediating antiin-flammatory responses. However, these findings do not eliminate the possibility that other protective factors may contribute, such as carbon monoxide contained in cigarettes (56). While future studies are required to investigate the effect of nicotine (and cholinergic stimulation) on placental inflammatory responses in vivo, one major advantage of our ex vivo model system is that it allowed mechanistic-based studies to better understand how nicotine and other cholinergic agonists exerts antiinflammatory effects on placenta cells.
Our observations of α7nAChR expression by placenta cells are consistent with previous studies reporting α7nAChR expression by the human placenta (57,58). While Kwon and coworkers found no change in α7nAChR gene expression within the placenta related to PIH, they did report that α7nAChR protein expression was significantly increased in placentas obtained from severe PIH patients (57). However, their conclusions are somewhat weakened by their semiquantitative data and small sample sizes. Regardless, this observation raises questions about the modulation of α7nAChR expression within the placenta and the function of the α7nAChR with reference to (a) acetylcholine, a potentially important placental signaling molecule; (b) nicotine, which may exert both positive and negative effects on fetal outcome, and (c) other cholinergic agonists. Further investigations to explore the effect of smoking on placenta α7nAChR expression and to assess both placental α7nAChR expression and acetylcholine levels during PIH are warranted.
Recent studies suggest the role of soluble VEGR-1 or sFlt in mediating the pathogenesis of PIH (9–11). sFlt is a secreted splice variant of Flt that binds angiogenic factors, VEGF and placenta growth factor (PlGF), preventing their binding to Flt in tissues and hence reducing angiogenesis. Numerous studies reveal higher sFlt expression by the placenta during PIH (9,59–61), and increased circulating levels of sFlt are observed during PIH (59). While the placenta was originally described as the sole source of sFlt, a recent study showed that peripheral blood mononuclear cells of pregnant women secrete high concentrations of sFlt (62). We investigated the effect of cholinergic stimulation on sFlt in our ex vivo model system using placenta explants because of its potential role in PIH, the link between sFlt and inflammation (63), and because smoking has been associated with reduced sFlt secretion (64). Our data suggest that sFlt levels are regulated, in part, by inflammation and that sFlt levels are not modulated by cholinergic agonists, such as nicotine. Based on previous studies showing the inhibitory effect of cigarette smoke on sFlt secretion by cultured placental explants (65), it is possible that other constituents in cigarette smoke regulate sFlt expression.
Numerous studies implicate a generalized phenomenon of maternal immune cell activation with excessive cytokine production with PIH. NFκB is family of transcription factors associated with inflammation. NFκB activation is triggered by LPS and proinflammatory cytokines, including TNF and IL-1β. Several studies by our group and others have linked the antiinflammatory function of cholinergic stimulation to the NFκB pathway (23,24,66,67). In addition, previous studies support the role of NFκB in labor (19). Further studies reveal the engagement of the NFκB pathway by circulating immune cells during PIH (20). Consistent with observed heightened expression of proinflammatory factors during PIH, antiinflammatory pharmacologics, namely potent glucocorticoids, are used, with some degree of success, to suppress maternal inflammatory responses and to improve maternal and fetal outcomes associated with HELLP Syndrome (50,68–70). Similarly, low-dose aspirin (alone or in combination with dipyridamole, another antiplatelet agent) may be associated with moderate reductions in the relative risk of PIH and birth prior to 34 weeks gestation (71). Together, these observations raise the question whether antiin-flammatory agents such as anti-TNF drugs might be useful for the treatment of PIH.
References
Klonoff-Cohen H, Edelstein A Savitz D. (1993) Cigarette smoking and preeclampsia. Obstet. Gynecol. 81:541–4.
Marcoux S, Brisson J, Fabia J. (1989) The effect of cigarette smoking on the risk of preeclampsia and gestational hypertension. Am. J. Epidemiol. 130:950–7.
Conde-Agudelo A, Althabe F, Belizan JM, Kafury-Goeta AC. (1999) Cigarette smoking during pregnancy and risk of preeclampsia: a systematic review. Am. J. Obstet. Gynecol. 181:1026–35.
Lindqvist PG, Marsal K. (1999) Moderate smoking during pregnancy is associated with a reduced risk of preeclampsia. Acta Obstet. Gynecol. Scand. 78:693–7.
Xiong X, Wang FL, Davidge ST, Demianczuk NN, Mayes DC, Olson DM, Saunders LD. (2000) Maternal smoking and preeclampsia. J. Reprod. Med. 45:727–32.
Mortensen JT, Thulstrup AM, Larsen H, Moller M, Sorensen HT. (2001) Smoking, sex of the offspring, and risk of placental abruption, placenta previa, and preeclampsia: a population-based cohort study. Acta Obstet. Gynecol. Scand. 80:894–8.
Sibai B, Dekker G, Kupferminc M. (2005) Pre-eclampsia. Lancet 365:785–99.
Granger JP, Alexander BT, Llinas MT, Bennett WA, Khalil RA. (2002) Pathophysiology of preeclampsia: linking placental ischemia/hypoxia with microvascular dysfunction. Microcirculation 9:147–60.
Levine RJ, Maynard SE, Qian C, et al. (2004) Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 350:672–83.
McKeeman GC, Ardill JE, Caldwell CM, Hunter AJ, McClure N. (2004) Soluble vascular endothelial growth factor receptor-1 (sFlt-1) is increased throughout gestation in patients who have preeclampsia develop. Am. J. Obstet. Gynecol. 191:1240–6.
Bdolah Y, Karumanchi SA, Sachs BP. (2005) Recent advances in understanding of preeclampsia. Croat. Med. J. 46:728–36.
Vince GS, Starkey PM, Austgulen R, Kwiatkowski D, Redman CW. (1995) Interleukin-6, tumor necrosis factor and soluble tumor necrosis factor receptors in women with pre-eclampsia. Br. J. Obstet. Gynaecol. 102:20–5.
Chen G, Wilson R, Wang SH, Zheng HZ, Walker JJ, McKillop JH. (1996) Tumour necrosis factor-alpha (TNF-alpha) gene polymorphism and expression in pre-eclampsia. Clin. Exp. Immunol. 104:154–9.
Wang Y, Walsh SW. (1996) TNF alpha concentrations and mRNA expression are increased in preeclamptic placentas. J. Reprod. Immunol. 32:157–69.
Kimya Y, Akdis C, Cengiz C, Ozan H, Tatlikazan S, Uncu G, Sengul F. (1997) Plasma interleukin-1alpha, interleukin-1beta and interleukin-1 receptor antagonist levels in pre-eclampsia. Eur.J.Obstet. Gynecol. Reprod. Biol. 73:17–21.
Velzing-Aarts FV, Muskiet FA, van der Dijs FP, Duits AJ. (2002) High serum interleukin-8 levels in afro-caribbean women with pre-eclampsia. Relations with tumor necrosis factor-alpha, duffy negative phenotype and von Willebrand factor. Am. J. Reprod. Immunol. 48:319–22.
Johnson MR, Anim-Nyame N, Johnson P, Sooranna SR, Steer PJ. (2002) Does endothelial cell activation occur with intrauterine growth restriction? BJOG. 109:836–9.
Conrad KP, Miles TM, Benyo DF. (1998) Circulating levels of immunoreactive cytokines in women with preeclampsia. Am. J. Reprod. Immunol. 40:102–11.
Lindstrom TM, Bennett PR. (2005) The role of nuclear factor kappa B in human labor. Reproduction 130:569–81.
Luppi P, Tse H, Lain KY, Markovic N, Piganelli JD, DeLoia JA. (2006) Preeclampsia activates circulating immune cells with engagement of the NF-kap-paB pathway. Am. J. Reprod. Immunol. 56:135–44.
Gallowitsch-Puerta M, Tracey KJ. (2005) Immunologic role of the cholinergic anti-inflammatory pathway and the nicotinic acetylcholine alpha 7 receptor. Ann. N. Y. Acad. Sci. 1062:209–19.
Sykes AP, Brampton C, Klee S, Chander CL, Whelan C, Parsons ME. (2000) An investigation into the effect and mechanisms of action of nicotine in inflammatory bowel disease. Inflamm. Res. 49:311–9.
Wang H, Liao H, Ochani M, et al. (2004) Cholin-ergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. 10:1216–21.
Saeed RW, Varma S, Peng-Nemeroff T, et al. (2005) Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J. Exp. Med. 201:1113–23.
Wang H, Yu M, Ochani M, et al. (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421:384–8.
Shytle RD, Mori T, Townsend K, et al. (2004) Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J. Neurochem. 89: 337–43.
Hesse DG, Tracey KJ, Fong Y, et al. (1988) Cytokine appearance in human endotoxemia and primate bacteremia. Surg. Gynecol. Obstet. 166: 147–53.
Simosky JK, Stevens KE, Kem WR, Freedman R. (2001) Intragastric DMXB-A, an alpha7 nicotinic agonist, improves deficient sensory inhibition in DBA/2 mice. Biol. Psychiatry 50:493–500.
Shimohama S, Greenwald DL, Shafron DH, et al. (1998) Nicotinic alpha 7 receptors protect against glutamate neurotoxicity and neuronal ischemic damage. Brain Res. 779:359–63.
O’Neill, HC, Rieger K, Kem WR, Stevens KE. (2003) DMXB, an alpha7 nicotinic agonist, normalizes auditory gating in isolation-reared rats. Psychopharmacology (Berl). 169:332–9.
HHS-CDC. (2004) Smoking during pregnancy— United States, 1990–2002. Morb. Mortal. Wkly. Rep. 53:911–5.
Srinivas SK, Macones GA. (2005) Preterm premature rupture of the fetal membranes: current concepts. Minerva Ginecol. 57:389–96.
Tikkanen M, Nuutila M, Hiilesmaa V, Paavonen J, Ylikorkala O. (2006) Clinical presentation and risk factors of placental abruption. Acta Obstet. Gynecol. Scand. 85:700–5.
Usta IM, Hobeika EM, Musa AA, Gabriel GE, Nassar H. (2005) Placenta previa-accreta: risk factors and complications. Am. J. Obstet. Gynecol. 193: 1045–9.
Salihu HM, Aliyu MH, Kirby RS. (2005) In utero nicotine exposure and fetal growth inhibition among twins. Am. J. Perinatol 22:421–7.
Hunt CE, Hauck FR. (2006) Sudden infant death syndrome. CMAJ. 174:1861–9.
England LJ, Levine RJ, Qian C, et al. (2002) Smoking before pregnancy and risk of gestational hypertension and preeclampsia. Am. J. Obstet. Gynecol. 186:1035–40.
de Jonge WJ, van der Zanden EP, The FO, et al. (2005) Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat. Immunol. 6:844–51.
van Westerloo DJ, Giebelen IA, Florquin S, et al. (2006) The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology. 130:1822–30.
Green JT, Thomas GA, Rhodes J, et al. (1997) Nicotine enemas for active ulcerative colitis—a pilot study. Aliment. Pharmacol. Ther. 11:859–63.
Guslandi M. (1999) Long-term effects of a single course of nicotine treatment in acute ulcerative colitis: remission maintenance in a 12-month follow-up study. Int. J. Colorectal Dis. 14:261–2.
Hauguel-de MS, Guerre-Millo M. (2006) The placenta cytokine network and inflammatory signals. Placenta. 27:794–8.
Borzychowski AM, Sargent IL, Redman CW. (2006) Inflammation and pre-eclampsia. Semin. Fetal Neonatal Med. 11:309–16.
Redman CW, Sargent IL. (2005) Latest advances in understanding preeclampsia. Science 308: 1592–4.
Sargent IL, Borzychowski AM, Redman CW. (2006) Immunoregulation in normal pregnancy and pre-eclampsia: an overview. Reprod. Biomed. Online 13:680–6.
Redman CW, Sargent IL. (2000) Placental debris, oxidative stress and pre-eclampsia. Placenta 21:597–602.
Redman CW, Sargent IL. (2004) Preeclampsia and the systemic inflammatory response. Semin. Nephrol. 24:565–70.
Matthiesen L, Berg G, Ernerudh J, Ekerfelt C, Jonsson Y, Sharma S. (2005) Immunology of preeclampsia. Chem. Immunol. Allergy 89:49–61.
Luppi P, DeLoia JA. (2006) Monocytes of preeclamptic women spontaneously synthesize pro-inflammatory cytokines. Clin. Immunol. 118: 268–75.
Feinberg BB. (2006) Preeclampsia: the death of Goliath. Am. J. Reprod. Immunol. 55:84–98.
Hu XL, Yang Y, Hunt JS. (1992) Differential distribution of interleukin-1 alpha and interleukin-1 beta proteins in human placentas. J. Reprod. Immunol. 22:257–68.
Chen HL, Yang YP, Hu XL, Yelavarthi KK, Fishback JL, Hunt JS. (1991) Tumor necrosis factor alpha mRNA and protein are present in human placental and uterine cells at early and late stages of gestation. Am. J. Pathol. 139:327–35.
Hunt JS. (1989) Cytokine networks in the utero-placental unit: macrophages as pivotal regulatory cells. J. Reprod. Immunol. 16:1–17.
Yelavarthi KK, Chen HL, Yang YP, Cowley BD, Fishback JL, Hunt JS. (1991) Tumor necrosis factor-alpha mRNA and protein in rat uterine and placental cells. J. Immunol. 146:3840–8.
Xu B, Makris A, Thornton A, Hennessy A. (2005) Glucocorticoids inhibit placental cytokines from cultured normal and preeclamptic placental explants. Placenta 26:654–60.
Bainbridge SA, Sidle EH, Smith GN. (2005) Direct placental effects of cigarette smoke protect women from pre-eclampsia: the specific roles of carbon monoxide and antioxidant systems in the placenta. Med. Hypotheses 64:17–27.
Kwon JY, Kim YH, Kim SH, Kang MH, Maeng YS, Lee KY, Park YW. (2006) Difference in the expression of alpha 7 nicotinic receptors in the placenta in normal versus severe preeclampsia pregnancies. Eur. J. Obstet. Gynecol. Reprod. Biol. 132:35–9.
Lips KS, Bruggmann D, Pfeil U, Vollerthun R, Grando SA, Kummer W. (2005) Nicotinic acetyl-choline receptors in rat and human placenta. Placenta 26:735–46.
Tsatsaris V, Goffin F, Munaut C, et al. (2003) Overexpression of the soluble vascular endothelial growth factor receptor in preeclamptic patients: pathophysiological consequences. J. Clin. Endocrinol. Metab. 88:5555–63.
Maynard SE, Min JY, Merchan J, et al. (2003) Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Invest. 111:649–58.
Zhou Y, McMaster M, Woo K, et al. (2002) Vascular endothelial growth factor ligands and receptors that regulate human cytotrophoblast survival are dysregulated in severe preeclampsia and hemolysis, elevated liver enzymes, and low platelets syndrome. Am. J. Pathol. 160:1405–23.
Rajakumar A, Michael HM, Rajakumar PA, et al. (2005) Extra-placental expression of vascular endothelial growth factor receptor-1, (Flt-1) and soluble Flt-1 (sFlt-1), by peripheral blood mononuclear cells (PBMCs) in normotensive and preeclamptic pregnant women. Placenta 26:563–73.
Clavel G, Bessis N, Lemeiter D, et al. (2007) An-giogenesis markers (VEGF, soluble receptor of VEGF and angiopoietin-1) in very early arthritis and their association with inflammation and joint destruction. Clin. Immunol. (epub, ahead of print).
Belgore FM, Lip GY, Blann AD. (2000) Vascular endothelial growth factor and its receptor, Flt-1, in smokers and non-smokers. Br. J. Biomed. Sci. 57:207–13.
Mehendale R, Hibbard JFALR. (2006) Placental angiogenesis marker, hypoxia, and cigarette smoke. Am. J. Obstet. Gynecol. 195:S134.
Pavlov VA, Ochani M, Yang LH, et al. (2007) Selective alpha7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endo-toxemia and severe sepsis. Crit. Care Med. 35: 1139–44.
Altavilla D, Guarini S, Bitto A, et al. (2006) Activation of the cholinergic anti-inflammatory pathway reduces NF-kappab activation, blunts TNF-alpha production, and protects against splanchic artery occlusion shock. Shock 25:500–6.
Martin JN, Rose CH, Briery CM. (2006) Understanding and managing HELLP syndrome: the integral role of aggressive glucocorticoids for mother and child. Am. J. Obstet. Gynecol. 195:914–34.
Matchaba P, Moodley J. (2004) Corticosteroids for HELLP syndrome in pregnancy. Cochrane. Database. Syst. Rev. CD002076.
Isler CM, Barrilleaux PS, Magann EF, Bass JD, Martin JN. (2001) A prospective, randomized trial comparing the efficacy of dexamethasone and betamethasone for the treatment of antepar-tum HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome. Am. J. Obstet. Gynecol. 184:1332–7.
Askie LM, Duley L, Henderson-Smart DJ, Stewart LA. (2007) Antiplatelet agents for prevention of pre-eclampsia: a meta-analysis of individual patient data. Lancet 369:1791–8.
Acknowledgments
We would like to thank Jeanne Woods, Jean Farrell, and the staff of The New York Blood Center for their assistance with the collection of placentas. We acknowledge the assistance of Xu Ping Wang with the Q-PCR studies. This work was funded by a Translational Research Award to CNM and BR and NIGMS RO1GM07027-02 (CNM).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Dowling, O., Rochelson, B., Way, K. et al. Nicotine Inhibits Cytokine Production by Placenta Cells via NFκB: Potential Role in Pregnancy-Induced Hypertension. Mol Med 13, 576–583 (2007). https://doi.org/10.2119/2007-00067.Dowling
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/2007-00067.Dowling